Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts



Abstract
1. Introduction
2. Results
2.1. Cytotoxic Effects of PM10 on Primary Dermal Fibroblasts
2.2. The Relationship between PM10 and Autophagy in HDFs (Western Blot, Immunochemistry and TEM)
2.3. Effect of PM10 on the Expression of Pro-Inflammatory Cytokines and Collagen Metabolism in HDFs (ELISA)
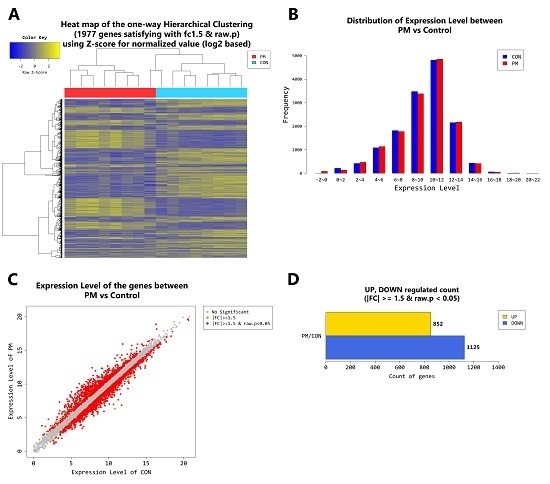
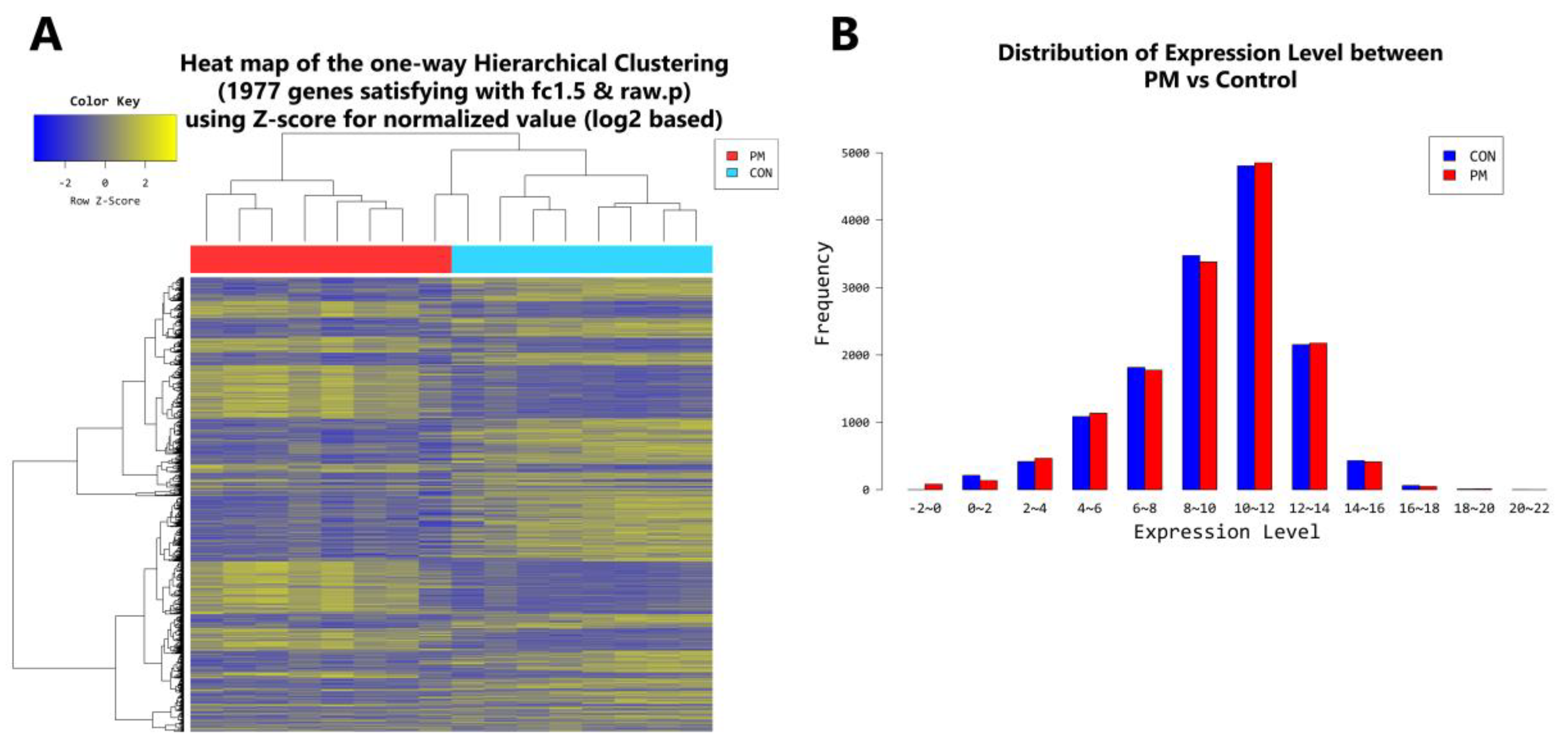
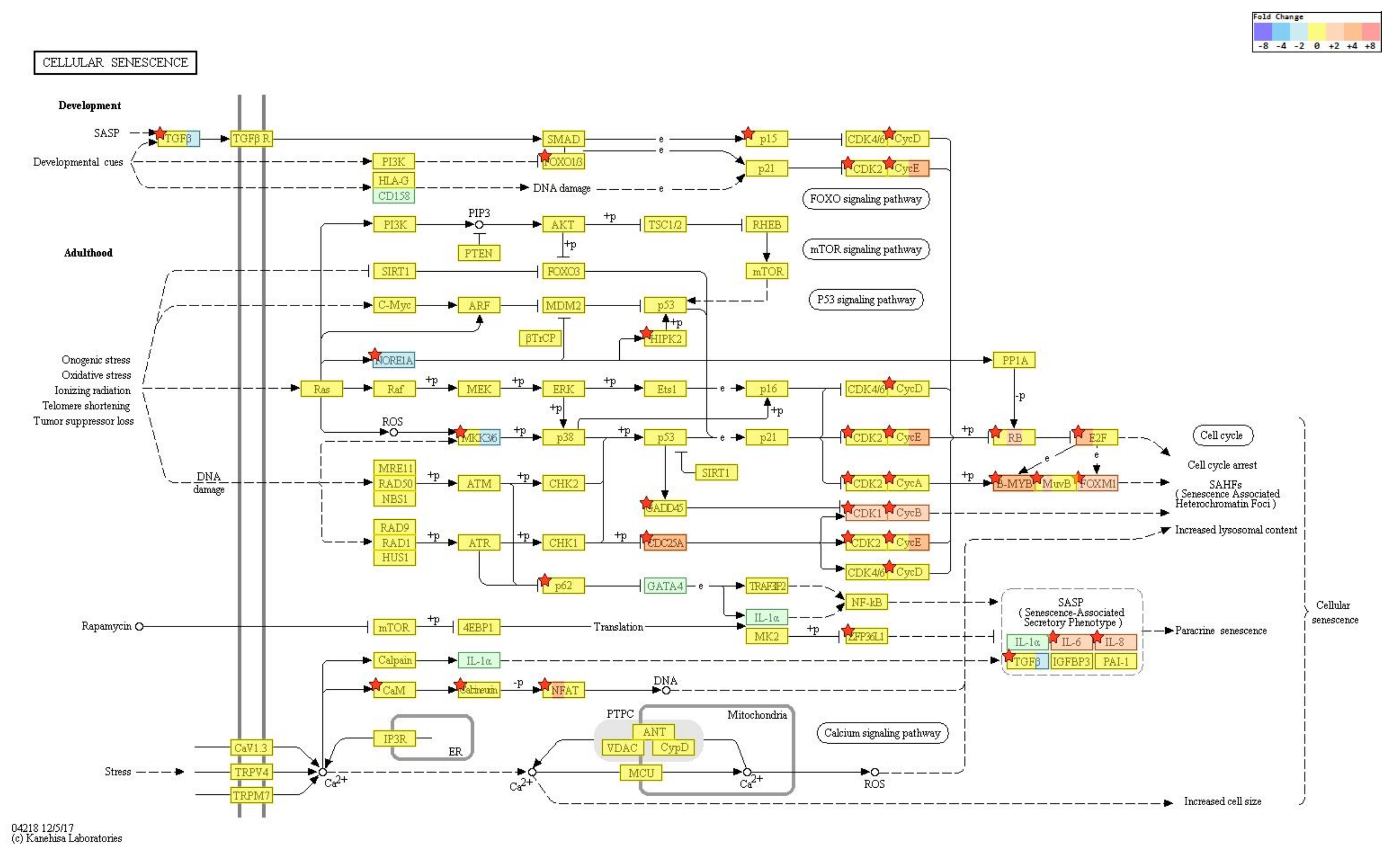
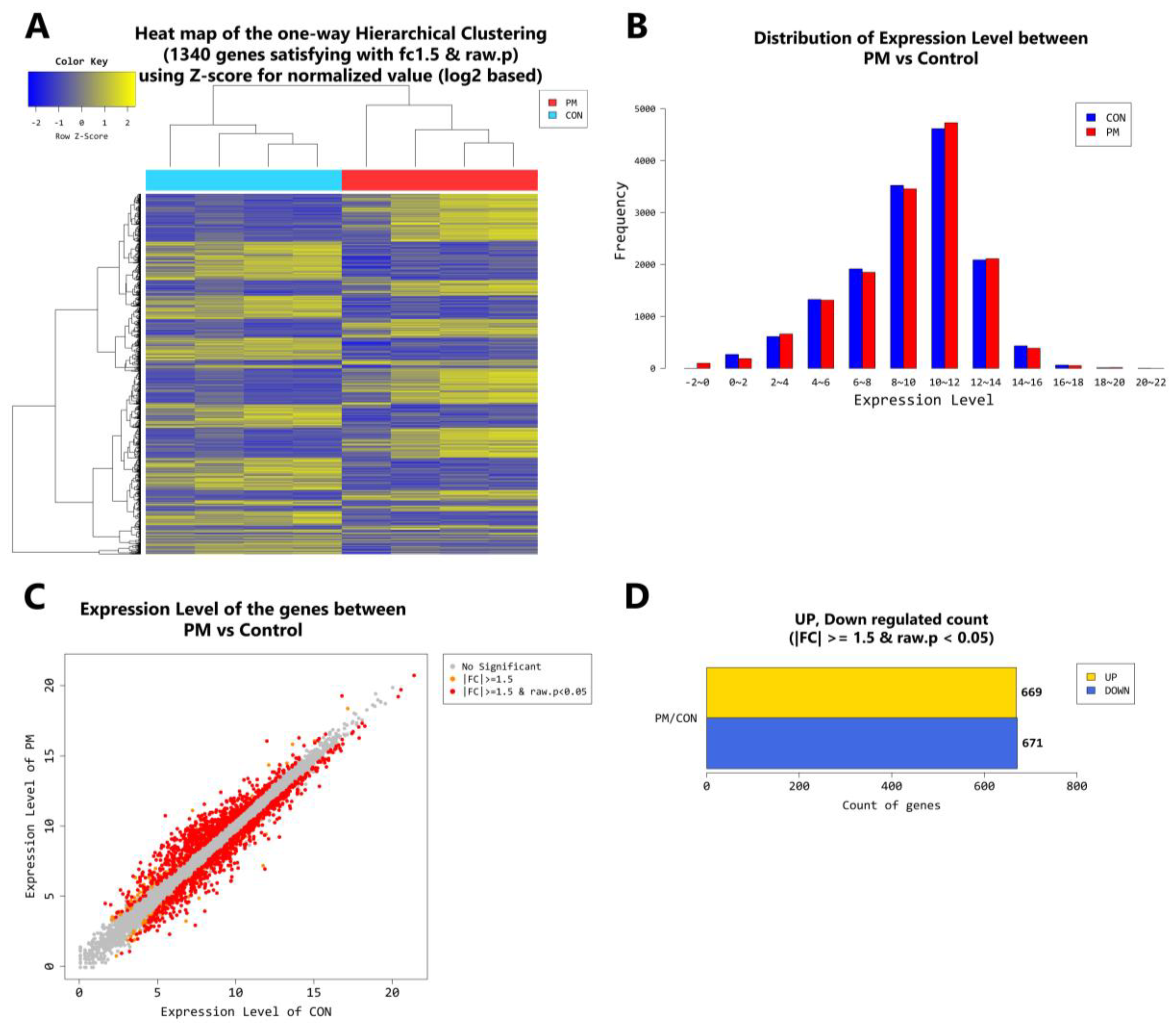
2.4. Gene Transcription Profile
3. Discussion
4. Materials and Methods
4.1. Skin Samples
4.2. Ethics
4.3. Cell Culture
4.4. Particulate Matter Preparation
4.5. Cell Viability Test
4.6. Cell Lysis and Western Blotting
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Immunochemistry and Confocal Microscopy
4.9. Transmission Electron Microscopy (TEM)
4.10. mRNA-Seq
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PM | Particulate matter |
| HDF | Human dermal fibroblast |
| IL | Interleukin |
| CYP1A1 | Cytochrome P450 family 1 subfamily A member 1 |
| CYP1B1 | Cytochrome P450 family 1 subfamily B member 1 |
| MMP | Matrix metalloproteinase |
| TGF | Transforming growth factor |
| COL1A1 | Collagen type 1 alpha 1 chain |
| COL1A2 | Collagen type 1 alpha 2 chain |
| ELN | Elastin |
| TEM | Transmission electron microscopy |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| CTSL | Cathepsin L |
| DAPK1 | Death associated protein kinase 1 |
| RRAGB | Ras related GTP binding B |
| DAPK2 | Death associated protein kinase 2 |
| AhR | Aryl hydrocarbon receptor |
| ROS | Reactive oxygen species |
| SNRPB | Small nuclear ribonucleoprotein polypeptides B and B1 |
| CHAF1B | Chromatin assembly factor 1 subunit B |
| KIAA1549L | KIAA 1549 like |
| CAPS | Calcyphosine |
| ADAMTS7 | ADAM metallopeptidase with thrombospondin type 1 motif 7 |
| SNRPD1 | Small nuclear ribonucleoprotein D1 polypeptide |
| ABL2 | ABL proto-oncogene 2, non-receptor tyrosine kinase |
| OBSCN | Obscurin, cytoskeletal calmodulin and titin-interacting RhoGEF |
| LMCD1 | LIM and cysteine rich domains 1 |
| SRPX | Sushi repeat containing protein, X-linked |
| LRRK2 | Leucine rich repeat kinase 2 |
| U2AF1 | U2 small nuclear RNA auxiliary factor 1 |
| HK2 | Hexokinase 2 |
| MAP1LC3A | Microtubule associated protein 1light chain 3 alpha |
| ITGB4 | Integrin subunit beta 4 |
| SQSTM1 | Sequestosome 1 |
| ZC3H12A | Zinc finger CCCH-type containing 12A |
| REP15 | RAB15 effector protein |
| RAB33A | RAB33A, member RAS oncogene family |
| MAP1A | Microtubule associated protein 1A |
| FAM131B | Family with sequence similarity 131 member B |
| FOXO1 | Forkhead box O1 |
| ALPK1 | Alpha kinase 1 |
| TMEM150C | Transmembrane protein 150C |
| ATP1B1 | ATPase NA+/K+ transporting subunit beta 1 |
| PRKN | Parkin RBR E3 ubiquitin protein ligase |
| PFKP | Phosphofructokinase, platelet |
| MTCL1 | Microtubule crosslinking factor 1 |
| DPF3 | Double PHD fingers 3 |
| PLOD2 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 |
| RFWD3 | Ring finger and WD repeat domain 3 |
| KIAA1324 | KIAA1324 |
References
- Schulze, C.; Wetzel, F.; Kueper, T.; Malsen, A.; Muhr, G.; Jaspers, S.; Blatt, T.; Wittern, K.P.; Wenck, H.; Kas, J.A. Stiffening of human skin fibroblasts with age. Biophys. J. 2010, 99, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Nandar, S.K.; Kathuria, S.; Ramesh, V. Effects of air pollution on the skin: A review. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 415–423. [Google Scholar] [PubMed]
- Morita, A.; Torii, K.; Maeda, A.; Yamaguchi, Y. Molecular basis of tobacco smoke-induced premature skin aging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Vierkotter, A.; Schikowski, T.; Ranft, U.; Sugiri, D.; Matsui, M.; Kramer, U.; Krutmann, J. Airborne particle exposure and extrinsic skin aging. J. Investig. Dermatol. 2010, 130, 2719–2726. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Vierkotter, A.; Schikowski, T.; Huls, A.; Ding, A.; Matsui, M.S.; Deng, B.; Ma, C.; Ren, A.; Zhang, J.; et al. Epidemiological evidence that indoor air pollution from cooking with solid fuels accelerates skin aging in Chinese women. J. Dermatol. Sci. 2015, 79, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Huls, A.; Vierkotter, A.; Gao, W.; Kramer, U.; Yang, Y.; Ding, A.; Stolz, S.; Matsui, M.; Kan, H.; Wang, S.; et al. Traffic-related air pollution contributes to development of facial lentigines: Further epidemiological evidence from Caucasians and Asians. J. Investig. Dermatol. 2016, 136, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Ferm, M.; Watt, J.; O’Hanlon, S.; De Santis, F.; Varotsos, C. Deposition measurement of particulate matter in connection with corrosion studies. Anal. Bioanal. Chem. 2006, 384, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Christodoulakis, J.; Varotsos, C.A.; Cracknell, A.P.; Kouremadas, G.A. The deterioration of materials as a result of air pollution as derived from satellite and ground based observations. Atmos. Environ. 2018, 185, 91–99. [Google Scholar] [CrossRef]
- Kim, J.; Kim, E.H.; Oh, I.; Jung, K.; Han, Y.; Cheong, H.K.; Ahn, K. Symptoms of atopic dermatitis are influenced by outdoor air pollution. J. Allergy Clin. Immunol. 2013, 132, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.O.; Kim, J.H.; Cho, S.I.; Chung, B.Y.; Ahn, I.S.; Lee, C.H.; Park, C.W. Improvement of atopic dermatitis severity after reducing indoor air pollutants. Ann. Dermatol. 2013, 25, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Magnani, N.D.; Muresan, X.M.; Belmonte, G.; Cervellati, F.; Sticozzi, C.; Pecorelli, A.; Miracco, C.; Marchini, T.; Evelson, P.; Valacchi, G. Skin damage mechanisms related to airborne particulate matter exposure. Toxicol. Sci. 2016, 149, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Rembiesa, J.; Ruzgas, T.; Engblom, J.; Holefors, A. The impact of pollutants on skin and proper efficacy testing for anti-pollution claims. Cosmetics 2018, 5. [Google Scholar] [CrossRef]
- Jin, S.P.; Li, Z.; Choi, E.K.; Lee, S.; Kim, Y.K.; Seo, E.Y.; Chung, J.H.; Cho, S. Urban particulate matter in air pollution penetrates into the barrier-disrupted skin and produces ROS-dependent cutaneous inflammatory response in vivo. J. Dermatol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Benson, H.A. Transdermal drug delivery: Penetration enhancement techniques. Curr. Drug Deliv. 2005, 2, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.L.; Wang, P.W.; Alijuffali, I.A.; Huang, C.T.; Lee, C.W.; Fang, J.Y. The impact of urban particulate pollution on skin barrier function and the subsequent drug absorption. J. Dermatol. Sci. 2015, 78, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Lin, Z.C.; Hu, S.C.; Chiang, Y.C.; Hsu, L.F.; Lin, Y.C.; Lee, I.T.; Tsai, M.H.; Fang, J.Y. Urban particulate matter down-regulates filaggrin via COX2 expression/PGE2 production leading to skin barrier dysfunction. Sci. Rep. 2016, 6, 27995. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Flutter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Beelen, R.; Hoek, G.; van den Brandt, P.A.; Goldbohm, R.A.; Fischer, P.; Schouten, L.J.; Armstrong, B.; Brunekreef, B. Long-term exposure to traffic-related air pollution and lung cancer risk. Epidemiology 2008, 19, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Castano-Vinyals, G.; Cantor, K.P.; Malats, N.; Tardon, A.; Garcia-Closas, R.; Serra, C.; Carrato, A.; Rothman, N.; Vermeulen, R.; Silverman, D.; et al. Air pollution and risk of urinary bladder cancer in a case-control study in Spain. Occup. Environ. Med. 2008, 65, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Cho, D.; Park, H.J. Air pollution and skin diseases: Adverse effects of airborne particulate matter on various skin diseases. Life Sci. 2016, 152, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Van Voorhis, M.; Knopp, S.; Julliard, W.; Fechner, J.H.; Zhang, X.; Schauer, J.J.; Mezrich, J.D. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS ONE 2013, 8, e82545. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Lim, H.K.; Hong, K.K.; Shin, M.K.; Lee, J.W.; Lee, S.W.; Kim, N.I. Cigarette smoke-induced interleukin-1 alpha may be involved in the pathogenesis of adult acne. Ann. Dermatol. 2014, 26, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Su, L.H.; Chen, T.H. Association of androgenetic alopecia with smoking and its prevalence among Asian men: A community-based survey. Arch. Dermatol. 2007, 143, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Puntoni, R.; Ceppi, M.; Gennaro, V.; Ugolini, D.; Puntoni, M.; La Manna, G.; Casella, C.; Merlo, D.F. Occupational exposure to carbon black and risk of cancer. Cancer Causes Control 2004, 15, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Morita, A.; Seite, S.; Haarmann-Stemmann, T.; Grether-Beck, S.; Krutmann, J. Environment-induced lentigines: Formation of solar lentigines beyond ultraviolet radiation. Exp. Dermatol. 2015, 24, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, J.S.; Kim, Y.C.; Chung, N.H.; Cho, M.H. Comparative study of PM2.5- and PM10-induced oxidative stress in rat lung epithelial cells. J. Vet. Sci. 2004, 5, 11–18. [Google Scholar] [PubMed]
- Huang, K.L.; Liu, S.Y.; Chou, C.C.; Lee, Y.H.; Cheng, T.J. The effect of size-segregated ambient particulate matter on Th1/Th2-like immune responses in mice. PLoS ONE 2017, 12, e0173158. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Liu, X.; Kobayashi, T.; Kohyama, T.; Wen, F.Q.; Romberger, D.J.; Conner, H.; Gilmour, P.S.; Donaldson, K.; MacNee, W.; et al. Ultrafine carbon black particles inhibit human lung fibroblast-mediated collagen gel contraction. Am. J. Respir. Cell Mol. Biol. 2003, 28, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Marchini, T.; Magnani, N.D.; Paz, M.L.; Vanasco, V.; Tasat, D.; Gonzalez Maglio, D.H.; Alvarez, S.; Evelson, P.A. Time course of systemic oxidative stress and inflammatory response induced by an acute exposure to residual oily fly ash. Toxicol. Appl. Pharmacol. 2014, 274, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role or aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Yun, J.W.; Park, J.H.; Park, B.W.; Kang, Y.H.; Hah, Y.S.; Hwang, S.C.; Woo, D.K.; Byun, J.H. Autophagy has a beneficial role in relieving cigarette-smoke induce apoptotic death in human gingival fibroblasts. Int. J. Med. Sci. 2016, 13, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Ramirez-Tortosa Mdel, C.; Gonzalez-Alonso, A.; Alfonsi, S.; Garcia-Marin, R.; de Miguel, M.; Battino, M. Autophagy in periodontitis patients and gingival fibroblasts: Unraveling the link between chronic disease and inflammation. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Lin, Z.Y.; Yang, L.W.; He, H.J.; Chen, T.; Xu, W.Y.; Li, C.Y.; Zhou, X.; Li, D.M.; Song, Z.Q.; et al. PM2.5 exacerbate allergic asthma involved in autophagy signaling pathway in mice. Int. J. Clin. Exp. Pathol. 2016, 9, 12247–12261. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PM10 Exposed HDFs/PM Non-Exposed (Control) HDFs | p-Value | |
|---|---|---|
| IL-6 ratio | 1.12 ± 0.05 | p = 0.046 |
| IL-8 ratio | 1.17 ± 0.05 | p = 0.037 |
| MMP-1 ratio | 2.46 ± 0.89 | p = 0.027 |
| Procollagen ratio | 0.77 ± 0.12 | p = 0.001 |
| TGF-β ratio | 0.58 ± 0.25 | p = 0.002 |
| PM/Control. Log2 fc | PM/Control. Log2 fc (Young) | PM/Control. Log2 fc (Old) | |
|---|---|---|---|
| IL-1β | 4.678166 | 8.732704 | 1.772352 |
| IL-6 | 2.593295 | 4.143132 | 1.463454 |
| IL-8 (CXCL8) | 3.634443 | 4.527864 | 2.403414 |
| IL-33 | 3.237957 | 3.337679 | 2.863740 |
| MMP-1 | 3.189445 | 2.380204 | 3.166181 |
| MMP-3 | 2.106478 | 6.295658 | 1.533143 |
| CYP1A1 (Cytochrome P450 1A1) | 4.844863 | 5.736596 | 2.106431 |
| CYP1B1 (Cytochrome P450 1B1) | 3.452584 | 2.917612 | 3.750746 |
| TGF-β | −2.772363 | −1.168725 | −2.549271 |
| COL1A1 (Collagen type I alpha 1 chain) | −2.022854 | −1.938849 | −1.915619 |
| COL1A2 (Collagen type I alpha 2 chain) | −1.800164 | −1.818843 | −1.666592 |
| ELN (Elastin) | −2.644748 | −2.094849 | −3.379390 |
| RRAGB | SNRPB | DAPK2 | CHAF1B | KIAA1549L | CAPS |
| ADAMTS7 | SNRPD1 | ABL2 | OBSCN | LMCD1 | SRPX |
| LRRK2 | U2AF1 | HK2 | MAP1LC3A | ITGB4 | SQSTM1 |
| DAPK1 | ZC3H12A | REP15 | RAB33A | MAP1A | FAM131B |
| FOXO1 | ALPK1 | TMEM150C | ATP1B1 | PRKN | PFKP |
| MTCL1 | DPF3 | PLOD2 | RFWD3 | KIAA1324 | CHAF1B |
| DAPK2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Byun, E.J.; Lee, J.D.; Kim, S.; Kim, H.S. Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts. Int. J. Mol. Sci. 2018, 19, 2727. https://doi.org/10.3390/ijms19092727
Park S-Y, Byun EJ, Lee JD, Kim S, Kim HS. Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts. International Journal of Molecular Sciences. 2018; 19(9):2727. https://doi.org/10.3390/ijms19092727
Chicago/Turabian StylePark, Seo-Yeon, Eun Jung Byun, Jeong Deuk Lee, Sungjoo Kim, and Hei Sung Kim. 2018. "Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts" International Journal of Molecular Sciences 19, no. 9: 2727. https://doi.org/10.3390/ijms19092727
APA StylePark, S.-Y., Byun, E. J., Lee, J. D., Kim, S., & Kim, H. S. (2018). Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts. International Journal of Molecular Sciences, 19(9), 2727. https://doi.org/10.3390/ijms19092727