Proteomics and Lipidomics in Inflammatory Bowel Disease Research: From Mechanistic Insights to Biomarker Identification

 , , , , and
, , , , and
Abstract
1. Introduction
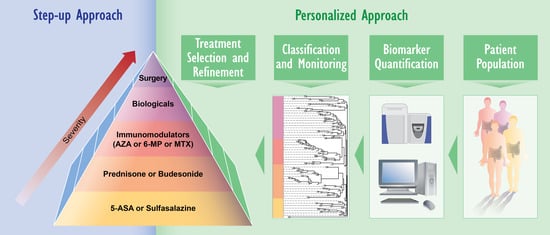
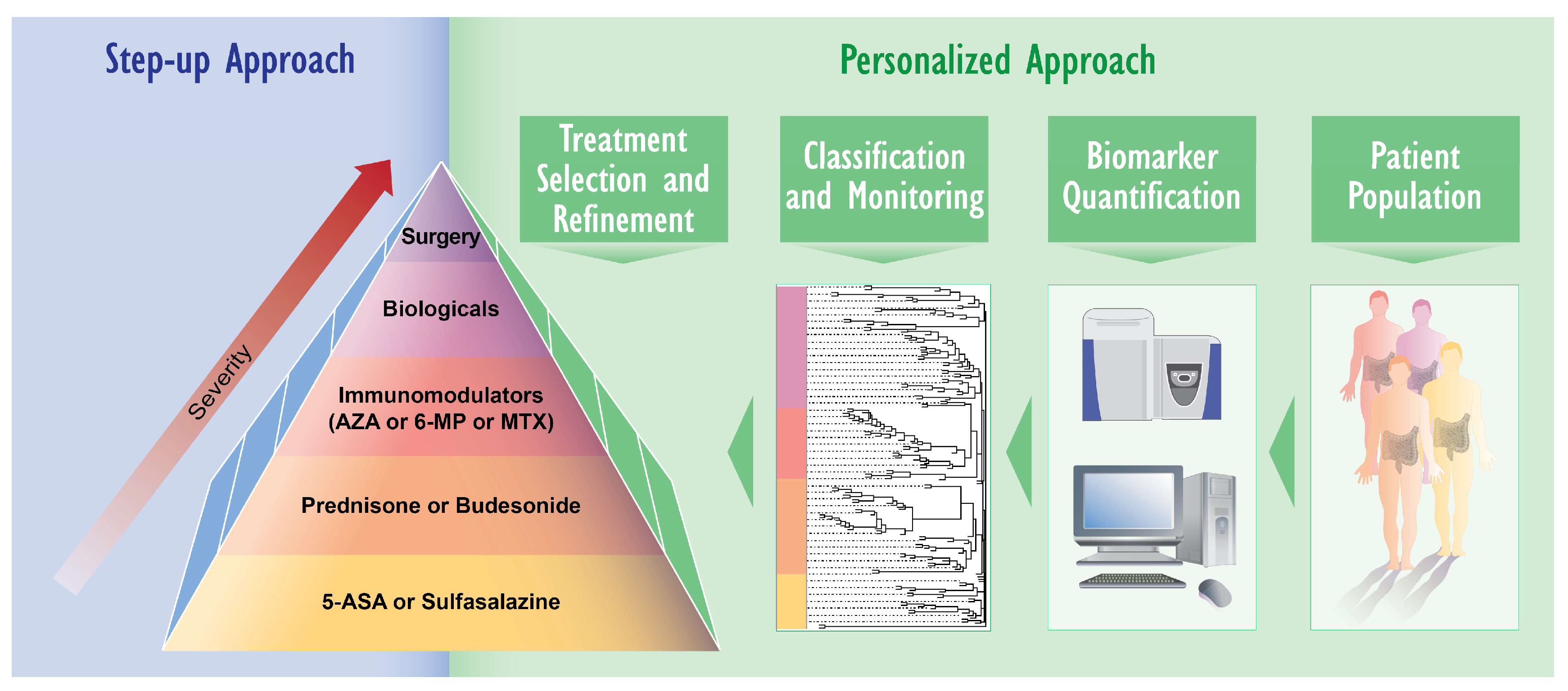
2. IBD Diagnosis and Treatment: Need for Novel Approaches
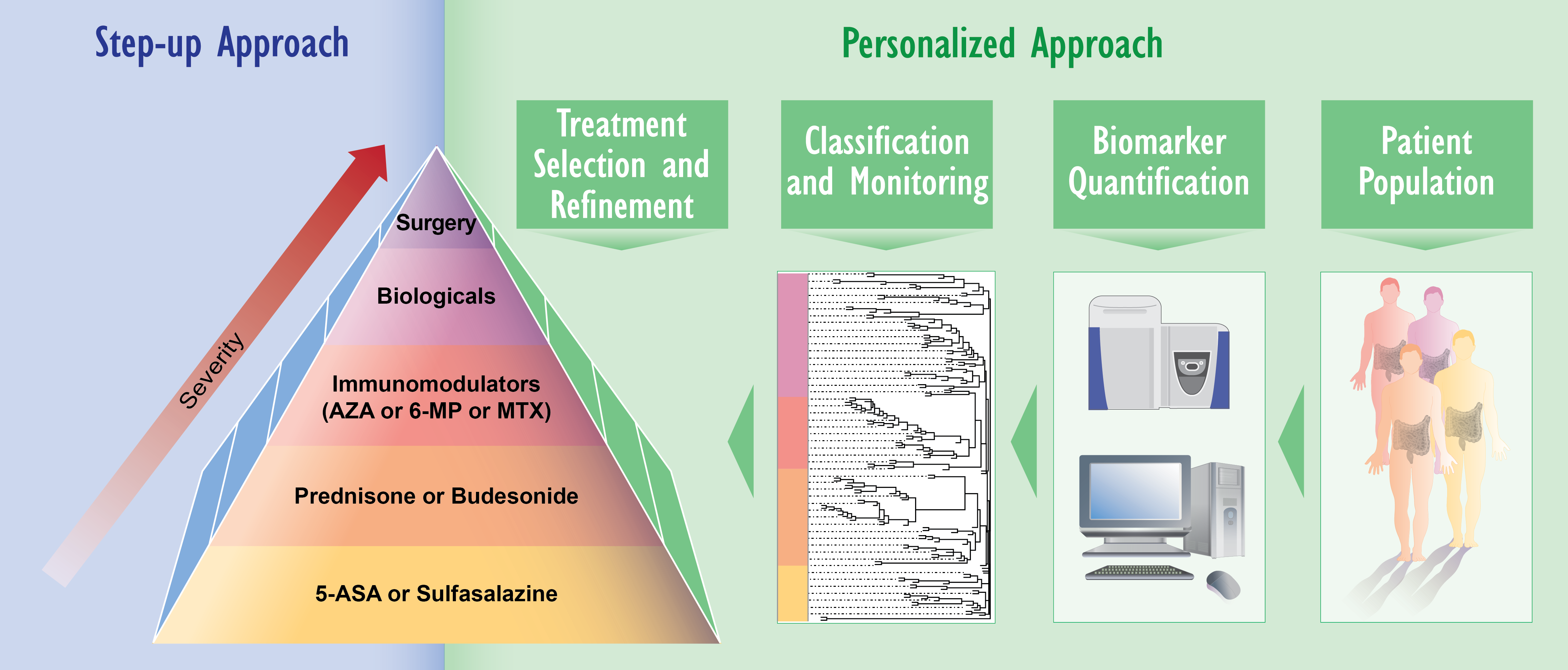
3. Applying Omics Analyses for IBD Research
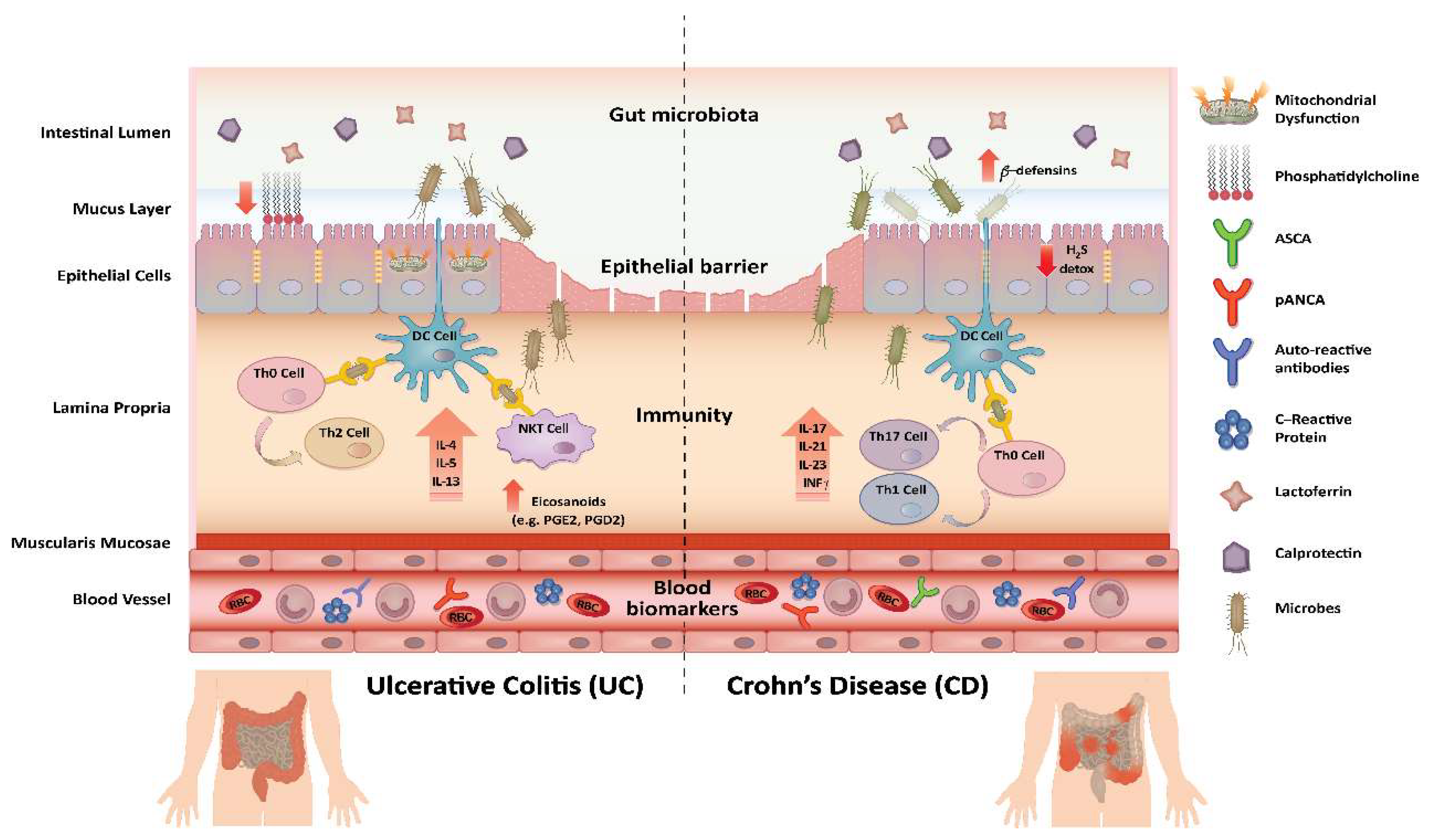
3.1. Mechanistic Insights Gained Using Proteomics and Lipidomics
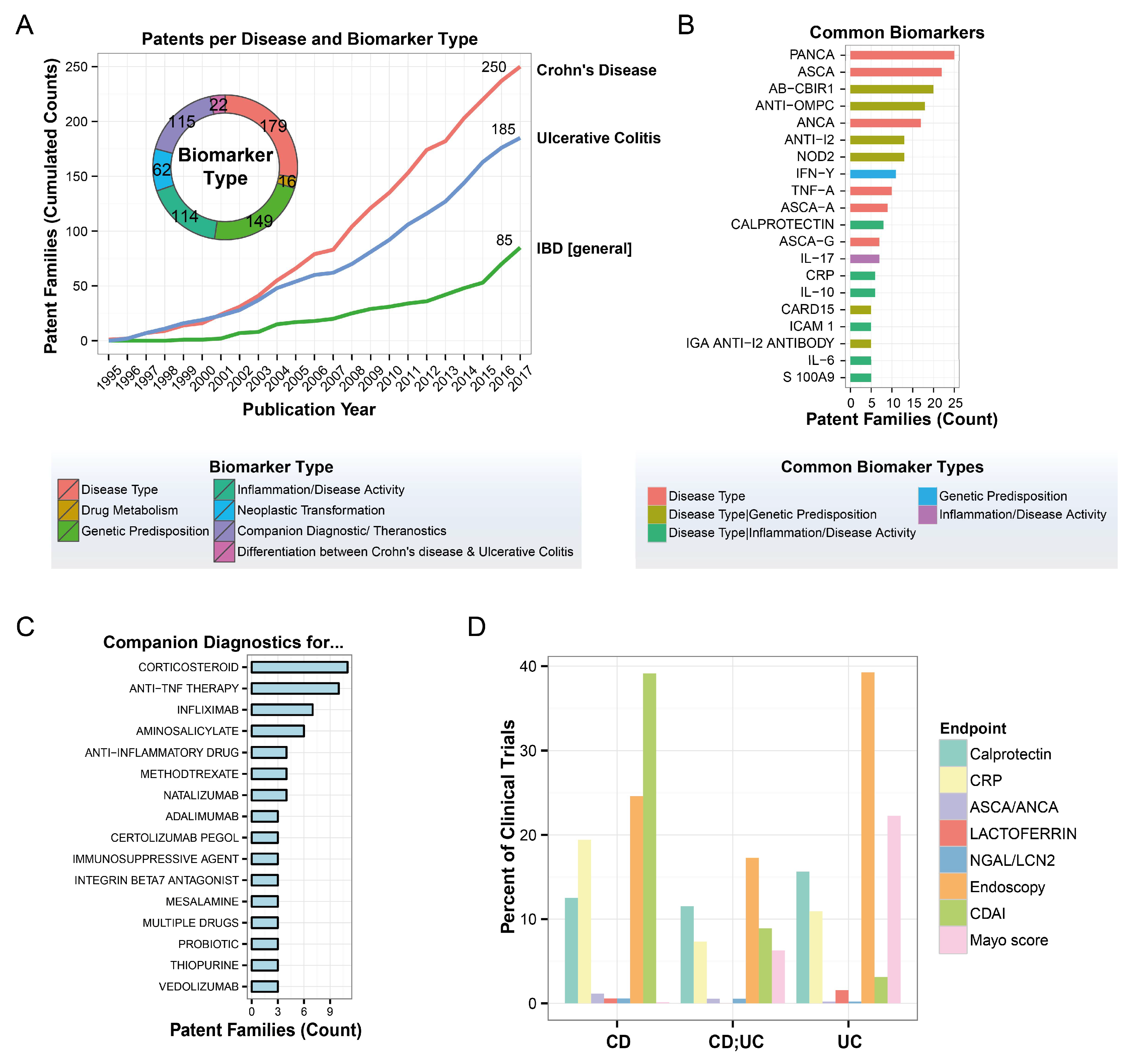
3.2. Biomarker Identification Using Proteomics and Lipidomics
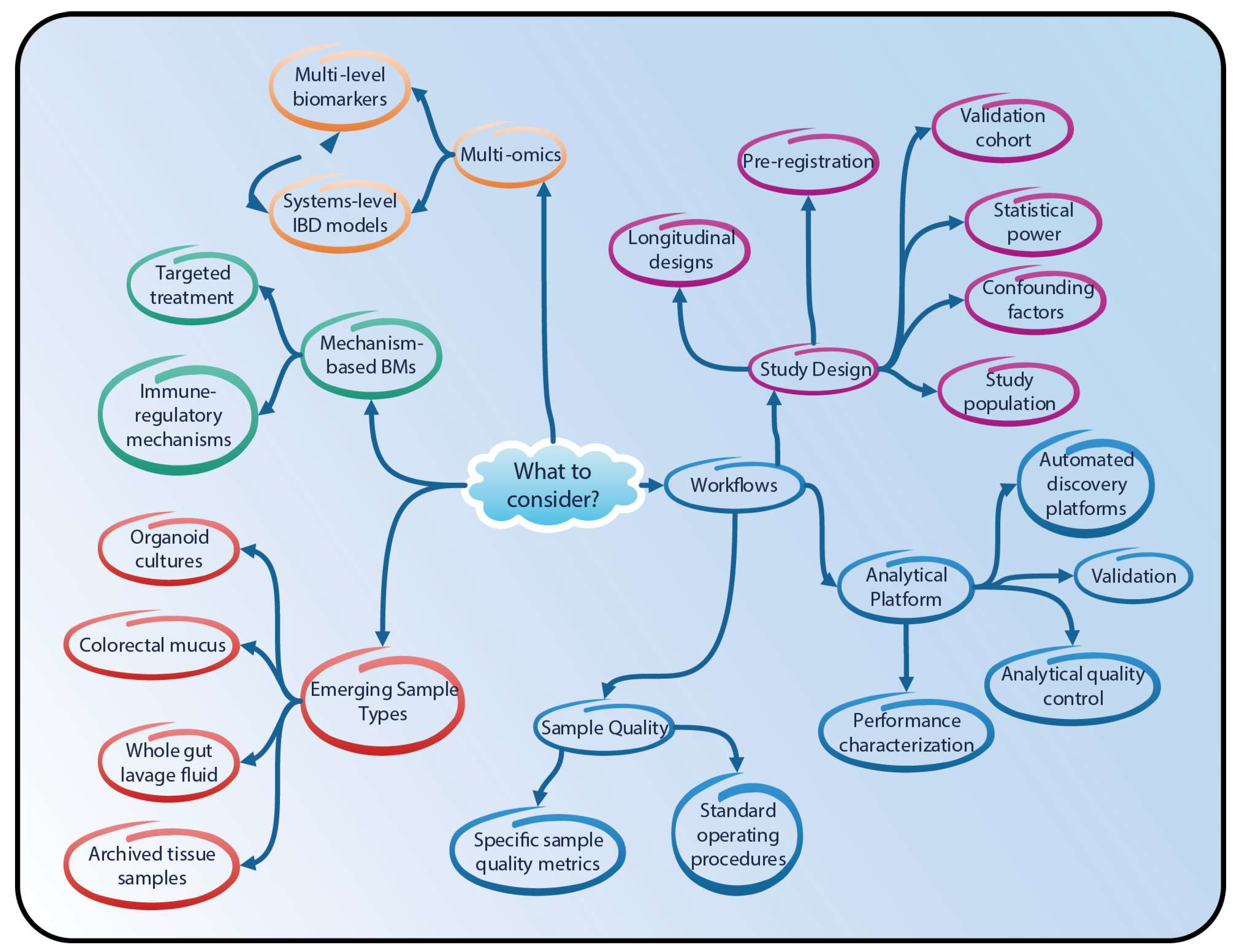
4. Proteomics and Lipidomics in IBD Research—Going Forward
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IBD | inflammatory bowel disease |
| MS | mass-spectrometry |
| UC | ulcerative colitis |
| CD | Crohn’s disease |
Appendix A
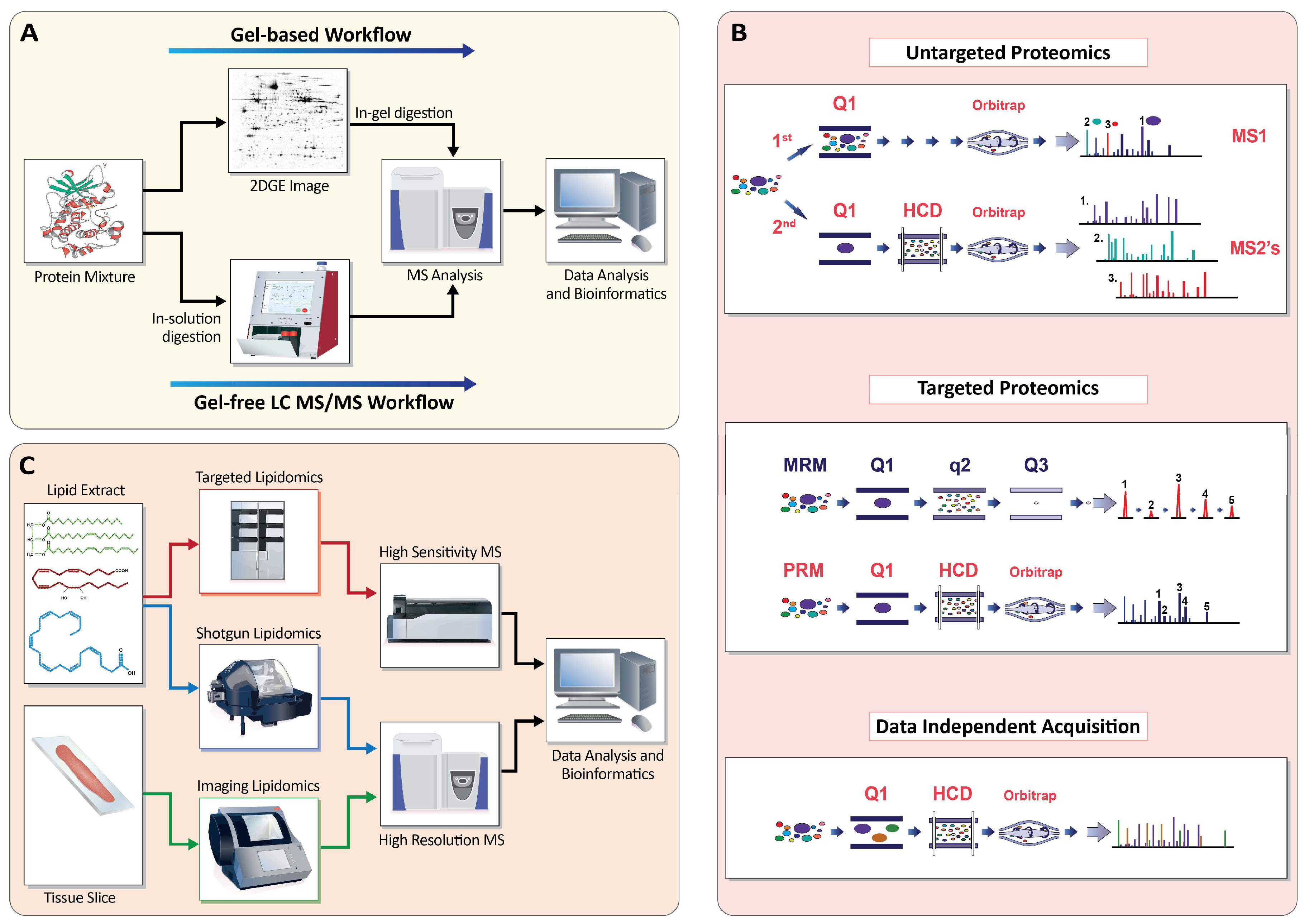
Appendix A.1. Profiling the Proteome
Appendix A.1.1. Gel-Based Approaches (2D-PAGE)
Appendix A.1.2. Gel-Free Approaches (LC-MS/MS)
Appendix A.1.3. Targeted MS
Appendix A.1.4. Data-Independent Acquisition
Appendix A.1.5. Beyond Expression Changes: Post-Translational Modifications
Appendix A.1.6. Non-MS-Based Proteomics Methods
Appendix A.2. Profiling the Lipidome
Appendix A.2.1. Shotgun Lipidomics
Appendix A.2.2. Targeted Lipidomics
Appendix A.2.3. FLUX Lipidomics
Appendix A.2.4. Imaging Lipidomics
References
- Bernstein, C.N.; Fried, M.; Krabshuis, J.; Cohen, H.; Eliakim, R.; Fedail, S.; Gearry, R.; Goh, K.; Hamid, S.; Khan, A.G. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm. Bowel Dis. 2010, 16, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, M.S.; Satsangi, J.; Ahmad, T.; Arnott, I.D.; Bernstein, C.N.; Brant, S.R.; Caprilli, R.; Colombel, J.F.; Gasche, C.; Geboes, K.; et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005, 19, 5a–36a. [Google Scholar] [CrossRef] [PubMed]
- Lennard-Jones, J. Classification of inflammatory bowel disease. Scand. J. Gastroenterol. 1989, 24, 2–6. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Tontini, G.E.; Vecchi, M.; Pastorelli, L.; Neurath, M.F.; Neumann, H. Differential diagnosis in inflammatory bowel disease colitis: State of the art and future perspectives. World J. Gastroenterol. 2015, 21, 21–46. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; De La Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn′s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar] [PubMed]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Nguyen, D.D.; Mizoguchi, E. Animal models of ulcerative colitis and their application in drug research. Drug Des. Devel. Ther. 2013, 7, 1341. [Google Scholar] [PubMed]
- Spencer, D.M.; Veldman, G.M.; Banerjee, S.; Willis, J.; Levine, A.D. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology 2002, 122, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn′s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Ordas, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef]
- Xu, X.-R.; Liu, C.-Q.; Feng, B.-S.; Liu, Z.-J. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- Bruewer, M.; Luegering, A.; Kucharzik, T.; Parkos, C.A.; Madara, J.L.; Hopkins, A.M.; Nusrat, A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol. 2003, 171, 6164–6172. [Google Scholar] [CrossRef] [PubMed]
- Simms, L.A.; Doecke, J.D.; Walsh, M.D.; Huang, N.; Fowler, E.V.; Radford-Smith, G.L. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn′s disease. Gut 2008, 57, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Molecular basis of epithelial barrier regulation: From basic mechanisms to clinical application. Am. J. Pathol. 2006, 169, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cell alpha-defensins in ileal Crohn′s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [PubMed]
- Ponder, A.; Long, M.D. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. J. Clin. Epidemiol. 2013, 5, 237–247. [Google Scholar]
- Montbarbon, M.; Pichavant, M.; Langlois, A.; Erdual, E.; Maggiotto, F.; Neut, C.; Mallevaey, T.; Dharancy, S.; Dubuquoy, L.; Trottein, F. Colonic inflammation in mice is improved by cigarette smoke through iNKT cells recruitment. PLoS ONE 2013, 8, e62208. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Integrating omics: The future of IBD? Dig. Dis. 2014, 32, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Fiocchi, C. Inflammatory Bowel Disease: Complexity and Variability Need Integration. Front. Med. 2018, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Cavenagh, M.M.; Lively, T.G.; Eberhard, D.A.; Bigbee, W.L.; Williams, P.M.; Mesirov, J.P.; Polley, M.-Y.C.; Kim, K.Y.; Tricoli, J.V. Criteria for the use of omics-based predictors in clinical trials: Explanation and elaboration. BMC Med. 2013, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Biedermann, L. Clinical Utility of Biomarkers in IBD. Curr. Gastroenterol. Rep. 2015, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Kochhar, G.; Lashner, B. Utility of Biomarkers in the Management of Inflammatory Bowel Disease. Curr. Treat. Options Gastroenterol. 2017, 15, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Soubieres, A.A.; Poullis, A. Emerging role of novel biomarkers in the diagnosis of inflammatory bowel disease. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.S.; Aldred, S.F.; Lee, P.K.; Tracy, R.P.; Schwartz, S.M.; Rieder, M.; Liu, K.; Williams, O.D.; Iribarren, C.; Lewis, E.C. Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am. J. Hum. Genet. 2005, 77, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Wasinger, V.C.; Leong, R.W. Current application of proteomics in biomarker discovery for inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2016, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Bennike, T.; Birkelund, S.; Stensballe, A.; Andersen, V. Biomarkers in Inflamm. Bowel Dis.: Current status and proteomics identification strategies. World J. Gastroenterol. 2014, 20, 3231–3244. [Google Scholar] [CrossRef] [PubMed]
- Gerich, M.E.; McGovern, D.P. Towards personalized care in IBD. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Viennois, E.; Zhao, Y.; Merlin, D. Biomarkers of IBD: From classical laboratory tools to personalized medicine. Inflamm. Bowel Dis. 2015, 21, 2467–2474. [Google Scholar] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, C.; Han, R.H.; Han, X. Novel advances in shotgun lipidomics for biology and medicine. Prog. Lipid Res. 2016, 61, 83–108. [Google Scholar] [CrossRef] [PubMed]
- Eustace, G.J.; Melmed, G.Y. Therapy for Crohn′s Disease: A Review of Recent Developments. Curr. Gastroenterol. Rep. 2018, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Vanga, R.; Long, M.D. Contemporary Management of Ulcerative Colitis. Curr. Gastroenterol. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Pouillon, L.; Bossuyt, P.; Peyrin-Biroulet, L. Considerations, challenges and future of anti-TNF therapy in treating inflammatory bowel disease. Expert Opin. Biol. Ther. 2016, 16, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef] [PubMed]
- Kane, S.V. Systematic review: Adherence issues in the treatment of ulcerative colitis. Aliment. Pharmacol. Therapeut. 2006, 23, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Flamant, M.; Roblin, X. Inflammatory bowel disease: Towards a personalized medicine. Therap. Adv. Gastroenterol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Coskun, M.; Vermeire, S.; Nielsen, O.H. Novel Targeted Therapies for Inflammatory Bowel Disease. Trends Pharmacol. Sci. 2017, 38, 127–142. [Google Scholar] [CrossRef] [PubMed]
- O′Toole, A.; Moss, A.C. Optimizing biologic agents in ulcerative colitis and Crohn′s disease. Curr. Gastroenterol. Rep. 2015, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Drucker, E.; Krapfenbauer, K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J. 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Barabási, A.-L.; Silverman, E.K. Network Medicine: Complex Systems in Human Disease and Therapeutics; Harvard University Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Li-Pook-Than, J.; Snyder, M. iPOP goes the world: Integrated personalized Omics profiling and the road toward improved health care. Chem. Biol. 2013, 20, 660–666. [Google Scholar] [CrossRef] [PubMed]
- de Souza, H.S.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.N.; Joung, J.G.; Bae, J.S.; Lee, C.S.; Koo, J.S.; Park, S.J.; Im, J.P.; Kim, Y.S.; Kim, J.W.; Park, W.Y.; et al. RNA-seq Reveals Transcriptomic Differences in Inflamed and Noninflamed Intestinal Mucosa of Crohn′s Disease Patients Compared with Normal Mucosa of Healthy Controls. Inflamm. Bowel Dis. 2017, 23, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Schubert, O.T.; Röst, H.L.; Collins, B.C.; Rosenberger, G.; Aebersold, R. Quantitative proteomics: Challenges and opportunities in basic and applied research. Nat. Protoc. 2017, 12, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Titz, B.; Elamin, A.; Martin, F.; Schneider, T.; Dijon, S.; Ivanov, N.V.; Hoeng, J.; Peitsch, M.C. Proteomics for systems toxicology. Comput. Struct. Biotechnol. J. 2014, 11, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.R.; Sylvänne, T.; Koistinen, K.M.; Tarasov, K.; Kauhanen, D.; Ekroos, K. High throughput quantitative molecular lipidomics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Han, X. Lipidomics: Techniques, applications, and outcomes related to biomedical sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Bennike, T.B.; Carlsen, T.G.; Ellingsen, T.; Bonderup, O.K.; Glerup, H.; Bogsted, M.; Christiansen, G.; Birkelund, S.; Stensballe, A.; Andersen, V. Neutrophil Extracellular Traps in Ulcerative Colitis: A Proteome Analysis of Intestinal Biopsies. Inflamm. Bowel Dis. 2015, 21, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, M.; Pearl, D.S.; Eiden, M.; Shute, J.K.; Brown, J.F.; Calder, P.C.; Trebble, T.M. Altered colonic mucosal Polyunsaturated Fatty Acid (PUFA) derived lipid mediators in ulcerative colitis: New insight into relationship with disease activity and pathophysiology. PLoS ONE 2013, 8, e76532. [Google Scholar] [CrossRef] [PubMed]
- Catana, C.S.; Berindan Neagoe, I.; Cozma, V.; Magdas, C.; Tabaran, F.; Dumitrascu, D.L. Contribution of the IL-17/IL-23 axis to the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 5823–5830. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Riaz, T.; Sollid, L.M.; Olsen, I.; de Souza, G.A. Quantitative Proteomics of Gut-Derived Th1 and Th1/Th17 Clones Reveal the Presence of CD28+ NKG2D- Th1 Cytotoxic CD4+ T cells. Mol. Cell. Proteom. 2016, 15, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Duguet, F.; Locard-Paulet, M.; Marcellin, M.; Chaoui, K.; Bernard, I.; Andreoletti, O.; Lesourne, R.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Saoudi, A. Proteomic analysis of regulatory T cells reveals the importance of Themis1 in the control of their suppressive function. Mol. Cell. Proteom. 2017, 16, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Sewell, G.W.; Hannun, Y.A.; Han, X.; Koster, G.; Bielawski, J.; Goss, V.; Smith, P.J.; Rahman, F.Z.; Vega, R.; Bloom, S.L.; et al. Lipidomic profiling in Crohn′s disease: Abnormalities in phosphatidylinositols, with preservation of ceramide, phosphatidylcholine and phosphatidylserine composition. Int. J. Biochem. Cell Biol. 2012, 44, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Shkoda, A.; Werner, T.; Daniel, H.; Gunckel, M.; Rogler, G.; Haller, D. Differential protein expression profile in the intestinal epithelium from patients with inflammatory bowel disease. J. Proteome Res. 2007, 6, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Nanni, P.; Mezzanotte, L.; Roda, G.; Caponi, A.; Levander, F.; James, P.; Roda, A. Differential proteomic analysis of HT29 Cl.16E and intestinal epithelial cells by LC ESI/QTOF mass spectrometry. J. Proteom. 2009, 72, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Wagenblast, J.; Erben, G.; Lehmann, W.D.; Hinz, U.; Merle, U.; Stremmel, W. Phosphatidylcholine and lysophosphatidylcholine in intestinal mucus of ulcerative colitis patients. A quantitative approach by nanoElectrospray-tandem mass spectrometry. Scand. J. Gastroenterol. 2004, 39, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Treede, I.; Gotthardt, D.; Tietje, A.; Zahn, A.; Ruhwald, R.; Schoenfeld, U.; Welsch, T.; Kienle, P.; Erben, G. Alterations of phospholipid concentration and species composition of the intestinal mucus barrier in ulcerative colitis: A clue to pathogenesis. Inflamm. Bowel Dis. 2009, 15, 1705–1720. [Google Scholar] [CrossRef] [PubMed]
- Karner, M.; Kocjan, A.; Stein, J.; Schreiber, S.; Von Boyen, G.; Uebel, P.; Schmidt, C.; Kupcinskas, L.; Dina, I.; Zuelch, F. First multicenter study of modified release phosphatidylcholine “LT-02” in ulcerative colitis: A randomized, placebo-controlled trial in mesalazine-refractory courses. Am. J. Gastroenterol. 2014, 109, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Kokkinidis, D.G.; Bosdelekidou, E.E.; Iliopoulou, S.M.; Tassos, A.G.; Texakalidis, P.T.; Economopoulos, K.P.; Kousoulis, A.A. Emerging treatments for ulcerative colitis: A systematic review. Scand. J. Gastroenterol. 2017, 52, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, J.S.; Sonnenburg, J.L.; Elias, J.E. Monitoring host responses to the gut microbiota. ISME J. 2015, 9, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Mottawea, W.; Chiang, C.-K.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S. Altered intestinal microbiota–host mitochondria crosstalk in new onset Crohn′s disease. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Yau, Y.Y.; Leong, R.W.L.; Pudipeddi, A.; Redmond, D.; Wasinger, V.C. Serological Epithelial Component Proteins Identify Intestinal Complications in Crohn′s Disease. Mol. Cell. Proteom. 2017, 16, 1244–1257. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Mundra, P.A.; Fang, L.; Galvin, A.; Moore, X.L.; Weir, J.M.; Wong, G.; White, D.A.; Chin-Dusting, J.; Sparrow, M.P.; et al. Lipidomic Profiling in Inflammatory Bowel Disease: Comparison Between Ulcerative Colitis and Crohn′s Disease. Inflamm. Bowel Dis. 2015, 21, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Deeke, S.A.; Starr, A.E.; Ning, Z.; Ahmadi, S.; Zhang, X.; Mayne, J.; Chiang, C.K.; Singleton, R.; Benchimol, E.I.; Mack, D.R.; et al. Mucosal-luminal interface proteomics reveals biomarkers of pediatric inflammatory bowel disease-associated colitis. Am. J. Gastroenterol. 2018, 113, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; M’Koma, A. Molecular differentiation of ulcerative colitis and Crohn′s colitis: Is it achievable? Clin. Lab. Int. 2015, 33–37. [Google Scholar]
- Korolkova, O.Y.; Myers, J.N.; Pellom, S.T.; Wang, L.; M′Koma, A.E. Characterization of serum cytokine profile in predominantly colonic inflammatory bowel disease to delineate ulcerative and Crohn′s colitides. Clin. Med. Insights Gastroenterol. 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Seeley, E.H.; Washington, M.K.; Caprioli, R.M.; M′Koma, A.E. Proteomic patterns of colonic mucosal tissues delineate Crohn′s colitis and ulcerative colitis. Proteom.-Clin. Appl. 2013, 7, 541–549. [Google Scholar] [CrossRef] [PubMed]
- M′koma, A.E.; Seeley, E.H.; Washington, M.K.; Schwartz, D.A.; Muldoon, R.L.; Herline, A.J.; Wise, P.E.; Caprioli, R.M. Proteomic profiling of mucosal and submucosal colonic tissues yields protein signatures that differentiate the inflammatory colitides. Inflamm. Bowel Dis. 2011, 17, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Starr, A.E.; Deeke, S.A.; Ning, Z.; Chiang, C.-K.; Zhang, X.; Mottawea, W.; Singleton, R.; Benchimol, E.I.; Wen, M.; Mack, D.R. Proteomic analysis of ascending colon biopsies from a paediatric inflammatory bowel disease inception cohort identifies protein biomarkers that differentiate Crohn′s disease from UC. Gut 2016, 2015, 33–37. [Google Scholar]
- Sofia, M.A.; Rubin, D.T. The Impact of Therapeutic Antibodies on the Management of Digestive Diseases: History, Current Practice, and Future Directions. Digest. Dis. Sci. 2017, 62, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.K.; Strid, H.; Isaksson, S.; Bajor, A.; Lasson, A.; Ung, K.A.; Ohman, L. Response to infliximab therapy in ulcerative colitis is associated with decreased monocyte activation, reduced CCL2 expression and downregulation of Tenascin C. J. Crohns. Colitis. 2015, 9, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Fiorillo, A.A.; Chaisson, E.; Gordish-Dressman, H.; Hathout, Y.; Damsker, J.M.; Hoffman, E.P.; Conklin, L.S. Identification of Pathway-Specific Serum Biomarkers of Response to Glucocorticoid and Infliximab Treatment in Children with Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2016, 7, e192. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Noble, W.S.; Wolf-Yadlin, A. Technical advances in proteomics: New developments in data-independent acquisition. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Greco, V.; Piras, C.; Pieroni, L.; Urbani, A. Direct Assessment of Plasma/Serum Sample Quality for Proteomics Biomarker Investigation. Serum/Plasma Proteom. Methods Protoc. 2017, 169, 3–21. [Google Scholar]
- Forshed, J. Experimental Design in Clinical ′Omics Biomarker Discovery. J. Proteome Res. 2017, 16, 3954–3960. [Google Scholar] [CrossRef] [PubMed]
- Tuck, M.K.; Chan, D.W.; Chia, D.; Godwin, A.K.; Grizzle, W.E.; Krueger, K.E.; Rom, W.; Sanda, M.; Sorbara, L.; Stass, S. Standard operating procedures for serum and plasma collection: Early detection research network consensus statement standard operating procedure integration working group. J. Proteome Res. 2008, 8, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.; Petzold, A.; Bennett, J.; Berven, F.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.; Fleming, J.; Furlan, R. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Mischak, H.; Allmaier, G.; Apweiler, R.; Attwood, T.; Baumann, M.; Benigni, A.; Bennett, S.E.; Bischoff, R.; Bongcam-Rudloff, E.; Capasso, G. Recommendations for biomarker identification and qualification in clinical proteomics. Sci. Transl. Med. 2010, 2, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.E.; Borchers, C.H. Mass spectrometry based biomarker discovery, verification, and validation—quality assurance and control of protein biomarker assays. Mol. Oncol. 2014, 8, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Surinova, S.; Schiess, R.; Hüttenhain, R.; Cerciello, F.; Wollscheid, B.; Aebersold, R. On the development of plasma protein biomarkers. J. Proteome Res. 2010, 10, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Bereman, M.S.; Beri, J.; Sharma, V.; Nathe, C.; Eckels, J.; MacLean, B.; MacCoss, M.J. An Automated Pipeline to Monitor System Performance in Liquid Chromatography–Tandem Mass Spectrometry Proteomic Experiments. J. Proteome Res. 2016, 15, 4763–4769. [Google Scholar] [CrossRef] [PubMed]
- Dogu, E.; Mohammad-Taheri, S.; Abbatiello, S.E.; Bereman, M.S.; MacLean, B.; Schilling, B.; Vitek, O. MSstatsQC: Longitudinal system suitability monitoring and quality control for targeted proteomic experiments. Mol. Cell. Proteom. 2017, 16, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Jeon, S.Y.; Park, J.-S.; Yun, J.Y.; Kil, H.N.; Hong, W.K.; Lee, M.-H.; Kim, J.-W.; Jeon, J.P.; Han, B.G. Identification of Clinical Biomarkers for Pre-Analytical Quality Control of Blood Samples. Biopreserv. Biobank. 2013, 11, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-E.; Hong, M.; Park, S.-K.; Yu, J.-I.; Wang, J.-S.; Shin, H.; Kim, J.-W.; Han, B.-G.; Shin, S.-Y. Inorganic phosphorus and potassium are putative indicators of delayed separation of whole blood. Osong Public Health Res. Perspect. 2016, 7, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.; Van Deerlin, V.M.; Gulley, M.L. Recommended principles and practices for validating clinical molecular pathology tests. Arch. Pathol. Lab. Med. 2009, 133, 743–755. [Google Scholar] [PubMed]
- US Department of Health Human Services. FDA Guidance for Industry, Bioanalytical Method Validation; US Department of Health Human Services: Washington, WA, USA, 2013. [Google Scholar]
- Magnusson, B. The Fitness for Purpose of Analytical Methods: A Laboratory Guide to Method Validation and Related Topics; Eurachem: Roma, Italy, 2014. [Google Scholar]
- Bringans, S.D.; Ito, J.; Stoll, T.; Winfield, K.; Phillips, M.; Peters, K.; Davis, W.A.; Davis, T.M.; Lipscombe, R.J. Comprehensive mass spectrometry based biomarker discovery and validation platform as applied to diabetic kidney disease. EuPA Open Proteom. 2017, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Kulak, N.A.; Pichler, G.; Holdt, L.M.; Teupser, D.; Mann, M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2016, 2, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Cominetti, O.; Núñez Galindo, A.; Cortheésy, J.; Oller Moreno, S.; Irincheeva, I.; Valsesia, A.; Astrup, A.; Saris, W.H.; Hager, J.R.; Kussmann, M. Proteomic biomarker discovery in 1000 human plasma samples with mass spectrometry. J. Proteome Res. 2015, 15, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Frantzi, M.; Bhat, A.; Latosinska, A. Clinical proteomic biomarkers: Relevant issues on study design & technical considerations in biomarker development. Clin. Transl. Med. 2014, 3, 7. [Google Scholar] [PubMed]
- Maes, E.; Cho, W.C.; Baggerman, G. Translating clinical proteomics: The importance of study design. Expert Rev. Proteom. 2015, 12, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Oberg, A.L.; Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. J. Proteome Res. 2009, 8, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Levin, Y. The role of statistical power analysis in quantitative proteomics. Proteomics 2011, 11, 2565–2567. [Google Scholar] [CrossRef] [PubMed]
- Abbaraju, N.V.; Cai, Y.; Rees, B.B. Protein recovery and identification from the gulf killifish, Fundulus grandis: Comparing snap-frozen and RNAlater® preserved tissues. Proteomics 2011, 11, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.; Ducret, A.; Tille, J.C.; Thomas, M.; McKee, T.A.; Rubbia-Brandt, L.; Scherl, A.; Lescuyer, P.; Cutler, P. Applications of mass spectrometry for quantitative protein analysis in formalin-fixed paraffin-embedded tissues. Proteomics 2014, 14, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.T. Proteomic analysis of FFPE tissue: Barriers to clinical impact. Expert Rev. Proteom. 2016, 13, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, O.J.; Arentz, G.; Hoffmann, P. Proteomic developments in the analysis of formalin-fixed tissue. BBA-Proteins Proteom. 2015, 1854, 559–580. [Google Scholar] [CrossRef] [PubMed]
- Bennike, T.B.; Kastaniegaard, K.; Padurariu, S.; Gaihede, M.; Birkelund, S.; Andersen, V.; Stensballe, A. Comparing the proteome of snap frozen, RNAlater preserved, and formalin-fixed paraffin-embedded human tissue samples. EuPA Open Proteom. 2016, 10, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dahele, A.; Drummond, H.E.; Hoque, S.S.; Humphreys, K.; Arnott, I.D. Whole gut lavage fluid analysis: A minimally invasive method for study of mucosal immunity and inflammation. In Celiac Disease: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 257–277. [Google Scholar]
- Kayazawa, M.; Saitoh, O.; Kojima, K.; Nakagawa, K.; Tanaka, S.; Tabata, K.; Matsuse, R.; Uchida, K.; Hoshimoto, M.; Hirata, I.; et al. Lactoferrin in whole gut lavage fluid as a marker for disease activity in inflammatory bowel disease: Comparison with other neutrophil-derived proteins. Am. J. Gastroenterol. 2002, 97, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Arnott, I.D.R.; Drummond, H.E.; Ghosh, S. Gut Mucosal Secretion of Interleukin 1β and Interleukin-8 Predicts Relapse in Clinically Inactive Crohn′s Disease. Digest. Dis. Sci. 2001, 46, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Bandaletova, T.; Chhaya, V.; Poullis, A.; Loktionov, A. Colorectal mucus non-invasively collected from patients with inflammatory bowel disease and its suitability for diagnostic cytology. Apmis 2016, 124, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Loktionov, A.; Chhaya, V.; Bandaletova, T.; Poullis, A. Inflammatory bowel disease detection and monitoring by measuring biomarkers in non-invasively collected colorectal mucus. J. Gastroenterol. Hepatol. 2017, 32, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, O.; Mazza, T.; Castellana, S.; Panza, A.; Latiano, T.; Corritore, G.; Andriulli, A.; Latiano, A. Inflammatory Bowel Disease Meets Systems Biology: A Multi-Omics Challenge and Frontier. OMICS 2016, 20, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Polytarchou, C.; Koukos, G.; Iliopoulos, D. Systems biology in Inflamm. Bowel Dis.: Ready for prime time. Curr. Opin. Gastroenterol. 2014, 30, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C.; Panes, J. Drug development in IBD: From novel target identification to early clinical trials. Gut 2016, 65, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Gligorijević, V.; Pržulj, N. Methods for biological data integration: Perspectives and challenges. J. R. Soc. Interface 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Aloi, M.; Nuti, F.; Stronati, L.; Cucchiara, S. Advances in the medical management of paediatric IBD. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Kolkman, A.; Dirksen, E.H.; Slijper, M.; Heck, A.J. Double standards in quantitative proteomics direct comparative assessment of difference in gel electrophoresis and metabolic stable isotope labeling. Mol. Cell. Proteom. 2005, 4, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Arentz, G.; Weiland, F.; Oehler, M.K.; Hoffmann, P. State of the art of 2D DIGE. Proteom.-Clin. Appl. 2015, 9, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Pütz, S.M.; Boehm, A.M.; Stiewe, T.; Sickmann, A. iTRAQ Analysis of a Cell Culture Model for Malignant Transformation, Including Comparison with 2D-PAGE and SILAC. J. Proteome Res. 2012, 11, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Elamin, A.; Titz, B.; Dijon, S.; Merg, C.; Geertz, M.; Schneider, T.; Martin, F.; Schlage, W.K.; Frentzel, S.; Talamo, F. Quantitative proteomics analysis using 2D-PAGE to investigate the effects of cigarette smoke and aerosol of a prototypic modified risk tobacco product on the lung proteome in C57BL/6 mice. J. Proteom. 2016, 145, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kelleher, N.L. Precision proteomics: The case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. USA 2008, 105, 18132–18138. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.W.; Cagney, G. An overview of label-free quantitation methods in proteomics by mass spectrometry. In Proteome Bioinformatics; Humana: New York, NY, USA, 2010; pp. 273–283. [Google Scholar]
- Li, Z.; Adams, R.M.; Chourey, K.; Hurst, G.B.; Hettich, R.L.; Pan, C. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Oda, Y.; Huang, K.; Cross, F.R.; Cowburn, D.; Chait, B.T. Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. USA 1999, 96, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Pan, S.; Gu, S.; Bradbury, E.M.; Chen, X. Amino acid residue specific stable isotope labeling for quantitative proteomics. Rapid Commun. Mass Spectrom. 2002, 16, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Soste, M.; Hrabakova, R.; Wanka, S.; Melnik, A.; Boersema, P.; Maiolica, A.; Wernas, T.; Tognetti, M.; von Mering, C.; Picotti, P. A sentinel protein assay for simultaneously quantifying cellular processes. Nat. Methods 2014, 11, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteom. 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.R.; Trelle, M.B.; Thingholm, T.E.; Jensen, O.N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Biotechniques 2006, 40, 790–798. [Google Scholar] [CrossRef] [PubMed]
- von Stechow, L.; Francavilla, C.; Olsen, J.V. Recent findings and technological advances in phosphoproteomics for cells and tissues. Expert Rev. Proteomics 2015, 12, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Halim, A.; Anonsen, J.H. Microbial glycoproteomics. Curr. Opin. Struct. Biol. 2017, 44, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Thaysen-Andersen, M.; Packer, N.H.; Schulz, B.L. Maturing glycoproteomics technologies provide unique structural insights into the N-glycoproteome and its regulation in health and disease. Mol. Cell. Proteom. 2016, 15, 1773–1790. [Google Scholar] [CrossRef] [PubMed]
- Houser, B. Bio-Rad′s Bio-Plex® suspension array system, xMAP technology overview. Arch. Physiol. Biochem. 2012, 118, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Heiskanen, L.A.; Suoniemi, M.; Ta, H.X.; Tarasov, K.; Ekroos, K. Long-term performance and stability of molecular shotgun lipidomic analysis of human plasma samples. Anal. Chem. 2013, 85, 8757–8763. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, K. Lipidomics perspective: From molecular lipidomics to validated clinical diagnostics. In Lipidomics, Technologies and Applications; Wiley-VCH: Weinheim, Germany, 2012; pp. 1–19. [Google Scholar]
- Weir, J.M.; Wong, G.; Barlow, C.K.; Greeve, M.A.; Kowalczyk, A.; Almasy, L.; Comuzzie, A.G.; Mahaney, M.C.; Jowett, J.B.M.; Shaw, J.; et al. Plasma lipid profiling in a large population-based cohort. J. Lipid Res. 2013, 54, 2898–2908. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Ekroos, K.; Kavaliauskiene, S.; Bergan, J.; Kauhanen, D.; Lintonen, T.; Sandvig, K. Determining the Turnover of Glycosphingolipid Species by Stable-Isotope Tracer Lipidomics. J. Mol. Biol. 2016, 428, 4856–4866. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.L.; Lee, Y.J. High-Spatial Resolution Mass Spectrometry Imaging: Toward Single Cell Metabolomics in Plant Tissues. Chem. Rec. 2017, 18, 66–77. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantification Approach | Abundant Protein Depletion | Separation | Scale(#Proteins/#Samples) | Precision | Comment | References |
|---|---|---|---|---|---|---|
| iTRAQ (discovery) | Yes | SCX C18 | Pool of 20 samples | Biomarker panel for diabetic kidney disease developed | [95] | |
| MRM (validation) | Yes | C18 | 8/572 | CV ~8% | ||
| Label-free | No | C18 | ~437/319 | CV <20% for 71% of proteins | Applied within weight-loss study | [96] |
| TMT | Yes | C18 | ~190/~1000 | Average CV of 12% for internal standard | Applied within multicentered dietary interventionstudy | [97] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titz, B.; Gadaleta, R.M.; Lo Sasso, G.; Elamin, A.; Ekroos, K.; Ivanov, N.V.; Peitsch, M.C.; Hoeng, J. Proteomics and Lipidomics in Inflammatory Bowel Disease Research: From Mechanistic Insights to Biomarker Identification. Int. J. Mol. Sci. 2018, 19, 2775. https://doi.org/10.3390/ijms19092775
Titz B, Gadaleta RM, Lo Sasso G, Elamin A, Ekroos K, Ivanov NV, Peitsch MC, Hoeng J. Proteomics and Lipidomics in Inflammatory Bowel Disease Research: From Mechanistic Insights to Biomarker Identification. International Journal of Molecular Sciences. 2018; 19(9):2775. https://doi.org/10.3390/ijms19092775
Chicago/Turabian StyleTitz, Bjoern, Raffaella M. Gadaleta, Giuseppe Lo Sasso, Ashraf Elamin, Kim Ekroos, Nikolai V. Ivanov, Manuel C. Peitsch, and Julia Hoeng. 2018. "Proteomics and Lipidomics in Inflammatory Bowel Disease Research: From Mechanistic Insights to Biomarker Identification" International Journal of Molecular Sciences 19, no. 9: 2775. https://doi.org/10.3390/ijms19092775
APA StyleTitz, B., Gadaleta, R. M., Lo Sasso, G., Elamin, A., Ekroos, K., Ivanov, N. V., Peitsch, M. C., & Hoeng, J. (2018). Proteomics and Lipidomics in Inflammatory Bowel Disease Research: From Mechanistic Insights to Biomarker Identification. International Journal of Molecular Sciences, 19(9), 2775. https://doi.org/10.3390/ijms19092775