1. Introduction
Genomic imprinting is a phenomenon of epigenetic gene regulation, whereby mono-allelic expression of a gene occurs, in a parent-of-origin specific manner in placental mammals and some plants [
1,
2].
Cyclin dependent kinase inhibitor 1c (
Cdkn1c) is a maternally expressed imprinted gene found on the IC2 imprinting cluster on mouse chromosome 7 [
3]. The gene functions both in limiting cell proliferation, primarily as a G1/S phase inhibitor [
4,
5], and in directing differentiation of certain select lineages. These include regulating the differentiation of skeletal muscle [
6], amacrine cells in the retina [
7], and the maintenance of adult quiescent neural stem cell populations [
8,
9]. Imprinted expression of
Cdkn1c is achieved through two distinct differentially methylated regions (DMRs). A germline differentially methylated region termed
KvDMR is located >200 kb upstream of
Cdkn1c within the
Kcnq1 gene, and this region acquires DNA methylation in the maternal germline in mice and humans to establish imprinted expression of an extensive genomic region on mouse distal chromosome 7/human chromosome 11p15, called the IC2 cluster [
10,
11,
12]. The
Cdkn1c somatic DMR (
sDMR) is located at the 5′ terminus of the
Cdkn1c coding region, and is required for silencing maintenance, as opposed to establishment, through methylation of the silent paternally inherited allele [
13]. This region has been shown to only possess methylation in somatic tissue in vivo, acquired during early embryonic development and after allelic expression is established [
14,
15,
16]. Our recent work using an imaging-based model of
Cdkn1c expression reported that the
Cdkn1c sDMR is sensitive to gestational protein restriction, with offspring exposed to the diet found to have reduced embryonic and post-natal methylation at this region, with an accompanying loss of imprinting of
Cdkn1c [
17].
Elevated expression of
Cdkn1c has been implicated as a cause of two similar but distinct childhood growth restriction disorders, Silver–Russell (SRS) and IMAGe syndrome [
18], while a loss-of-function of
Cdkn1c is present in familial cases of the childhood overgrowth disorder Beckwith–Wiedemann syndrome [
19]. In order to model SRS, our lab developed transgenic mouse lines carrying additional copies of
Cdkn1c, on a bacterial artificial chromosome (BAC) spanning
Cdkn1c and two other imprinted genes
Phlda2 and
Slc22a18. To distinguish between the effects of elevated
Cdkn1c expression and the effect of increasing the dosage of the other two genes, we developed a transgenic line containing a modified version of this same BAC where
Cdkn1c expression is disrupted by a
β-galactosidase insertion [
20,
21]. These lines provide an alternative route for studying imprinted genes, focusing on dosage rather than gene function
per se. Classical knockout models, used to study the gene function of imprinted genes, frequently present with high levels of lethality associated with heterozygous deletion models presenting as a phenotypic homozygous deletion, due to the mono-allelic expression [
22]. Using the BAC transgenic lines, we have demonstrated a dosage-dependent fetal growth restriction due to elevated
Cdkn1c [
20], which was maintained post-natally and into early adulthood. Further to this, a new function of the gene in regulating brown adipose tissue formation was recently described by our group, with elevated markers of mitochondrial uncoupling also uncovered in the white adipose tissue [
23]. In SRS, children are born small and fail to catch up, with excessive thinness being an additional characteristic. Some SRS children are also reported to be fussy eaters, which has been suggested to contribute to their failure to gain weight. However, we showed that young mice carrying an extra copy of
Cdkn1c were also thin with little subcutaneous fat despite consuming similar calories to controls [
23]. Some SRS children also have night sweats which could suggest dysfunctional thermoregulation, consistent with the increase in brown adipose tissue we observed in our mouse model [
23].
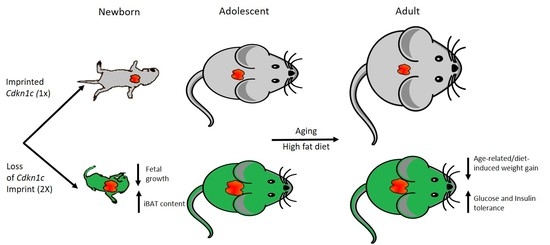
In this study, we further explored the impact of increased
Cdkn1c dosage in adult mice, focusing on the predicted role for
Cdkn1c in influencing adult weight gain through regulating the development of brown fat depots. Imprinted gene function has classically been thought to be predominantly restricted to regulating embryonic and placental development; however, recent work has highlighted important post-natal functions for this class of gene [
23,
24,
25]. Therefore, further elucidation of both gene and imprint function, in adulthood, will enable a clearer understanding of this method of epigenetic gene regulation.
2. Results
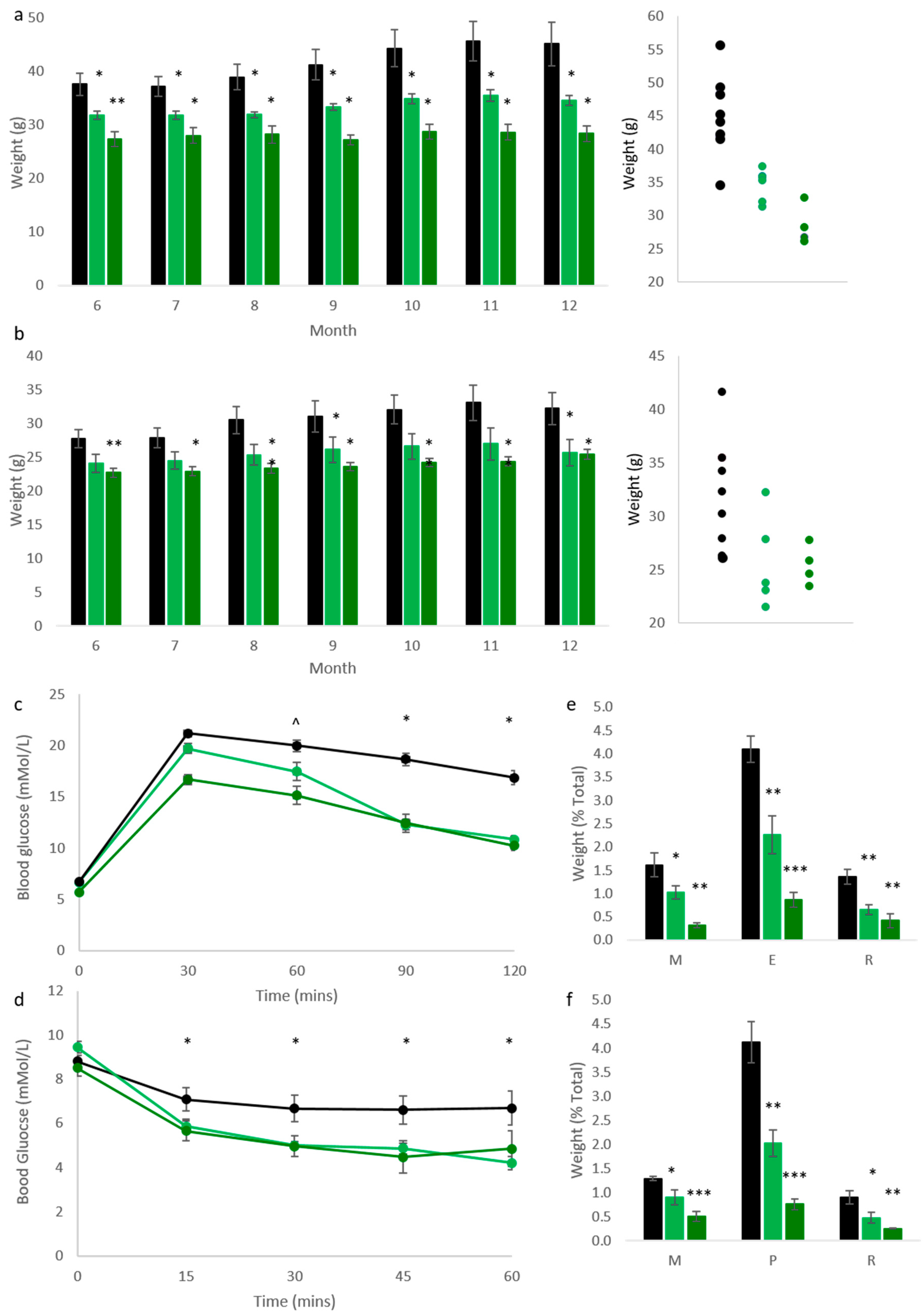
We have previously reported that mice bearing one (Cdkn1c
BACx1) or two copies (Cdkn1c
BACx2) of a BAC spanning the
Cdkn1c locus were significantly lighter than wild type (WT) litter mates at 10 weeks of age, with relative increases in expression of
Cdkn1c in white and brown adipose tissue [
23]. This phenotype was attributed to excess
Cdkn1c, causing jointly a fetal growth restriction phenotype that was not resolved post-natally and reduced white adipose tissue (WAT) stores due to elevated brown adipose tissue (BAT) formation. To further understand the effect of elevated
Cdkn1c into adulthood, mice were aged to 1 year, co-housed with sex-matched WT littermates. Monthly weighing found significantly reduced weights in both male (
Figure 1a) and female (
Figure 1b) Cdkn1c
BACx1 (light green) and Cdkn1c
BACx2 (dark green) mice when compared to wild type (black). In fact, all four transgenic groups increased in weight by less than 3 g over this 6-month period, in contrast to an average 7.5 g increase in male, and 4.5 g increase in female WT. Glucose and insulin tolerance testing was performed on the male cohort at 1 year of age, and found a significantly improved clearance of glucose after challenge (
Figure 1c) in both Cdkn1c
BACx1 and Cdkn1c
BACx2 lines, in addition to heightened response to insulin testing, compared to age-matched WT males (
Figure 1d). White adipose deposits were weighed in culled mice and found to be significantly smaller in both males (
Figure 1e) and females (
Figure 1f), as a percentage of total body weight, in order to control for growth restriction. As had been observed in the whole-body weights (
Figure 1a,b), this reduction was found to be dosage-dependent, with Cdkn1c
BACx2 mice showing a greater reduction in fat pad weights when compared to Cdkn1c
BACx1.
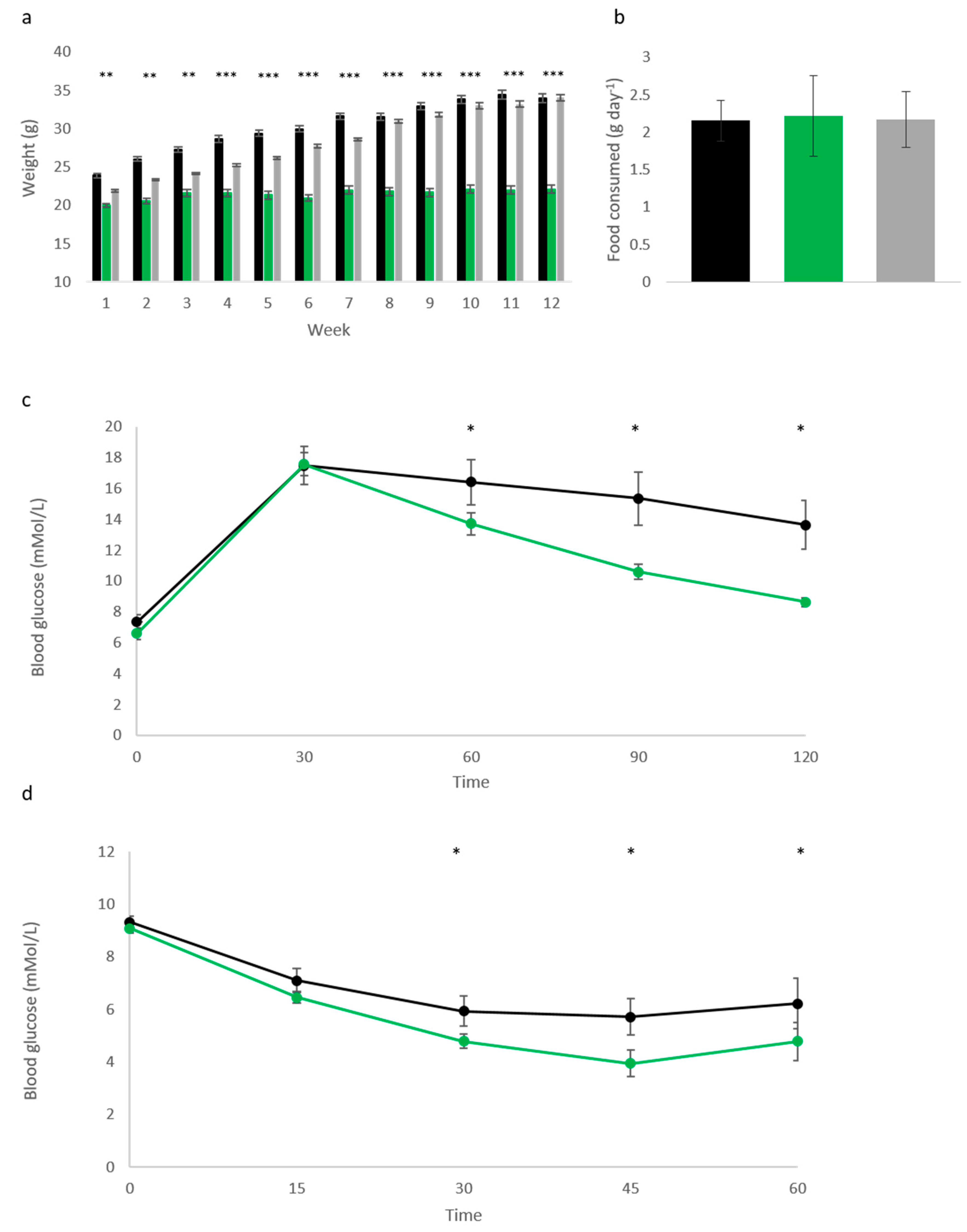
Following the observation that elevated
Cdkn1c could protect against age-related metabolic syndrome, we challenged Cdkn1c
BACx1 male mice and litter-matched WT controls with a high fat diet (58V8, TestDiet) from 10 weeks of age for 12 weeks. Cdkn1c
BACx1 males were found to be highly resistant to weight gain during the dietary challenge (
Figure 2a). The average weight gain was 12.4% of the original body weight (2.1 g) over the 12-week period in contrast to the average 41.2% (10.1 g) gained by wild type male controls. In this experiment, we also included a third transgenic line to isolate the contribution of
Cdkn1c to this resistance against weight gain. This third line of mice called Cdkn1c
BAC-LacZ carries one copy of the same BAC transgene but without over-expression of
Cdkn1c. Any phenotypes observed in Cdkn1c
BACx1 mice and Cdkn1c
BACx2 mice that are absent in Cdkn1c
BAC-LacZ mice must be due to elevated
Cdkn1c. Cdkn1c
BAC-LacZ male mice gained weight on the high fat diet with the same profile as WT controls (
Figure 2b) demonstrating that this protection against weight gain was due to
Cdkn1c alone. To explore the extent of this phenotype, glucose tolerance testing was performed on these animals, with Cdkn1c
BACx1 males displaying significantly improved glucose clearance (
Figure 2c), compared to wild type mice. Further to this, insulin tolerance testing revealed a heightened response to insulin challenge in Cdkn1c
BACx1 males (
Figure 2d), correlating with the effects observed in aging animals (
Figure 1c,d).
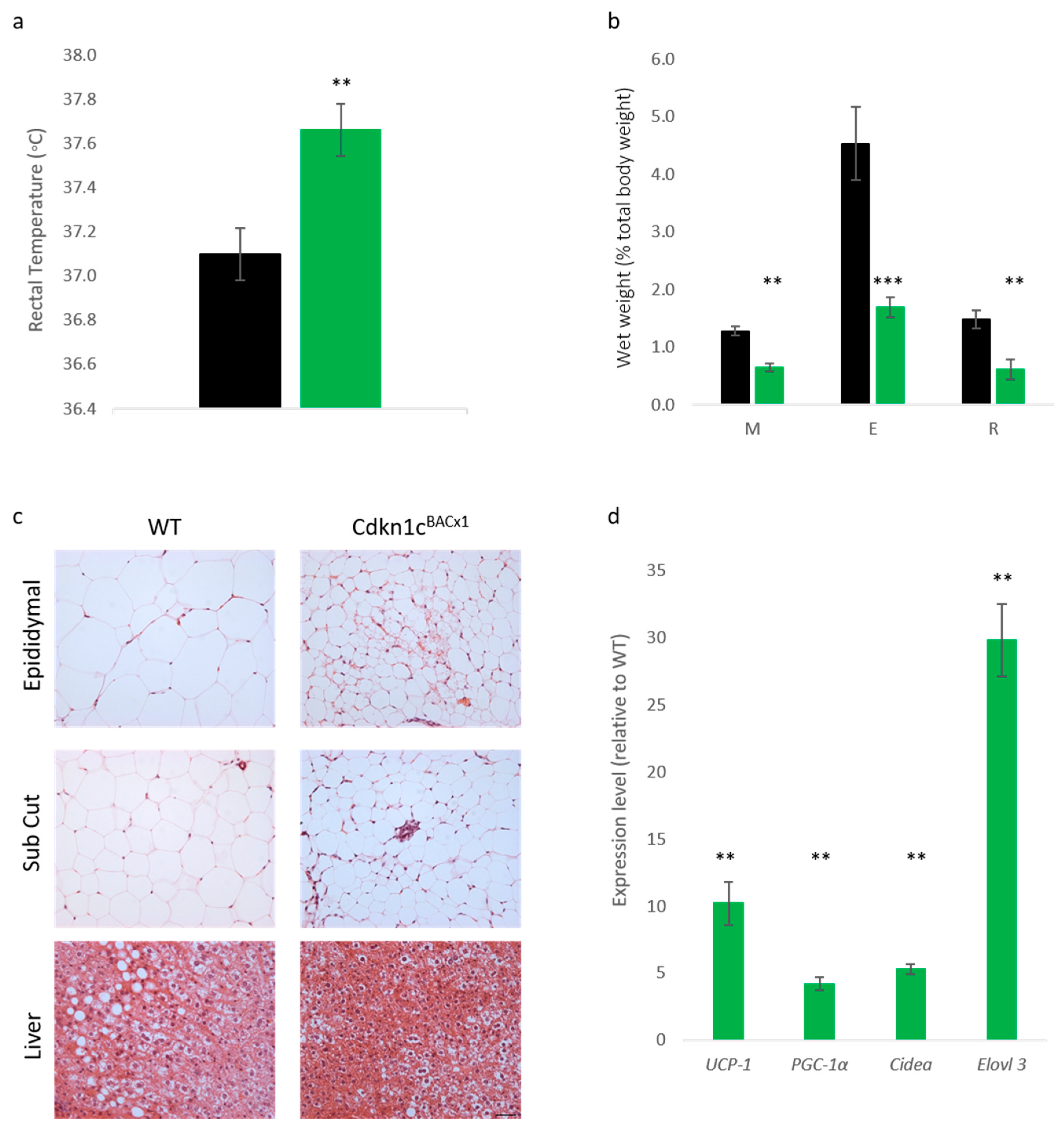
Reduced weight in adult mice possessing excess
Cdkn1c may, in part,2 be attributed to increased brown adipose tissue content, both in the classic interscapular brown adipose depot but also within white adipose depots, and therefore elevated mitochondrial uncoupling [
23]. In order to assess whether the protective phenotype observed in Cdkn1c
BACx1 mice challenged with a high-fat diet could be attributed to increased BAT content, core temperature was first checked after the 12-week dietary challenge. Cdkn1c
BACx1 males were found to be significantly hotter than wild type mice (
Figure 3a). Upon culling, the wet weights of selected white adipose depots were found to be significantly reduced in the Cdkn1c
BACx1 mice (
Figure 3b), with epididymal (E) and retroperitoneal (R) depots more than 50% smaller than wild type, as a percentage of total body weight. Qualitatively, hematoxylin and eosin (H+E) staining revealed differences in white adipocytes (
Figure 3c) with WT epididymal and anterior sub-cutaneous depots containing predominantly larger uni-locular adipocytes relative to the corresponding depots from Cdkn1c
BACx1 males, which contained areas of multi-locular adipocytes most closely associated with brown adipocytes. In the liver, there appeared to be reduced lipid accumulation (white regions) in Cdkn1c
BACx1 compared to WT samples. We previously reported the increased expression of a number of BAT markers downstream of the BAT-determining gene
Prdm16 in WAT from young mice, and increased PRDM16 protein but not mRNA, suggesting a function for
Cdkn1c in the post-transcriptional regulation of
Prdm16 [
23]. QRT-PCR analysis of retroperitoneal adipose tissue revealed a higher mRNA expression of
mitochondrial uncoupling protein 1 (
Ucp-1) and the brown adipose markers
PGC-1α,
Cidea and
Elovl3 in Cdkn1c
BACx1 retroperitoneal depots relative to WT (
Figure 3d). While we have not excluded the contribution of other sites of
Cdkn1c over-expression to the phenotype, taken together, these results are consistent with the increased BAT driven by elevated
Cdkn1c dosage providing a protective effect against weight gain.
Recent work has shown that the epigenetic marks which silence the paternal allele of
Cdkn1c can be disrupted through protein restriction during gestation [
17]. Further to this, the stability of imprints during aging has recently come into focus, with several reports of altered DNA methylation in aged samples [
26,
27]. We examined
Cdkn1c expression in retroperitoneal white adipose tissue (R) and interscapular brown adipose tissue (iBAT) in 10-week old WT mice compared to 12-month old mice from the same colony. Older mice showed significantly reduced expression of
Cdkn1c in white adipose and slightly elevated expression in brown adipose tissue compared to younger mice (
Figure 4a). Two regions of differential methylation (
Cdkn1c sDMR and
KvDMR) that regulate
Cdkn1c expression were examined through bisulphite sequencing of genomic DNA from the same individuals to try and account for these observed changes in transcription. Methylation of the
Cdkn1c sDMR and
KvDMR in early life adipose tissue has previously been shown to be similar to that seen in other tissue [
23]. A qualitative analysis of 10-week (left column) and 1-year old (right column) wild type retroperitoneal adipose tissue revealed a similar pattern of methylation at both
Cdkn1c sDMR and the
KvDMR (
Figure 4b). No gross differences were observed in brown adipose tissue (
Figure 4c), where expression had been found to be modestly elevated in 1-year old samples (
Figure 4a). Finally, samples were analyzed from the high-fat diet cohort, with no gross differences in methylation detected after 12 weeks of dietary challenge in retroperitoneal tissue (
Figure 4d).
3. Discussion
In this study, we show that mice with a single extra copy of
Cdkn1c are able to resist both age and diet-driven weight gain and maintain a healthier management of glucose than matched controls. While we have not defined the mechanism underlying this phenotype, the gains in brown adipose tissue that we have shown in younger mice [
23], alongside the higher body temperature and increased expression of markers of brown fat, suggest that increased thermogenesis may, at least in part, play a contributory role.
Using both loss-of-expression and gain-in-expression animal models, alongside ex-vivo approaches, we previously identified an important and intrinsic role for
Cdkn1c in the development of both classic brown adipose tissue and the brown-like adipose tissue that develops post-natally within white adipose depots [
23]. We showed that mice with a 2–3-fold elevated expression of
Cdkn1c possessed substantially more brown adipose tissue and substantially less white adipose tissues at 10 weeks of age. In this new study, we show that mice from these same transgenic lines do not gain significant weight as they age, unlike their wild type littermates, and this was true for both males and females. We also show that these older mice are better able to manage their glucose levels. A caveat to this observation is the high blood glucose values relative to other studies on this strain background [
28]. This may be explained by the standard chow on which we routinely maintain mice, Labdiet 5008, which provides a relatively higher proportion of energy from fat and carbohydrate compared to other standard chow diets and may induce a diabetic state when fed over a prolonged period. An alternative and potentially more interesting explanation is that the WT mice were inherently stressed, a factor known to impact blood glucose levels [
29]. Higher stress levels in the WT littermate controls is suggested by our previous observation that male Cdkn1c
BACx1 mice disrupt social stability when co-housed with WT mice, resulting in an increased incidence of fighting and wounding within these groups [
30]. Further studies including single housing animals will be required to fully explore these differences.
We further show that younger mice exposed to a high-fat diet for 12 weeks gain almost no weight despite consuming a similar amount of HFD as their WT counterparts under the same protocol. As with the older mice, the high-fat diet-exposed
Cdkn1c transgenic mice were also better able to manage their glucose levels than the controls. Using a control line of mice (Cdkn1c
BAC-LacZ) which carried the same BAC transgene but without over-expression of
Cdkn1c, we were able to attribute this weight resistance phenotype to elevated
Cdkn1c. Taken together, these data highlight a previously unappreciated role for
Cdkn1c in weight regulation later in life, at least in mice. The loss or gain in function of
CDKN1C in humans is already implicated in the childhood growth disorders Beckwith-Wiedemann Syndrome, IMAGe syndrome and SRS, recapitulated to some extent in animal models. This new data highlights a role for
Cdkn1c in regulating adiposity in the adult mouse. Quite remarkably, only a two-fold elevation in expression of
Cdkn1c was able to protect mice against both age and diet-induced weight gain. It should be noted that the changes we observed were between 6 months and 1 year of age, which is thought to represent middle age in mice. Our analyses did not extend to cover the full aging period. It will be interesting to follow these mice for an extended period into old age to ask whether they maintain this metabolic advantage, and whether they have an extended lifespan. Further to this, while a reduction in body weight was observed in both genders, a dosage dependent phenotype was not so clearly observed in females. Variation in the individual weights of the transgenic groups was larger at 12 months than those observed in male mice (
Figure 1a, b). It will be important to explore this potential difference between genders in a larger cohort over a longer time period.
The inflexibility of the
Cdkn1c imprint to either aging or post-natal diet modification contrasts with data seen from
in utero exposure [
17]. We found that erosion of the
Cdkn1c sDMR occurred in offspring exposed to gestational low protein diet, with these changes maintained post-natally. Our current data would therefore lend support to the hypothesis that altering the dosage of
Cdkn1c in response to gestational environmental factors can be achieved, but once laid down, this methylation is resistant to post-natal adjustment. However, we analyzed DNA methylation in a small number of individuals, so we cannot exclude more modest changes in DNA methylation with time or changes in other tissues or other strains of mice not analyzed. This stubbornness to change likely provides a protective effect in mice, as has been highlighted by the differences in weight observed in our loss of imprinting model (Cdkn1c
BACx1). A single extra copy of
Cdkn1c was sufficient to restrict mice in both aging and diet models to only modest weight gain, in addition to the embryonic and post-natal growth restriction previously described [
20]. In a laboratory setting, such responses would appear advantageous. However, outside of food available
ad libitum, such excessive uncoupling may in fact reduce survival chances, due to the challenges in laying down sufficient adipose stores. By restricting changes in imprinted status to an embryonic window, rather than a constant environmental sensitivity, this might be prevented.
Cdkn1c has only recently been identified as having a role in adipose tissue development [
23]. Interestingly, the response to glucose and insulin tolerance testing in aging mice was found to be largely comparable between the Cdkn1c
BACx1 and Cdkn1c
BACx2 line, whereas whole body and adipose depots weights were found to be dosage dependent, with the Cdkn1c
BACx2 line having even greater reductions. This dosage-based phenotype is also present at early post-natal and fetal time points [
20]. The lack of a dosage-based phenotype for glucose and insulin tolerance may be attributed to simply avoiding the age-related metabolic syndrome that is observed in wild type mice, due to their leanness, rather than substantial changes to insulin signaling. However, epigenetic dysregulation at the IC2 cluster has previously been demonstrate to affect β-cell mass [
31], and therefore a direct role cannot be excluded.
In summary, this is the first report linking
Cdkn1c dosage to weight gain in adult mice. Our findings, if they translate to humans, have potentially monumental implications for human health. In addition to the potential relevance to the rare human disorders that involve either elevated expression of
Cdkn1c or gain in function mutations of this gene, SRS and IMAGe syndrome [
18], worldwide obesity has nearly tripled in the last 40 years, resulting in over 650 million obese adults and an estimated 41 million children under the age of 5 years being overweight or obese (WHO). Unlike many other genes implicated in weight regulation,
Cdkn1c is epigenetically regulated. This suggests the possibility that epidrugs could be developed that activate the silent
Cdkn1c allele with therapeutic benefit.
Cdkn1c encodes a cyclin-dependent kinase inhibitor and, while we have not identified the molecular mechanism through which
Cdkn1c drives the development of brown adipose, it may be possible to develop designer drugs to mimic this activity. Ultimately, drugs aimed at
Cdkn1c may provide a way to increase BAT thermogenesis in adult humans to increase whole-body energy expenditure and address the global epidemic of obesity.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



