A Novel Approach Based on a Weighted Interactive Network to Predict Associations of MiRNAs and Diseases



Abstract
1. Introduction
2. Results and Case Studies
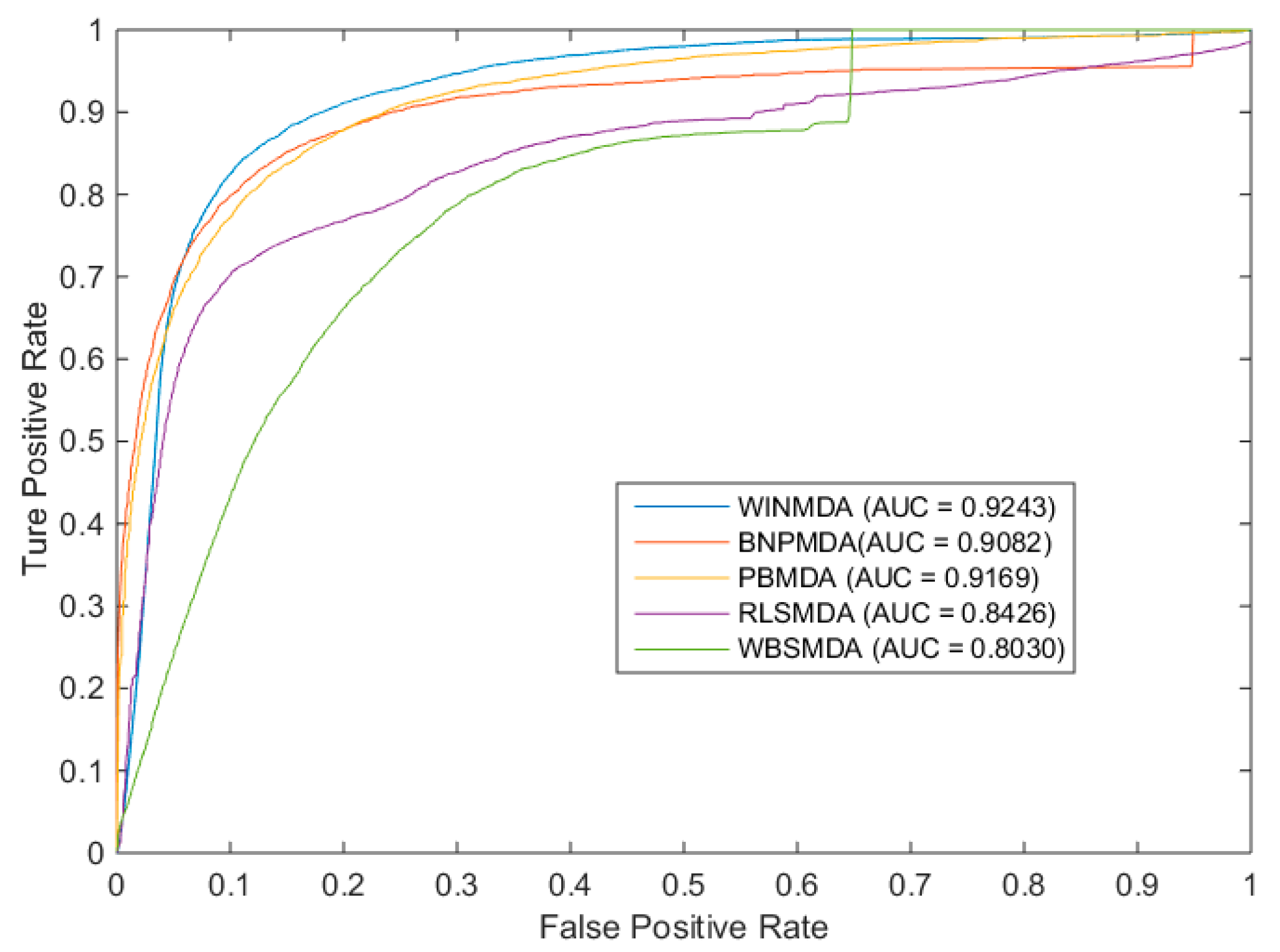
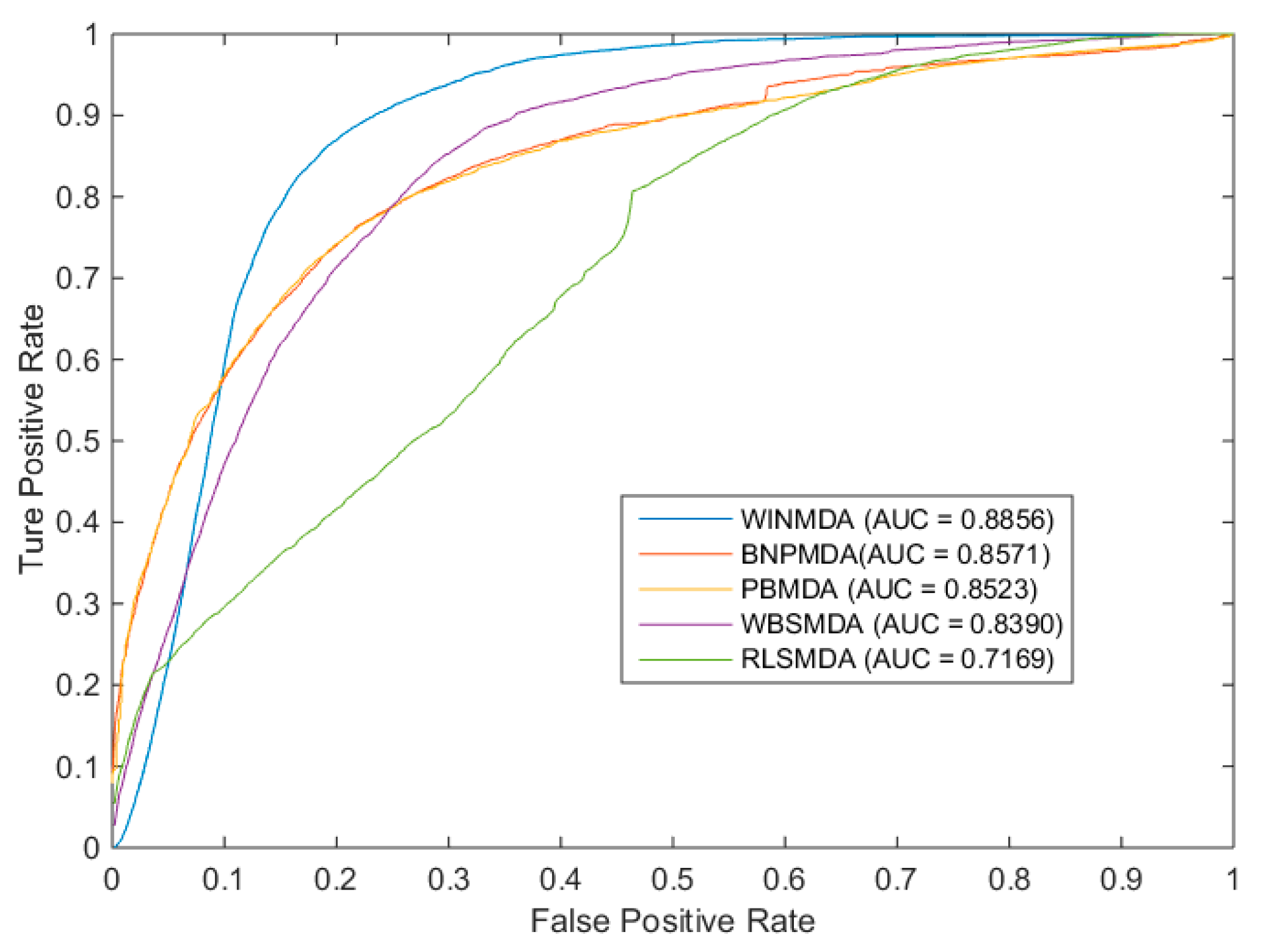
2.1. Comparison with Existing State-of-the-Art Methods
2.2. Evaluation of the Effects of Parameters
2.2.1. Effects of Parameter T
2.2.2. Effects of Parameter w
2.3. Case Studies
3. Discussion
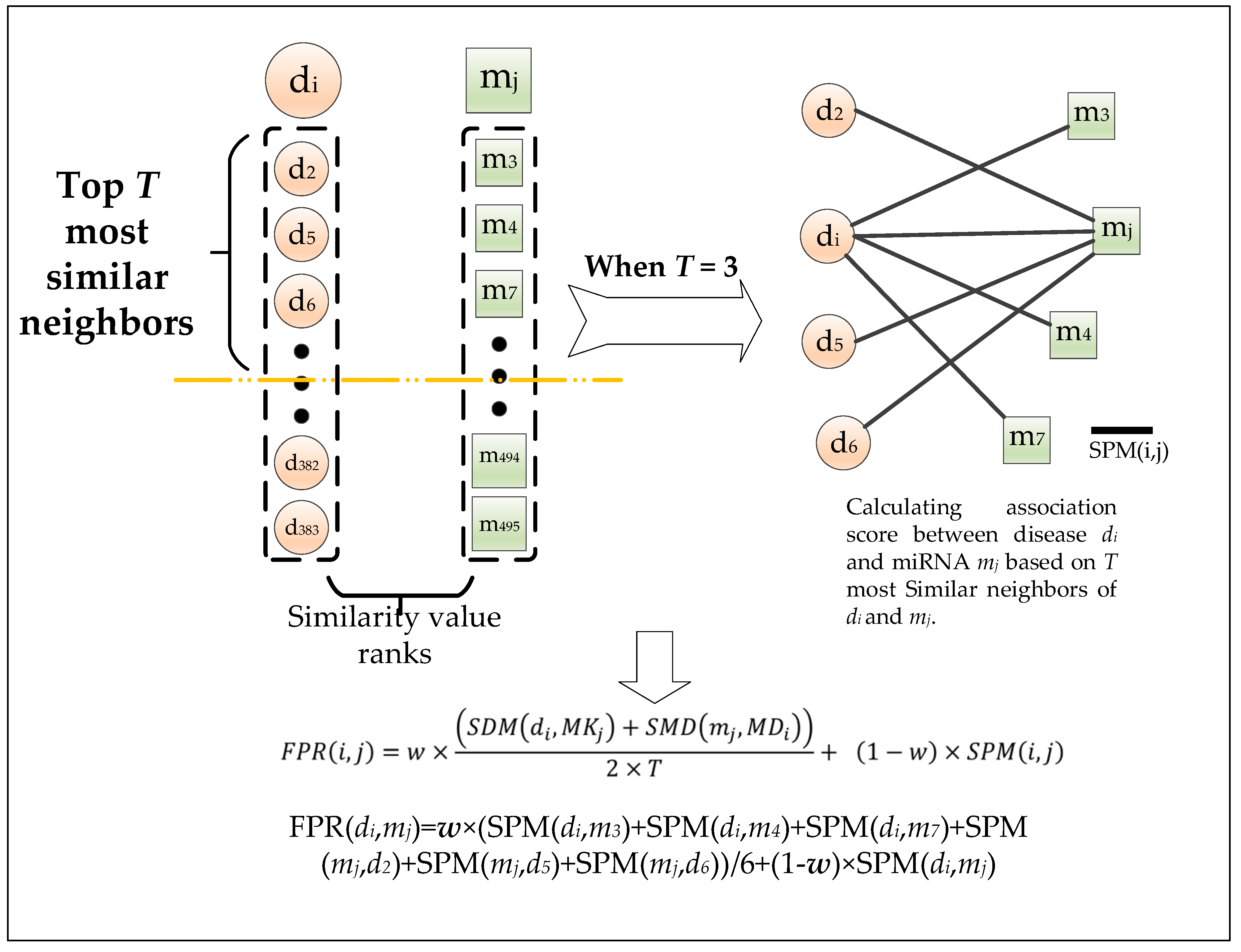
4. Materials and Methods
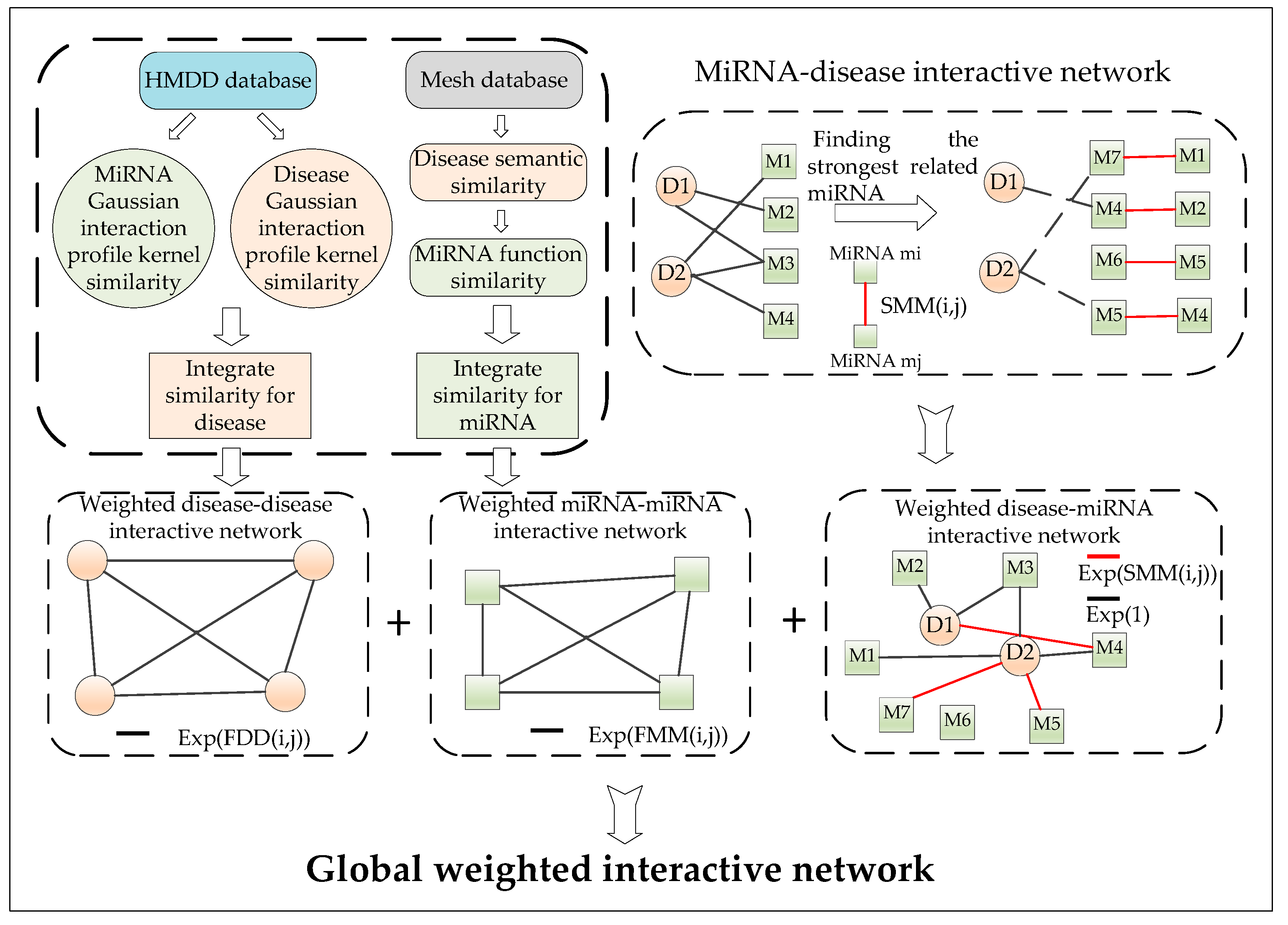
4.1. Construction of the miRNA–Disease Interactive Network
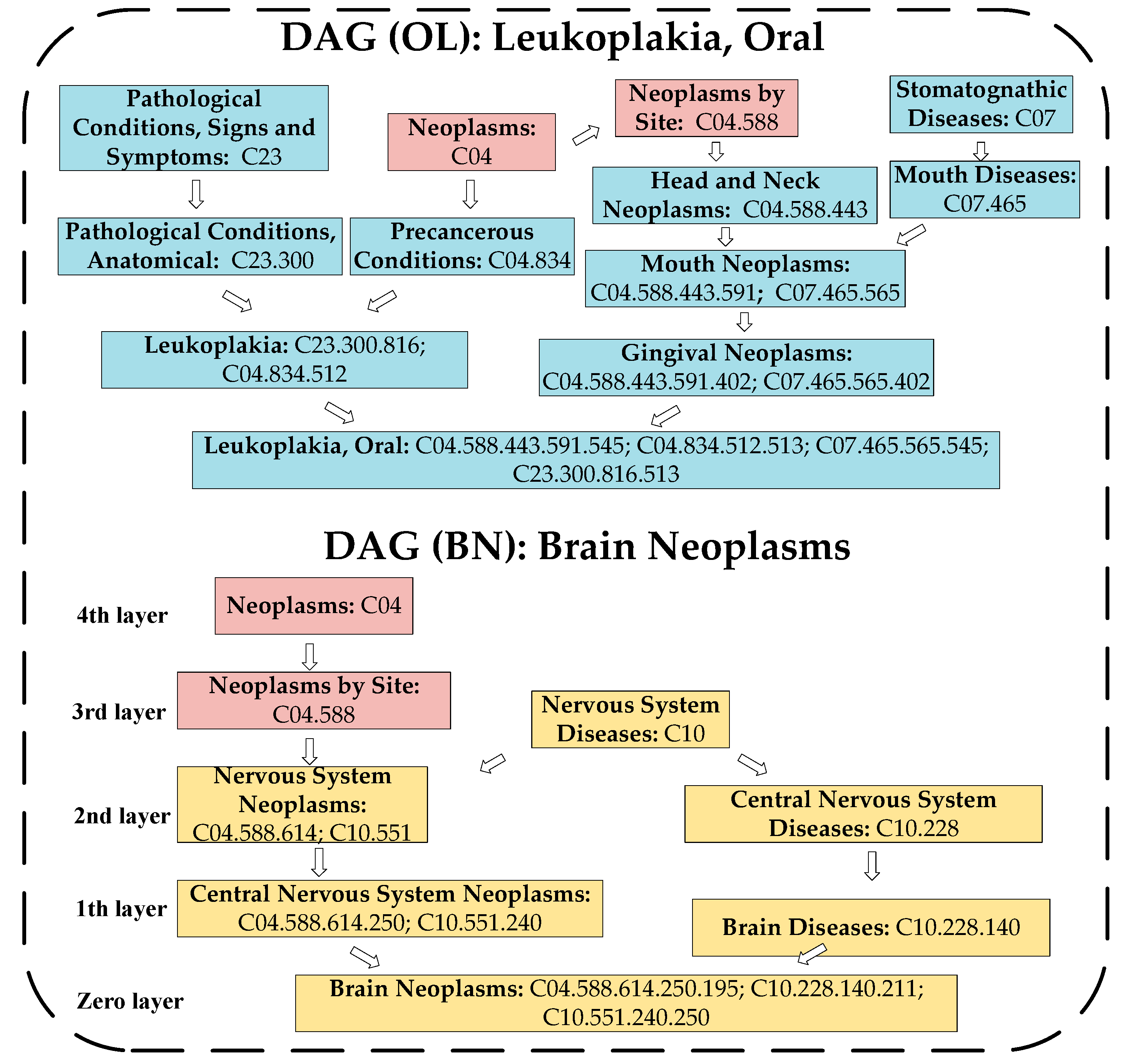
4.2. Calculation of the Disease Semantic Similarity
4.3. Calculation of the miRNA Functional Similarity
4.4. Disease Gaussian Interaction Profile Kernel Similarity Measurement
4.5. MicroRNA Gaussian Interaction Profile Kernel Similarity Measurement
4.6. Calculation of the Integrated Similarity
4.7. Construction of the Weighted Interactive Network
4.8. Calculation of the Shortest Path Based on the Weighted Interactive Network
4.9. Calculation of the Shortest Path Based on the Weighted Interactive Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| miRNAs | MicroRNAs |
| WINMDA | Weighted interactive network for discovering potential miRNA–disease associations |
| CXCL8 | C–X–C ligand 8 |
| ROC | receiver operating characteristics |
| AUC | Area of ROC under the curve |
| LOOCV | Leave-one-out cross-validation |
| OC | Ovarian cancer |
| ncRNAs | Non-coding RNAs |
| MeSH | Medical Subject Headings |
| DAGs | Direct acyclic graphs |
| HMDD | Human microRNA Disease Database |
| LncRNA | long non-coding RNA |
| NEAT 1 | nuclear paraspeckle assembly transcript 1 |
| SNPs | single-nucleotide polymorphisms |
| dbDEMC | Differentially Expressed miRNAs in Human Cancers |
References
- Chen, X.; Yan, G.Y. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, J.; Song, L.; Zeng, X.; Wang, G. Similarity computation strategies in the microRNA-disease network: A survey. Brief. Funct. Genom. 2016, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Khella, H.W.; Scorilas, A.; Mozes, R.; Mirham, L.; Lianidou, E.; Krylov, S.N.; Lee, J.Y.; Ordon, M.; Stewart, R.; Jewett, M.A. Low expression of miR-126 is a prognostic marker for metastatic clear cell renal cell carcinoma. Am. J. Pathol. 2015, 185, 693–703. [Google Scholar] [CrossRef]
- Nan, D.; Wu, H.; Tao, T.; Peng, E. NEAT1 regulates cell proliferation and apoptosis of ovarian cancer by miR-34a-5p/BCL2. Oncotargets Ther. 2017, 10, 4905–4915. [Google Scholar] [CrossRef]
- Xu, X.; Wu, X.; Jiang, Q.; Sun, Y.; Liu, H.; Chen, R.; Wu, S. Downregulation of microRNA-1 and microRNA-145 contributes synergistically to the development of colon cancer. Int. J. Mol. Med. 2015, 36, 1630–1638. [Google Scholar] [CrossRef]
- Chen, B.; Duan, L.; Yin, G.; Jing, T.; Jiang, X. Simultaneously expressed miR-424 and miR-381 synergistically suppress the proliferation and survival of renal cancer cells—Cdc2 activity is up-regulated by targeting WEE1. Clinics 2013, 68, 825–833. [Google Scholar] [CrossRef]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2017, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Liu, L.; Lü, L.; Zou, Q. Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 2018, 34, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Ping, P.; Wang, L.; Kuang, L.; Ye, S.; Mfb, I.; Pei, T. A Novel method for lncRNA-disease association prediction based on an lncRNA-disease association network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ping, P.; Wang, L.; Kuang, L.; Li, X.; Wu, Z. A Novel probability model for lncRNA⁻disease association prediction based on the Naïve Bayesian Classifier. Genes 2018, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffithsjones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.; Li, J. Predicting miRNA–disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z.H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2017, 18, 558. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, Z. ELLPMDA: Ensemble learning and link prediction for miRNA-disease association prediction. RNA Biol. 2018, 15, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, D.H.; You, Z.H. A heterogeneous label propagation approach to explore the potential associations between miRNA and disease. J. Transl. Med. 2018, 16, 348. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, C.C.; Yin, J.; You, Z.H. Novel human miRNA-disease association inference based on random forest. Mol. Ther. Nucleic Acids 2018, 13, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cheng, J.Y.; Yin, J. Predicting microRNA-disease associations using bipartite local models and hubness-aware regression. RNA Biol. 2018, 15, 1192–1205. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.X.; Yan, G.Y. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef]
- Chen, X.; Yan, G.Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014, 4, 5501. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and between score for miRNA-disease association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef]
- Zou, Q.; Li, J.; Hong, Q.; Lin, Z.; Wu, Y.; Shi, H.; Ying, J. Prediction of microRNA-disease associations based on social network analysis methods. Biomed. Res. Int. 2015, 2015, 810514. [Google Scholar] [CrossRef]
- You, Z.H.; Huang, Z.A.; Zhu, Z.; Yan, G.Y.; Li, Z.W.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L. LRSSLMDA: Laplacian regularized sparse subspace learning for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005912. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Kuang, L.; Wang, L.; Ping, P.; Xuan, Z.; Pei, T.; Wu, Z. Prediction of microRNA-disease associations based on distance correlation set. BMC Bioinform. 2018, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, L.; Xie, D.; Zhao, Q. EGBMMDA: Extreme gradient boosting machine for miRNA-disease association prediction. Cell Death Dis. 2018, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.H.; Liu, H. BNPMDA: Bipartite network projection for miRNA-disease association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.D.; Jiang, L.H.; Sun, D.W.; Li, J.; Ji, Z.L. The role of mir-130a in cancer. Breast Cancer 2017, 24, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Depke, J.L.; Onitilo, A.A. Coalition building and the intervention wheel to address breast cancer screening in hmong women. Clin. Med. Res. 2011, 9, 1–6. [Google Scholar] [CrossRef]
- Finley, J.W.; Davis, C.D.; Feng, Y. Selenium from high selenium broccoli protects rats from colon cancer. J. Nutr. 2000, 130, 2384. [Google Scholar] [CrossRef]
- Bakitas, M.; Ahles, T.A.; Skalla, K.; Brokaw, F.C.; Byock, I.; Hanscom, B.; Lyons, K.D.; Hegel, M.T. Proxy perspectives about end-of-life care for Person’s with cancer. Cancer 2010, 112, 1854–1861. [Google Scholar] [CrossRef]
- Heath, D.D.; Devlin, R.H.; Heath, J.W.; Iwama, G.K. Genetic, environmental and interaction effects on the incidence of jacking in Oncorhynchus tshawytscha (chinook salmon). Heredity 1994, 72, 146–154. [Google Scholar] [CrossRef]
- Hollis, M.; Nair, K.; Vyas, A.; Chaturvedi, L.S.; Gambhir, S.; Vyas, D. MicroRNAs potential utility in colon cancer: Early detection, prognosis, and chemosensitivity. World J. Gastroenterol. 2015, 21, 8284–8292. [Google Scholar] [CrossRef]
- Gomes, S.E.; Simões, A.E.S.; Pereira, D.M.; Rui, E.C.; Rodrigues, C.M.P.; Borralho, P.M. miR-143 or miR-145 overexpression increases cetuximab-mediated antibody-dependent cellular cytotoxicity in human colon cancer cells. Oncotarget 2016, 7, 9368–9387. [Google Scholar] [CrossRef] [PubMed]
- Signs, S.A.; Fisher, R.C.; Tran, U.; Chakrabarti, S.; Sarvestani, S.K.; Xiang, S.; Liska, D.; Roche, V.; Lai, W.; Gittleman, H.R. Stromal miR-20a controls paracrine CXCL8 secretion in colitis and colon cancer. Oncotarget 2018, 9, 13048–13059. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ge, F.; Hu, B.; Wu, L.; Yang, H.; Wang, H. rs35301225 polymorphism in miR-34a promotes development of human colon cancer by deregulation of 3′UTR in E2F1 in Chinese population. Cancer Cell Int. 2017, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Eric, B.R.; Chen, Y.; Scull, B.; Kay, L.P.; Morgan, D. Molecular Imaging of Gastric Neoplasia with Near Infrared Fluorescent (NIRF) Activatable Probes. Mol. Imaging 2012, 11, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, X.; Han, Y.; Lu, Y.; Shang, Y.; Liu, C.; Li, T.; Jin, Z.; Fan, D.; Wu, K. Regulation of UHRF1 by miR-146a/b modulates gastric cancer invasion and metastasis. FASEB J. 2013, 27, 4929–4939. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Shi, Q.; Zhang, X.; Ding, Y.; Shan, L.; Shan, X.; Qian, J.; Zhou, X.; Huang, Z.; Zhu, W. Involvement of miR-143 in cisplatin resistance of gastric cancer cells via targeting IGF1R and BCL2. Tumour Biol. 2015, 36, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Marech, I.; Vacca, A.; Ranieri, G.; Gnoni, A.; Dammacco, F. Novel strategies in the treatment of castration-resistant prostate cancer (Review). Int. J. Oncol. 2012, 40, 1313–1320. [Google Scholar] [CrossRef]
- Salinas, C.A.; Tsodikov, A.; Ishakhoward, M.; Cooney, K.A. Prostate Cancer in Young Men: An Important Clinical Entity. Nat. Rev. Urol. 2014, 11, 317–323. [Google Scholar] [CrossRef]
- Chu, H.; Zhong, D.; Tang, J.; Li, J.; Xue, Y.; Tong, N.; Qin, C.; Yin, C.; Zhang, Z.; Wang, M. A functional variant in miR-143 promoter contributes to prostate cancer risk. Arch. Toxicol. 2016, 90, 403–414. [Google Scholar] [CrossRef]
- Wang, D.; Lu, G.; Shao, Y.; Xu, D. MiR-182 promotes prostate cancer progression through activating Wnt/β-catenin signal pathway. Biomed. Pharmacother. 2018, 99, 334–339. [Google Scholar] [CrossRef]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R. Senescent stroma promotes prostate cancer progression: The role of miR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; You, Z.H.; Yan, G.Y.; Gong, D.W. IRWRLDA: Improved random walk with restart for lncRNA-disease association prediction. Oncotarget 2016, 7, 57919–57931. [Google Scholar] [CrossRef] [PubMed]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LOOCV | 5-Fold Cross-Validation | 10-Fold Cross-Validation |
|---|---|---|
| 0.9243 | 0.9183 ± 0.0007 | 0.9200 ± 0.0004 |
| T | AUC | T | AUC |
|---|---|---|---|
| 1 | 0.9145 | 12 | 0.9241 |
| 2 | 0.9160 | 16 | 0.9243 |
| 5 | 0.9222 | 18 | 0.9242 |
| 8 | 0.9244 | 20 | 0.9188 |
| w | AUC | w | AUC |
|---|---|---|---|
| 0 | 0.9135 | 0.6 | 0.9243 |
| 0.1 | 0.9188 | 0.7 | 0.9239 |
| 0.2 | 0.9209 | 0.8 | 0.9222 |
| 0.3 0.4 0.5 | 0.9216 0.9235 0.9241 | 0.9 1 | 0.9189 0.9160 |
| Top 1–25 miRNAs | Evidence | Top 26–50 miRNAs | Evidence |
|---|---|---|---|
| hsa-mir-143 | dbDEMC and miR2Disease | hsa-let-7e | dbDEMC |
| hsa-mir-20a | dbDEMC and miR2Disease | hsa-mir-486 | 26895105 |
| hsa-mir-34a | dbDEMC and miR2Disease | hsa-mir-133b | dbDEMC and miR2Disease |
| hsa-mir-210 | dbDEMC | hsa-mir-200a | unconfirmed |
| hsa-mir-21 | dbDEMC and miR2Disease | hsa-mir-141 | dbDEMC and miR2Disease |
| hsa-mir-155 | dbDEMC and miR2Disease | hsa-let-7f | dbDEMC and miR2Disease |
| hsa-mir-95 | dbDEMC and miR2Disease | hsa-mir-29a | dbDEMC and miR2Disease |
| hsa-mir-146a | dbDEMC | hsa-mir-181a | dbDEMC and miR2Disease |
| hsa-mir-16 | dbDEMC | hsa-mir-9 | dbDEMC and miR2Disease |
| hsa-mir-125b | dbDEMC | hsa-mir-29b | dbDEMC and miR2Disease |
| hsa-mir-92a | unconfirmed | hsa-let-7c | dbDEMC |
| hsa-mir-31 | dbDEMC and miR2Disease | hsa-let-7d | dbDEMC |
| hsa-mir-223 | dbDEMC and miR2Disease | hsa-mir-196a | dbDEMC and miR2Disease |
| hsa-mir-221 | dbDEMC and miR2Disease | hsa-let-7i | dbDEMC |
| hsa-mir-222 | dbDEMC | hsa-mir-142 | 23619912 |
| hsa-let-7a | dbDEMC and miR2Disease | hsa-mir-1 | dbDEMC and miR2Disease |
| hsa-mir-19b | dbDEMC and miR2Disease | hsa-mir-133a | dbDEMC and miR2Disease |
| hsa-mir-15a | dbDEMC | hsa-mir-192 | dbDEMC and miR2Disease |
| hsa-mir-18a | dbDEMC and miR2Disease | hsa-mir-150 | 26455323 |
| hsa-mir-200b | dbDEMC | hsa-mir-203 | dbDEMC and miR2Disease |
| hsa-mir-19a | dbDEMC and miR2Disease | hsa-mir-451a | 25484364 |
| hsa-let-7b | dbDEMC and miR2Disease | hsa-let-7g | dbDEMC and miR2Disease |
| hsa-mir-24 | miR2Disease | hsa-mir-124 | dbDEMC |
| hsa-mir-199a | unconfirmed | hsa-mir-224 | dbDEMC and miR2Disease |
| hsa-mir-200c | dbDEMC and miR2Disease | hsa-mir-146b | 28466779 |
| Top 1–25 miRNAs | Evidence | Top 26–50 miRNAs | Evidence |
|---|---|---|---|
| hsa-mir-146b | 26673617 | hsa-mir-20a | 29450946 |
| hsa-mir-130a | 25834316 | hsa-mir-375 | 21343377 |
| hsa-mir-21 | miR2Disease | hsa-mir-17 | 30024601 |
| hsa-mir-146a | 28922434 | hsa-mir-222 | miR2Disease |
| hsa-mir-155 | 26950485 | hsa-mir-101 | 28944848 |
| hsa-mir-145 | miR2Disease | hsa-mir-199a | 24655788 |
| hsa-mir-143 | miR2Disease | hsa-mir-22 | 28482669 |
| hsa-mir-200a | 25740983 | hsa-mir-196a | 24527072 |
| hsa-mir-200b | 25740983 | hsa-mir-223 | 22270966 |
| hsa-mir-126 | 26464628 | hsa-mir-7 | 26261179 |
| hsa-mir-200c | 27766962 | hsa-mir-34c | 18803879 |
| hsa-let-7a | miR2Disease | hsa-mir-122 | 29509059 |
| hsa-mir-141 | miR2Disease | hsa-mir-218 | 27696291 |
| hsa-mir-34a | 25834316 | hsa-mir-34b | unconfirmed |
| hsa-mir-142 | 21343377 | hsa-mir-10b | 25190020 |
| hsa-mir-31 | 19598010 | hsa-mir-103a | 29754469 |
| hsa-mir-16 | miR2Disease | hsa-mir-27a | miR2Disease |
| hsa-mir-192 | 24981590 | hsa-mir-150 | 20067763 |
| hsa-mir-486 | 26895105 | hsa-mir-18a | 26950485 |
| hsa-mir-221 | miR2Disease | hsa-mir-19a | 22802949 |
| hsa-mir-107 | miR2Disease | hsa-mir-106a | miR2Disease |
| hsa-let-7f | 21533124 | hsa-mir-9 | 28418879 |
| hsa-let-7g | 25972194 | hsa-mir-451a | unconfirmed |
| hsa-mir-133b | 23296701 | hsa-mir-124 | 27041578 |
| hsa-mir-125b | 24846940 | hsa-mir-1 | 25874496 |
| Top 1–25 miRNAs | Evidence | Top 26–50 miRNAs | Evidence |
|---|---|---|---|
| hsa-mir-143 | dbDEMC and miR2Disease | hsa-mir-15a | dbDEMC and miR2Disease |
| hsa-mir-182 | dbDEMC and miR2Disease | hsa-mir-181b | dbDEMC and miR2Disease |
| hsa-mir-96 | dbDEMC and miR2Disease | hsa-mir-375 | dbDEMC and miR2Disease |
| hsa-mir-34a | dbDEMC and miR2Disease | hsa-mir-200a | dbDEMC |
| hsa-mir-210 | miR2Disease | hsa-mir-34b | dbDEMC |
| hsa-mir-150 | dbDEMC | hsa-mir-34c | dbDEMC |
| hsa-mir-92a | Unconfirmed | hsa-let-7b | dbDEMC and miR2Disease |
| hsa-mir-141 | miR2Disease | hsa-mir-218 | dbDEMC and miR2Disease |
| hsa-mir-21 | dbDEMC and miR2Disease | hsa-mir-101 | dbDEMC and miR2Disease |
| hsa-mir-222 | dbDEMC and miR2Disease | hsa-mir-124 | dbDEMC |
| hsa-mir-31 | dbDEMC and miR2Disease | hsa-mir-223 | dbDEMC and miR2Disease |
| hsa-mir-146b | 25712341 | hsa-let-7a | dbDEMC and miR2Disease |
| hsa-mir-221 | dbDEMC and miR2Disease | hsa-mir-224 | dbDEMC and miR2Disease |
| hsa-mir-203 | 26499781 | hsa-mir-205 | dbDEMC and miR2Disease |
| hsa-mir-126 | dbDEMC and miR2Disease | hsa-let-7d | dbDEMC and miR2Disease |
| hsa-mir-200b | Unconfirmed | hsa-mir-1 | dbDEMC |
| hsa-mir-200c | dbDEMC | hsa-let-7c | dbDEMC and miR2Disease |
| hsa-mir-146a | miR2Disease | hsa-mir-127 | dbDEMC and miR2Disease |
| hsa-mir-17 | miR2Disease | hsa-mir-135b | dbDEMC |
| hsa-mir-100 | dbDEMC and miR2Disease | hsa-mir-214 | dbDEMC and miR2Disease |
| hsa-mir-16 | dbDEMC and miR2Disease | hsa-mir-93 | 26124181 |
| hsa-mir-199a | dbDEMC and miR2Disease | hsa-mir-708 | 22552290 |
| hsa-mir-20a | miR2Disease | hsa-mir-155 | dbDEMC |
| hsa-mir-133b | dbDEMC | hsa-mir-133a | dbDEMC |
| hsa-mir-27b | dbDEMC and miR2Disease | hsa-mir-195 | dbDEMC and miR2Disease |
| Disease | WINMDA | BNPMDA | PBMDA | WBSMDA | RLSMDA |
|---|---|---|---|---|---|
| Breast neoplasms | 44 | 48 | 46 | 36 | 42 |
| Colon neoplasms | 47 | 45 | 47 | 45 | 46 |
| Gastric neoplasms | 48 | 43 | 46 | 43 | 44 |
| Kidney neoplasms | 45 | 43 | 42 | 42 | 45 |
| Liver neoplasms | 48 | 45 | 45 | 46 | 46 |
| Prostate neoplasms | 48 | 44 | 45 | 42 | 44 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Kuang, L.; Feng, X.; Zou, Q.; Wang, L. A Novel Approach Based on a Weighted Interactive Network to Predict Associations of MiRNAs and Diseases. Int. J. Mol. Sci. 2019, 20, 110. https://doi.org/10.3390/ijms20010110
Zhao H, Kuang L, Feng X, Zou Q, Wang L. A Novel Approach Based on a Weighted Interactive Network to Predict Associations of MiRNAs and Diseases. International Journal of Molecular Sciences. 2019; 20(1):110. https://doi.org/10.3390/ijms20010110
Chicago/Turabian StyleZhao, Haochen, Linai Kuang, Xiang Feng, Quan Zou, and Lei Wang. 2019. "A Novel Approach Based on a Weighted Interactive Network to Predict Associations of MiRNAs and Diseases" International Journal of Molecular Sciences 20, no. 1: 110. https://doi.org/10.3390/ijms20010110
APA StyleZhao, H., Kuang, L., Feng, X., Zou, Q., & Wang, L. (2019). A Novel Approach Based on a Weighted Interactive Network to Predict Associations of MiRNAs and Diseases. International Journal of Molecular Sciences, 20(1), 110. https://doi.org/10.3390/ijms20010110