Proteomic Studies Reveal Disrupted in Schizophrenia 1 as a Player in Both Neurodevelopment and Synaptic Function


 ,
, 

Abstract
1. Introduction
2. Results
2.1. Proteomic Analysis
2.2. Ingenuity Pathway
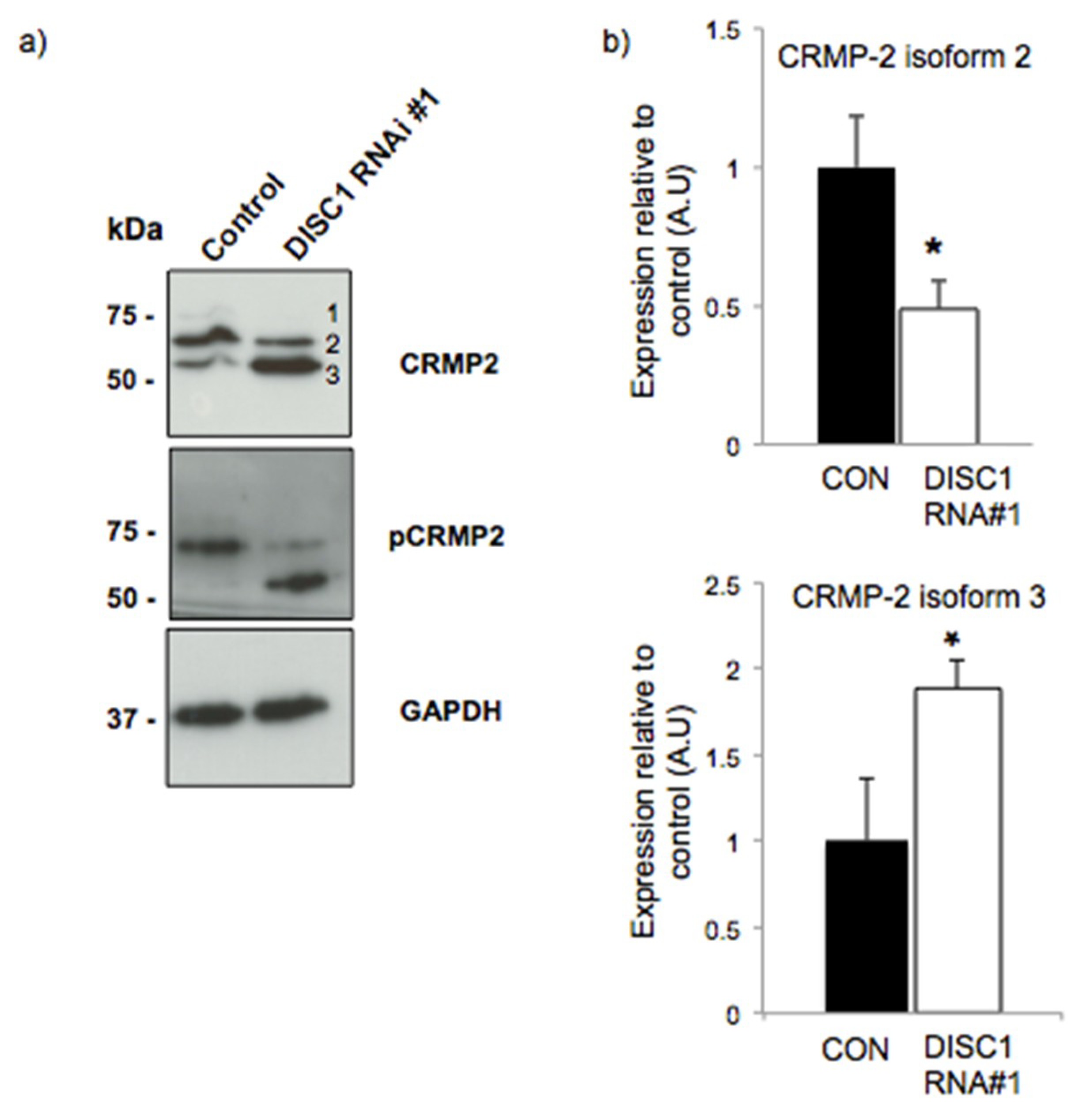
2.3. DISC1 Alters the Expression of Neurodevelopmental Related Proteins
2.4. DISC1 Alters the Expression of Synaptic Function Related Proteins
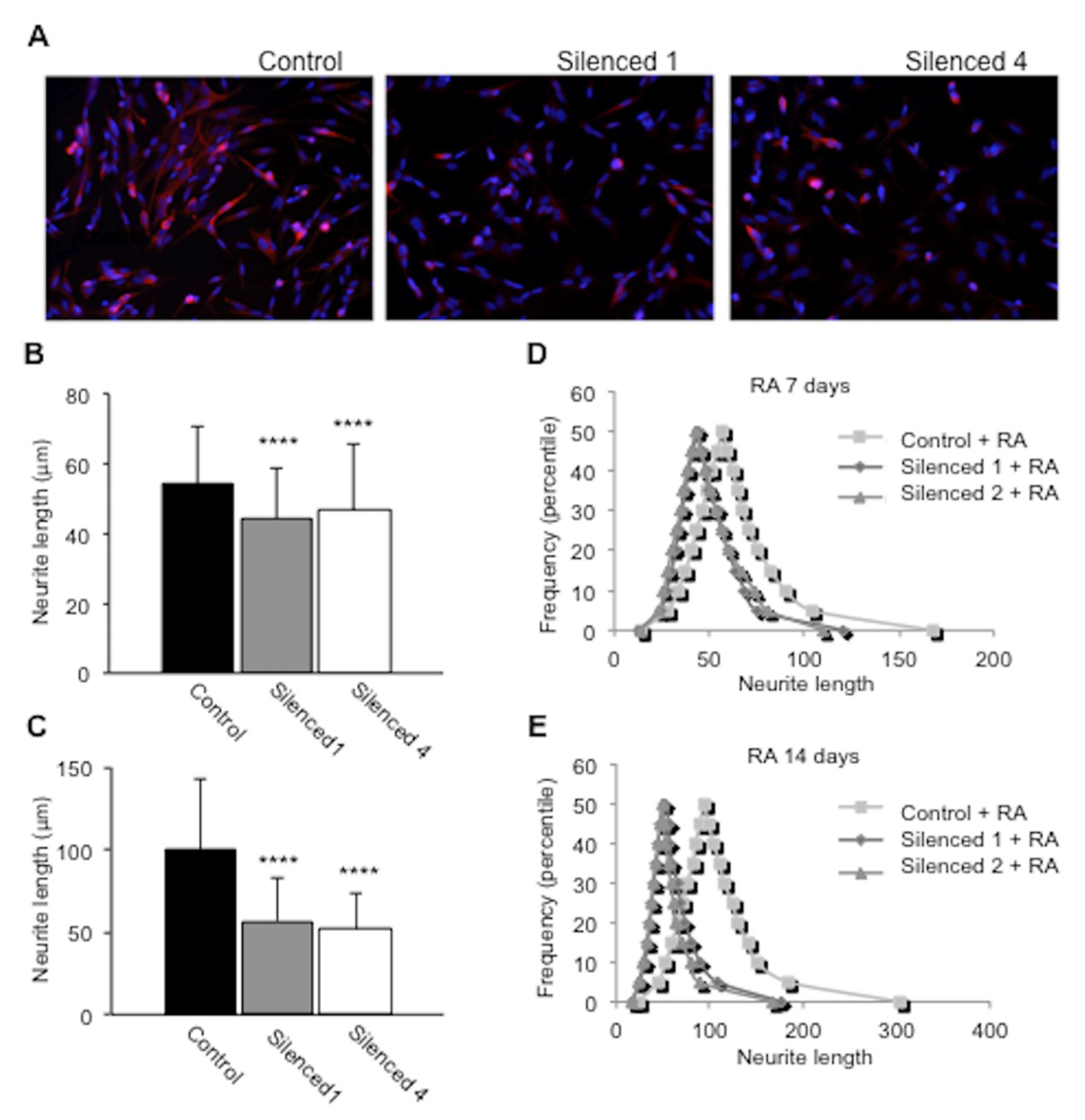
2.5. DISC1 Silenced SH-SY5Y Cells Show Impaired Neurite Outgrowth
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Culture
4.3. Ethics Statement
4.4. DISC1 Silencing
4.5. Sample Preparation for Proteomic Studies
4.6. Proteomic Studies
4.7. Differential Image Analysis
4.8. Mass Spectrometric Analysis
4.9. SDS-PAGE and Western Blotting
4.10. Ingenuity Pathway
4.11. Neurite Outgrowth Assays
4.12. Immunocytochemistry of SH-SY5Y Cells
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-DE | Two-dimensional electrophoresis |
| CRMP-2 | Collapsing response mediator protein 2 |
| DISC1 | Disrupted in Schizophrenia 1 |
| MAP1B | Microtubule associated binding protein 1 |
| MUNC18 | Mammalian uncoordinated-18 |
| MALDI | Matrix-assisted laser desorption ionization |
| MS | Mass Spectrometry |
| NCS-1 | Neural calcium sensor 1 |
| SM | Sec1/Munc18-like proteins |
| SNARE | (Soluble NSF Attachment Protein) Receptor |
| WB | Western blot |
References
- St. Clair, D.; Blackwood, D.; Muir, W.; Walker, M.; Carothers, A.; Spowart, G.; Gosden, C.; Evans, H.J. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990, 336, 13–16. [Google Scholar] [CrossRef]
- Millar, J.K. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000, 9, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.A.; Sawa, A.; Holmes, S.E.; Ross, C.A.; DeLisi, L.E.; Margolis, R.L. A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol. Psychiatry 2005, 10, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Liu, C.Y.; Zhang, F.; Duan, X.; Wen, Z.; Song, J.; Feighery, E.; Lu, B.; Rujescu, D.; St Clair, D.; et al. Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 2012, 148, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Ge, X.; Frank, C.L.; Madison, J.M.; Koehler, A.N.; Doud, M.K.; Tassa, C.; Berry, E.M.; Soda, T.; Singh, K.K.; et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 2009, 136, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Lepagnol-Bestel, A.M.; Kvajo, M.; Karayiorgou, M.; Simonneau, M.; Gogos, J.A. A Disc1 mutation differentially affects neurites and spines in hippocampal and cortical neurons. Mol. Cell. Neurosci. 2013, 54, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, W.; Huang, S.; Song, J.; Kim, J.Y.; Tian, X.; Kang, E.; Sano, Y.; Liu, C.; Balaji, J.; et al. MTOR Inhibition Ameliorates Cognitive and Affective Deficits Caused by Disc1 Knockdown in Adult-Born Dentate Granule Neurons. Neuron 2013, 77, 647–654. [Google Scholar] [CrossRef]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef]
- Wang, Q.; Charych, E.I.; Pulito, V.L.; Lee, J.B.; Graziane, N.M.; Crozier, R.A.; Revilla-Sanchez, R.; Kelly, M.P.; Dunlop, A.J.; Murdoch, H.; et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol. Psychiatry 2011, 16, 1006–1023. [Google Scholar] [CrossRef]
- Tsuboi, D.; Kuroda, K.; Tanaka, M.; Namba, T.; Iizuka, Y.; Taya, S.; Shinoda, T.; Hikita, T.; Muraoka, S.; Iizuka, M.; et al. Disrupted-in-schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat. Neurosci. 2015, 18, 698–707. [Google Scholar] [CrossRef]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.S.; Yoon, K.J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, Y.; Tomoda, T.; Kleiderlein, J.; Kamiya, A.; Bord, L.; Fujii, K.; Okawa, M.; Yamada, N.; Hatten, M.E.; Snyder, S.H.; et al. Disrupted-in-Schizophrenia-1 (DISC-1): Mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc. Natl. Acad. Sci. USA 2003, 100, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Ming, G.-L.; Song, H.; Waga, C.; Enomoto, A.; Kaibuchi, K.; Kohsaka, S.; Uchino, S. NMDA receptor regulates migration of newly generated neurons in the adult hippocampus via Disrupted-In-Schizophrenia 1 (DISC1). J. Neurochem. 2011, 118, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Kubo, K.; Tomoda, T.; Takaki, M.; Youn, R.; Ozeki, Y.; Sawamura, N.; Park, U.; Kudo, C.; Okawa, M.; et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 2005, 7, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, K.; Kamiya, A.; Oh, E.C.; Kanki, H.; Seshadri, S.; Robinson, J.F.; Murdoch, H.; Dunlop, A.J.; Kubo, K.I.; Furukori, K.; et al. DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 2011, 473, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Camargo, L.M.; Collura, V.; Rain, J.-C.; Mizuguchi, K.; Hermjakob, H.; Kerrien, S.; Bonnert, T.P.; Whiting, P.J.; Brandon, N.J. Disrupted in Schizophrenia 1 Interactome: Evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry 2007, 12, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.K.; Pickard, B.S.; Mackie, S.; James, R.; Christie, S.; Buchanan, S.R.; Malloy, M.P.; Chubb, J.E.; Huston, E.; Baillie, G.S.; et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 2005, 310, 1187–1191. [Google Scholar] [CrossRef]
- Enomoto, A.; Asai, N.; Namba, T.; Wang, Y.; Kato, T.; Tanaka, M.; Tatsumi, H.; Taya, S.; Tsuboi, D.; Kuroda, K.; et al. Roles of Disrupted-In-Schizophrenia 1-Interacting Protein Girdin in Postnatal Development of the Dentate Gyrus. Neuron 2009, 63, 774–787. [Google Scholar] [CrossRef]
- Brandon, N.J.; Sawa, A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 2011, 12, 707–722. [Google Scholar] [CrossRef]
- Sialana, F.J.; Wang, A.-L.; Fazari, B.; Kristofova, M.; Smidak, R.; Trossbach, S.V.; Korth, C.; Huston, J.P.; de Souza Silva, M.A.; Lubec, G. Quantitative Proteomics of Synaptosomal Fractions in a Rat Overexpressing Human DISC1 Gene Indicates Profound Synaptic Dysregulation in the Dorsal Striatum. Front. Mol. Neurosci. 2018, 11, 26. [Google Scholar] [CrossRef]
- Xia, M.; Broek, J.A.C.; Jouroukhin, Y.; Schoenfelder, J.; Abazyan, S.; Jaaro-Peled, H.; Sawa, A.; Bahn, S.; Pletnikov, M. Cell Type-Specific Effects of Mutant DISC1: A Proteomics Study. Mol. Neuropsychiatry 2016, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Taya, S.; Shinoda, T.; Tsuboi, D.; Asaki, J.; Nagai, K.; Hikita, T.; Kuroda, S.; Kuroda, K.; Shimizu, M.; Hirotsune, S.; et al. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J. Neurosci. 2007, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Nagai, J.; Baba, R.; Ohshima, T. CRMPs Function in Neurons and Glial Cells: Potential Therapeutic Targets for Neurodegenerative Diseases and CNS Injury. Mol. Neurobiol. 2017, 54, 4243–4256. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.F.; Strittmatter, S.M. The CRMP family of proteins and their role in Sema3A signaling. In Semaphorins: Receptor and Intracellular Signaling Mechanisms; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2007. [Google Scholar]
- Yamashita, N.; Goshima, Y. Collapsin response mediator proteins regulate neuronal development and plasticity by switching their phosphorylation status. Mol. Neurobiol. 2012, 45, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Ujike, H.; Sakai, A.; Takaki, M.; Imamura, T.; Tanaka, Y.; Kuroda, S. The Human Dihydropyrimidinase-Related Protein 2 Gene on Chromosome 8p21 Is Associated with Paranoid-Type Schizophrenia. Biol. Psychiatry 2003, 53, 571–576. [Google Scholar] [CrossRef]
- Johnston-Wilson, N.L.; Sims, C.D.; Hofmann, J.P.; Anderson, L.; Shore, A.D.; Torrey, E.F.; Yolken, R.H. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol. Psychiatry 2000, 5, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 2011, 43, 180–191. [Google Scholar] [CrossRef]
- Wakatsuki, S.; Saitoh, F.; Araki, T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 2011, 13, 1415–1423. [Google Scholar] [CrossRef]
- Zhang, Z.; Ottens, A.K.; Sadasivan, S.; Kobeissy, F.H.; Fang, T.; Hayes, R.L.; Wang, K.K. Calpain-mediated collapsin response mediator protein-1, -2, and -4 proteolysis after neurotoxic and traumatic brain injury. J. Neurotrauma 2007, 24, 460–472. [Google Scholar] [CrossRef]
- Zhang, J.-N.; Michel, U.; Lenz, C.; Friedel, C.C.; Köster, S.; d’Hedouville, Z.; Tönges, L.; Urlaub, H.; Bähr, M.; Lingor, P.; et al. Calpain-mediated cleavage of collapsin response mediator protein-2 drives acute axonal degeneration. Sci. Rep. 2016, 6, 37050. [Google Scholar] [CrossRef]
- Rogemond, V.; Auger, C.; Giraudon, P.; Becchi, M.; Auvergnon, N.; Belin, M.F.; Honnorat, J.; Moradi-Améli, M. Processing and nuclear localization of CRMP2 during brain development induce neurite outgrowth inhibition. J. Biol. Chem. 2008, 283, 14751–14761. [Google Scholar] [CrossRef] [PubMed]
- Alabi, A.A.; Tsien, R.W. Perspectives on Kiss-and-Run: Role in Exocytosis, Endocytosis, and Neurotransmission. Annu. Rev. Physiol. 2013, 75, 393–422. [Google Scholar] [CrossRef] [PubMed]
- Dulubova, I.; Khvotchev, M.; Liu, S.; Huryeva, I.; Su, T.C. Munc18-1 binds directly to the neuronal SNARE complex. Proc. Natl. Acad. Sci. USA 2007, 104, 2697–2702. [Google Scholar] [CrossRef]
- von Mollard, G.F.; Stahl, B.; Li, C.; Südhof, T.C.; Jahn, R. Rab proteins in regulated exocytosis. Trends Biochem. Sci. 1994, 19, 164–168. [Google Scholar] [CrossRef]
- Søgaard, M.; Tani, K.; Ye, R.R.; Geromanos, S.; Tempst, P.; Kirchhausen, T.; Rothman, J.E.; Söllner, T. A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell 1994, 78, 937–948. [Google Scholar] [CrossRef]
- Ramos, A.; Rodríguez-Seoane, C.; Rosa, I.; Trossbach, S.V.; Ortega-Alonso, A.; Tomppo, L.; Ekelund, J.; Veijola, J.; Järvelin, M.R.; Alonso, J.; et al. Neuropeptide precursor VGF is genetically associated with social anhedonia and underrepresented in the brain of major mental illness: Its downregulation by DISC1. Hum. Mol. Genet. 2014, 23, 5859–5865. [Google Scholar] [CrossRef]
- Niwa, M.; Cash-Padgett, T.; Kubo, K.; Saito, A.; Ishii, K.; Sumitomo, A.; Taniguchi, Y.; Ishizuka, K.; Jaaro-Peled, H.; Tomoda, T.; et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: No-More Disrupted-in-Schizophrenia. Mol. Psychiatry 2016, 21, 1488–1489. [Google Scholar] [CrossRef]
- Bader, V.; Tomppo, L.; Trossbach, S.V.; Bradshaw, N.J.; Prikulis, I.; Rutger Leliveld, S.; Lin, C.Y.; Ishizuka, K.; Sawa, A.; Ramos, A.; et al. Proteomic, genomic and translational approaches identify CRMP1 for a role in schizophrenia and its underlying traits. Hum. Mol. Genet. 2012, 21, 4406–4418. [Google Scholar] [CrossRef]
- McLean, C.K.; Narayan, S.; Lin, S.Y.; Rai, N.; Chung, Y.; Hipolito, M.M.S.; Cascella, N.G.; Nurnberger, J.I.; Ishizuka, K.; Sawa, A.S.; et al. Lithium-associated transcriptional regulation of CRMP1 in patient-derived olfactory neurons and symptom changes in bipolar disorder. Transl. Psychiatry 2018, 8, 81. [Google Scholar] [CrossRef]
- Pletnikov, M.V.; Ayhan, Y.; Nikolskaia, O.; Xu, Y.; Ovanesov, M.V.; Huang, H.; Mori, S.; Moran, T.H.; Ross, C.A. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 2008, 13, 173–186. [Google Scholar] [CrossRef]
- Miyoshi, K.; Honda, A.; Baba, K.; Taniguchi, M.; Oono, K.; Fujita, T.; Kuroda, S.; Katayama, T.; Tohyama, M. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol. Psychiatry 2003, 8, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Hirota, Y.; Armstrong, B.; Sawa, A.; Tomoda, T. DISC1 regulates synaptic vesicle transport via a lithium-sensitive pathway. Neurosci. Res. 2011, 71, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Ottis, P.; Bader, V.; Trossbach, S.V.; Kretzschmar, H.; Michel, M.; Leliveld, S.R.; Korth, C. Convergence of two independent mental disease genes on the protein level: Recruitment of dysbindin to cell-invasive disrupted-in-schizophrenia 1 aggresomes. Biol. Psychiatry 2011, 70, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Castaño, Z.; Gordon-Weeks, P.R.; Kypta, R.M. The neuron-specific isoform of glycogen synthase kinase-3beta is required for axon growth. J. Neurochem. 2010, 113, 117–130. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Function | Protein | Fold Change | p Value |
|---|---|---|---|
| Neurite outgrowth or neural migration | Dihydropyrimidinase-related protein 5 (CRMP-5) | 2.59 | 3.066 × 10−5 |
| Dihydropyrimidinase-related protein 3 (CRMP-3) | 3.12, 2.23 | 1.469 × 10−4, 2.894 × 10−4 | |
| Dihydropyrimidinase-relatedprotein 2 (CRMP-2) | 2.21, 2.03 | 2.457 × 10−4, 0.059 | |
| Dihydropyrimidinase-related protein 1 (CRMP-1) | 2.10 | 9.180 × 10−5 | |
| Tubulin alpha-1A chain (TBA1A) | 2.01,2.99,2.13 | 0.0043, 1.326 × 10−4, 4.067 × 10−4 | |
| Tubulin beta-2B chain (TBB2B) | Inf, 2.42 | 0.0065, 0.0156 | |
| Microtubule-associated protein (MAP1B) | 2.04, 2.20,3.03 | 3.661 × 10−7, 2.894 × 10−4, 2.717 × 10−5 | |
| 14-3-3 protein epsilon (14-3-3ε) | 2.67, 3.19 | 6.713 × 10−5, 1.854 × 10−4 | |
| 14-3-3 protein zeta/delta (14-3-3θ/Δ) | 8.42, 2.63, 3.26, 3.88, 3.89, 6.81, 3.82, 6.20 | 3.028 × 10−6, 2.334 × 10−4, 1.579 × 10−4 9.307 × 10−4, 0.021, 2.080 × 10−5, 0.0022, 2.572 × 10−6 | |
| 14-3-3 protein gamma (14-3-3γ) | 3.24, 2.30 | 6.104 × 10−5, 0.0084 | |
| Platelet-activating factor acetylhydrolase IB (Lis-1) | 2.48 | 0.0049 | |
| Stathmin (STMN) | 2.12, 4.64 | 8.206 × 10−4, 1.021 × 10−4 | |
| Syntaxin-7 (STX7) | 2.13 | 0.0022 | |
| Tropomyosin alpha-3 chain (TPM3) | 2.88 | 0.0115 | |
| Actin, cytoplasmic 2 (ACTG) | 4.94, 2.95 | 4.398 × 10−5, 1.081 × 10−5 | |
| Cadherin-13 (CAD13) | 2.31 | 0.0108 | |
| Calreticulin (CALR) | 2.60 | 0.0088 | |
| Septin-5 (SEPT5) | 2.15 | 0.0034 | |
| Apolipoprotein A-I (APOA1) | 2.41 | 9.215 × 10−5 | |
| Dynamin 1 (DYN1) | 4.33 | 1.440 × 10−4 | |
| Dynamin 1 (DYN1) | Dihydropyrimidinase-related protein 5 (CRMP-5) | 2.59 | 3.066 × 10−5 |
| Dihydropyrimidinase-relatedprotein 2 (CRMP-2) | 2.21, 2.03 | 2.457 × 10−4, 0.059 | |
| Microtubule-associated protein (MAP1B) | 2.04, 2.20, 3.03 | 3.661 × 10−7, 2.894 × 10−4, 2.717 × 10−5 | |
| Transitional endoplasmic Reticulum ATPase (TERA) | 2.21 | 5.537 × 10−4 | |
| Stathmin (STMN) | 2.12, 4.64 | 8.206 × 10−4, 1.021 × 10−4 | |
| Syntaxin-binding protein 1 (STXB1) | 3.43 | 0.0010 | |
| Syntaxin-7 (STX7) | 2.13 | 0.0022 | |
| Ras-related protein Rab-1A (RAB1A) | 2.01 | 0.0023 | |
| Ras-related protein Rab-2A (RAB2A) | 2.43 | 4.164 × 10−4 | |
| Ras-related protein Rab-11B (RB11B) | 3.25 | 0.0305 | |
| Ras-related protein Rab-18 (RAB18) | 3.23 | 5.527 × 10−4 | |
| Cadherin-13 (CAD13) | 2.31 | 0.0108 | |
| Rho GDP-dissociation inhibitor 2 (GDIR2) | 2.28 | 1.061 × 10−4 | |
| Phosphatidylethanolamine-binding protein 1 (HCNP) | 3.84, 6.49 | 4.081 × 10−4, 2.377 × 10−4 | |
| Calreticulin (CALR) | 2.60 | 0.0088 | |
| Adaptin ear-binding coat-associated protein 1 (NECP1) | 2.51 | 6.028 × 10−5 | |
| Neuronal calcium sensor 1 (NCS1) | 2.24 | 9.215 × 10−5 | |
| Dynamin 1 (DYN1) | 4.33 | 1.440 × 10−4 |
| Name | p Value | Proteins |
|---|---|---|
| 14-3-3 mediated signaling | 4.99 × 10−7 | TUBA1A, 14-3-3G, TUBB2B, PDIA3,1 4-3-3E, 14-3-3Z |
| Semaphorin signaling in neurons | 5.28 × 10−6 | CRMP3, CRMP1, CRMP2, CRMP5 |
| Remodeling of epithelial adherent junctions | 1.52 × 10−5 | DNM1L, TUBA1A, ACTG1, TUBB2B |
| Cell cycle: G2/M DNA damage checkpoint regulation | 1.75 × 10−4 | 14-3-3G, 14-3-3E, 14-3-3Z |
| PI3K/AKT signaling | 1.87 × 10−4 | 14-3-3G, 14-3-3E, HSP90AA1, 14-3-3Z |
| Name | p Value | Proteins |
|---|---|---|
| Outgrowth of cells | 3.94 × 10−8 | DNM1L, TUBA1A, HBA1/HBA2, CRMP3, MAP1B, SET, PDIA3, CRMP2, 14-3-3G, HSP90AA1, CRMP5 |
| Patterning of dendrites | 9.56 × 10−8 | CRMP1, CRMP2, GDA |
| Outgrowth of neurites | 1.94 × 10−7 | DNM1L, TUBA1A, HBA1/HBA2, DPYSL3, MAP1B, SET, PDIA3, CRMP2, 14-3-3Z, CRMP5 |
| Branching of neurons | 2.53 × 10−7 | DNM1L, HNRNPK, CRMP3, MAP1B, PDIA3, CRMP1, CRMP2, CRMP5, GDA |
| Organization of cytoplasm | 7.08 × 10−7 | CDH13, RAB2A, HNRNPK, CRMP1, CRMP2, CRMP5, STMN1, CALR, TPM3, DNM1L, ACTG1, PEX5, CRMP3, MAP1B, RAB1A, PDIA3, HSP90AA1, GDA |
| Fibrogenesis | 8.53 × 10−7 | CALR, CDH13, TPM3, ACTG1, CRMP3, MAP1B, APOA1, CRMP2, GDA, STMN1 |
| Endocytosis | 1.39 × 10−6 | CALR, CDH13, HNRNPK, MAP1B, RAB1A, APOA1, CRMP2, VCP, HSP90AA1, NECAP1 |
| Neuritogenesis | 2.09 × 10−6 | DNM1L, HNRNPK, CRMP3, MAP1B, PDIA3, CRMP1, CRMP2, HSP90AA1, CRMP5, GDA, STMN1 |
| Branching of neurites | 2.50 × 10−6 | DNM1L, HNRNPK, MAP1B, PDIA3, CRMP1, CRMP2, CRMP5, GDA |
| Microtubule dynamics | 3.39 × 10−6 | CDH13, RAB2A, HNRNPK, CRMP1, CRMP2, CRMP5, STMN1, TPM3, DNM1L, ACTG1, CRMP3, MAP1B, PDIA3, HSP90AA1, GDA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, A.; Rodríguez-Seoane, C.; Rosa, I.; Gorroño-Etxebarria, I.; Alonso, J.; Veiga, S.; Korth, C.; Kypta, R.M.; García, Á.; Requena, J.R. Proteomic Studies Reveal Disrupted in Schizophrenia 1 as a Player in Both Neurodevelopment and Synaptic Function. Int. J. Mol. Sci. 2019, 20, 119. https://doi.org/10.3390/ijms20010119
Ramos A, Rodríguez-Seoane C, Rosa I, Gorroño-Etxebarria I, Alonso J, Veiga S, Korth C, Kypta RM, García Á, Requena JR. Proteomic Studies Reveal Disrupted in Schizophrenia 1 as a Player in Both Neurodevelopment and Synaptic Function. International Journal of Molecular Sciences. 2019; 20(1):119. https://doi.org/10.3390/ijms20010119
Chicago/Turabian StyleRamos, Adriana, Carmen Rodríguez-Seoane, Isaac Rosa, Irantzu Gorroño-Etxebarria, Jana Alonso, Sonia Veiga, Carsten Korth, Robert M. Kypta, Ángel García, and Jesús R. Requena. 2019. "Proteomic Studies Reveal Disrupted in Schizophrenia 1 as a Player in Both Neurodevelopment and Synaptic Function" International Journal of Molecular Sciences 20, no. 1: 119. https://doi.org/10.3390/ijms20010119
APA StyleRamos, A., Rodríguez-Seoane, C., Rosa, I., Gorroño-Etxebarria, I., Alonso, J., Veiga, S., Korth, C., Kypta, R. M., García, Á., & Requena, J. R. (2019). Proteomic Studies Reveal Disrupted in Schizophrenia 1 as a Player in Both Neurodevelopment and Synaptic Function. International Journal of Molecular Sciences, 20(1), 119. https://doi.org/10.3390/ijms20010119