Histone Deacetylase Inhibitors and Diabetic Kidney Disease

Abstract
1. Introduction
2. A Note on Terminology that Reflects the Changing Face of Diabetic Kidney Disease
3. Diabetic Kidney Disease: Scope of the Problem
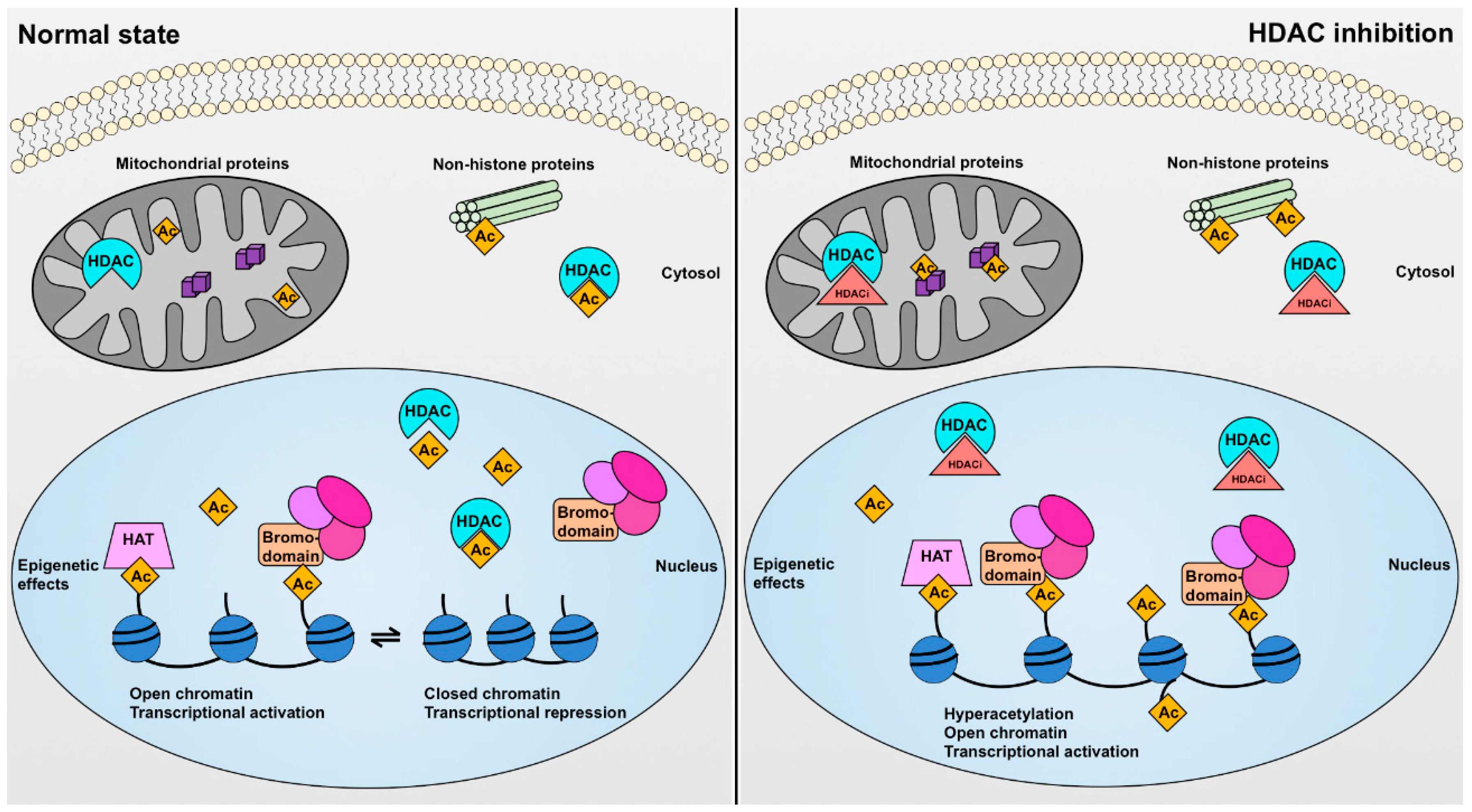
4. Histone Deacetylases and Their Biological Effects
5. Pharmacological HDAC Inhibitors
6. Evidence for Altered HDAC Activity in Diabetic Kidney Disease
7. HDAC Inhibitor Effects in Experimental Diabetic Kidney Disease
8. Hydroxamic Acids
8.1. Trichostatin A
8.2. Vorinostat
9. Short Chain Fatty Acids
9.1. Valproate
9.2. Sodium Butyrate
10. Future Directions
11. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Li, Y.; Wang, J.; Burrows, N.R.; Ali, M.K.; Rolka, D.; Williams, D.E.; Geiss, L. Changes in diabetes-related complications in the united states, 1990–2010. N. Engl. J. Med. 2014, 370, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Satija, A.; Manson, J.E. Curbing the diabetes pandemic: The need for global policy solutions. JAMA 2015, 313, 2319–2320. [Google Scholar] [CrossRef] [PubMed]
- Panchapakesan, U.; Pollock, C. Drug repurposing in kidney disease. Kidney Int. 2018, 94, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.; Huang, Q.; Thai, K.; Advani, S.L.; White, K.E.; Kelly, D.J.; Yuen, D.A.; Connelly, K.A.; Marsden, P.A.; Gilbert, R.E. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am. J. Pathol. 2011, 178, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Huang, Q.; Thai, K.; Advani, S.L.; Lee, K.; Yuen, D.A.; Connelly, K.A.; Advani, A. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: Potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011, 79, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Christensen, C.K.; Vittinghus, E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 1983, 32 (Suppl. 2), 64–78. [Google Scholar] [CrossRef] [PubMed]
- Perkins, B.A.; Ficociello, L.H.; Silva, K.H.; Finkelstein, D.M.; Warram, J.H.; Krolewski, A.S. Regression of microalbuminuria in type 1 diabetes. N. Engl. J. Med. 2003, 348, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.J.; Nguyen, Q.D.; Curhan, G.; Hsu, C.Y. Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. JAMA 2003, 289, 3273–3277. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on diabetic nephropathy: Core curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed. 2017. Available online: https://www.idf.org/e-library/epidemiology-research/diabetes-atlas.html (accessed on 24 January 2018).
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- United States Renal Data System. 2017 USRDS Annual Report: Epidemiology of Kidney Disease in the United States; National Instututes of Health, National Institute of Diabetes and Digesttive and Kidney Diseases: Bethesda, MD, USA, 2017. [Google Scholar]
- Eriksen, B.O.; Ingebretsen, O.C. The progression of chronic kidney disease: A 10-year population-based study of the effects of gender and age. Kidney Int. 2006, 69, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Penno, G.; Solini, A.; Orsi, E.; Bonora, E.; Fondelli, C.; Trevisan, R.; Vedovato, M.; Cavalot, F.; Lamacchia, O.; Scardapane, M.; et al. Non-albuminuric renal impairment is a strong predictor of mortality in individuals with type 2 diabetes: The renal insufficiency and cardiovascular events (riace) italian multicentre study. Diabetologia 2018. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The collaborative study group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Lehnert, H.; Brochner-Mortensen, J.; Gomis, R.; Andersen, S.; Arner, P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N. Engl. J. Med. 2001, 345, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of sglt2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhuang, S. Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front. Physiol. 2015, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.C.; Denis, G.V. Bet domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Yerra, V.G.; Advani, A. Histones and heart failure in diabetes. Cell. Mol. Life Sci. CMLS 2018. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [PubMed]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The smrt and n-cor corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.N.; Brijmohan, A.S.; Advani, A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin. Sci. 2016, 130, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.S.; Olsen, C.A. Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angew. Chem. Int. Ed. Engl. 2012, 51, 9083–9087. [Google Scholar] [CrossRef] [PubMed]
- Chun, P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch. Pharm. Res. 2018, 41, 162–183. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A phase ii trial of belinostat (pxd101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. Panorama 2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase ii multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous t-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Ning, Z.Q.; Xing, P.Y.; Xu, J.L.; Cao, H.X.; Dou, G.F.; Meng, Z.Y.; Shi, Y.K.; Lu, X.P.; Feng, F.Y. Phase i study of chidamide (cs055/hbi-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother. Pharmacol. 2012, 69, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jia, B.; Xu, W.; Li, W.; Liu, T.; Liu, P.; Zhao, W.; Zhang, H.; Sun, X.; Yang, H.; et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: A multicenter real-world study in china. J. Hematol. Oncol. 2017, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.M.; Naunton, M. Valproate: A simple chemical with so much to offer. J. Clin. Pharm. Ther. 2005, 30, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.S.; Chaudhary, D.K.; Mohan, A.; Kumar, P.; Chaturvedi, C.P.; Ecelbarger, C.M.; Godbole, M.M.; Tiwari, S. Greater efficacy of atorvastatin versus a non-statin lipid-lowering agent against renal injury: Potential role as a histone deacetylase inhibitor. Sci. Rep. 2016, 6, 38034. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Rue, T.C.; Cleary, P.A.; Lachin, J.M.; Molitch, M.E.; Steffes, M.W.; Sun, W.; Zinman, B.; Brunzell, J.D.; White, N.H.; et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: An analysis of the diabetes control and complications trial/epidemiology of diabetes interventions and complications cohort. Arch. Intern. Med. 2011, 171, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Physiol. Renal. Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhen, J.; Zhang, C.; Wan, Q.; Liu, G.; Wei, X.; Zhang, Y.; Wang, Z.; Han, H.; et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014, 86, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Bompada, P.; Atac, D.; Laakso, M.; Groop, L.; De Marinis, Y. Epigenetic regulation of glucose-stimulated osteopontin (opn) expression in diabetic kidney. Biochem. Biophys. Res. Commun. 2016, 469, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Kosanam, H.; Thai, K.; Zhang, Y.; Advani, A.; Connelly, K.A.; Diamandis, E.P.; Gilbert, R.E. Diabetes induces lysine acetylation of intermediary metabolism enzymes in the kidney. Diabetes 2014, 63, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Porter, L.E.; Gordon, M.; Fisher, N.D.; De’Oliveira, J.M.; Laffel, L.M.; Passan, D.R.; Williams, G.H.; Hollenberg, N.K. The paradox of the low-renin state in diabetic nephropathy. J. Am. Soc. Nephrol. 1999, 10, 2382–2391. [Google Scholar] [PubMed]
- Anderson, S.; Jung, F.F.; Ingelfinger, J.R. Renal renin-angiotensin system in diabetes: Functional, immunohistochemical, and molecular biological correlations. Am. J. Physiol. 1993, 265, F477–F486. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the mrl-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Nino, M.D.; Valino-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Y.; Cui, W.; Lou, Y.; Sun, G.; Zhang, D.; Miao, L. Role of epigenetic histone modifications in diabetic kidney disease involving renal fibrosis. J. Diabetes Res. 2017, 2017, 7242384. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G.; Tikoo, K. Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp. Mol. Pathol. 2015, 98, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G.; Tikoo, K.; Kumar, V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of nf-kappab/inos signaling in diabetic rat. Biochimie 2015, 110, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Qin, H.J.; Zhang, Z.; Xu, Y.; Yang, X.C.; Zhao, D.M.; Li, X.N.; Sun, L.K. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum stressinduced apoptosis. Mol. Med. Rep. 2016, 13, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jena, G. Sodium butyrate, a HDAC inhibitor ameliorates enos, inos and tgf-beta1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol. 2014, 73, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Jia, Y.; Liu, X.; Zhang, H.; Li, T.; Huang, W.; Chen, X.; Wang, F.; Sun, W.; Wu, H. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J. Endocrinol. 2017, 232, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Noh, H.; Seo, J.Y.; Yu, M.R.; Ha, H. Histone deacetylase inhibitors: A novel class of therapeutic agents in diabetic nephropathy. Kidney Int. Suppl. 2007, S61–S66. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Lee, J.Y.; Kim, J.S.; Ryu, J.H.; Choi, J.Y.; Lee, J.W.; Im, G.J.; Kim, T.K.; Seo, J.W.; Park, H.J.; et al. Synthesis and biological evaluation of 3-(4-substituted-phenyl)-n-hydroxy-2-propenamides, a new class of histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5745–5751. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.; Gilbert, R.E.; Thai, K.; Gow, R.M.; Langham, R.G.; Cox, A.J.; Connelly, K.A.; Zhang, Y.; Herzenberg, A.M.; Christensen, P.K.; et al. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J.Am. Soc. Nephrol. 2009, 20, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cdna from hl-60 cells treated with 1,25-dihydroxyvitamin d-3. Biochim. Biophys. Acta 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Majumder, S.; Thai, K.; Abdalla, M.; Hu, P.; Advani, S.L.; White, K.E.; Bowskill, B.B.; Guarna, G.; Dos Santos, C.C.; et al. The histone methyltransferase enzyme enhancer of zeste homolog 2 protects against podocyte oxidative stress and renal injury in diabetes. J. Am. Soc. Nephrol. 2016, 27, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, Y.; Cai, M.; Bompada, P.; Atac, D.; Kotova, O.; Johansson, M.E.; Garcia-Vaz, E.; Gomez, M.F.; Laakso, M.; Groop, L. Epigenetic regulation of the thioredoxin-interacting protein (txnip) gene by hyperglycemia in kidney. Kidney Int. 2016, 89, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, R.; Hou, Y.; Song, S.; Han, W.; Chen, N.; Du, Y.; Ren, Y.; Shi, Y. Thioredoxin-interacting protein deficiency ameliorates kidney inflammation and fibrosis in mice with unilateral ureteral obstruction. Lab. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.H.; Thornhill, B.A.; Forbes, M.S.; Coleman, C.M.; Marcinko, E.S.; Liaw, L.; Chevalier, R.L. Osteopontin regulates renal apoptosis and interstitial fibrosis in neonatal chronic unilateral ureteral obstruction. Kidney Int. 2006, 70, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Andersen, M.J. Increased kidney size and glomerular filtration rate in early juvenile diabetes. Diabetes 1973, 22, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Seyer-Hansen, K. Renal hypertrophy in streptozotocin-diabetic rats. Clin. Sci. Mol. Med. Suppl. 1976, 51, 551–555. [Google Scholar] [CrossRef]
- Seyer-Hansen, K. Renal hypertrophy in experimental diabetes mellitus. Kidney Int. 1983, 23, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Inomata, S. Renal hypertrophy as a prognostic index for the progression of diabetic renal disease in non-insulin-dependent diabetes mellitus. J. Diabetes Complicat. 1993, 7, 28–33. [Google Scholar] [CrossRef]
- Kleinman, K.S.; Fine, L.G. Prognostic implications of renal hypertrophy in diabetes mellitus. Diabetes Metab. Rev. 1988, 4, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Cox, A.; McNally, P.G.; Wu, L.L.; Dziadek, M.; Cooper, M.E.; Jerums, G. Increased epidermal growth factor in experimental diabetes related kidney growth in rats. Diabetologia 1997, 40, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Wassef, L.; Kelly, D.J.; Gilbert, R.E. Epidermal growth factor receptor inhibition attenuates early kidney enlargement in experimental diabetes. Kidney Int. 2004, 66, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Shaw, P.G.; Davidson, N.E. Inhibition of histone deacetylase suppresses egf signaling pathways by destabilizing egfr mrna in er-negative human breast cancer cells. Breast Cancer Res. Treat. 2009, 117, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Cipollone, F.; Mezzetti, A.; Chiarelli, F. The role of nitric oxide in the development of diabetic angiopathy. Horm. Metab. Res. 2004, 36, 319–335. [Google Scholar] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Chateauvieux, S.; Morceau, F.; Dicato, M.; Diederich, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 inhibition promotes transcription factor eb activation and is protective in experimental kidney disease. Front. Pharmacol. 2018, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. Tfeb links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Enami, Y.; Kurajyo, C.; Matsui-Yuasa, I.; Konishi, Y.; Kojima-Yuasa, A. The enhancement of phase 2 enzyme activities by sodium butyrate in normal intestinal epithelial cells is associated with nrf2 and p53. Mol. Cell. Biochem. 2012, 370, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, J.J.; Li, X.; Yang, Y.; Xie, X.F.; Hu, K. Post-occlusion administration of sodium butyrate attenuates cognitive impairment in a rat model of chronic cerebral hypoperfusion. Pharmacol. Biochem. Behav. 2015, 135, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D. Drug discovery for diabetic nephropathy: Trying the leap from mouse to man. Semin. Nephrol. 2012, 32, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Kothapally, J.; Mao, H.; Tolbert, E.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 2009, 297, F996–F1005. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Lee, S.M.; Kim, J.Y.; Kim, S.Y.; Kim, Y.H.; Kim, T.H.; Kang, M.S.; Jang, W.H.; Seo, S.K. Therapeutic activity of the histone deacetylase inhibitor sb939 on renal fibrosis. Int. Immunopharmacol. 2017, 42, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.P.; Tsai, Y.G.; Lin, T.Y.; Wu, M.J.; Lin, C.Y. The attenuation of renal fibrosis by histone deacetylase inhibitors is associated with the plasticity of FOXP3+IL-17+ T cells. BMC Nephrol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Thanh, T.; Kim, D.; Lee, S.; Kim, W.; Park, S.K.; Kang, K.P. Inhibition of histone deacetylase 1 ameliorates renal tubulointerstitial fibrosis via modulation of inflammation and extracellular matrix gene transcription in mice. Int J. Mol. Med. 2018, 41, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Song, J.H.; Kim, I.J.; Joo, S.Y.; Eom, G.H.; Kim, I.; Cha, H.; Cho, J.M.; Ma, S.K.; Kim, S.W.; et al. Histone deacetylase inhibitor, CG200745 attenuates renal fibrosis in obstructive kidney disease. Sci Rep. 2018, 8, 11546. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, F.; Noto, T.; Matsuoka, H.; Urano, Y.; Sudo, Y.; Takakura, S.; Mutoh, S. Prevention of renal interstitial fibrosis via histone deacetylase inhibition in rats with unilateral ureteral obstruction. Transpl. Immunol. 2010, 23, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Marumo, T.; Hishikawa, K.; Yoshikawa, M.; Hirahashi, J.; Kawachi, S.; Fujita, T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am. J. Physiol. Renal. Physiol. 2010, 298, F133–F141. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Dahllof, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R. Histone deacetylase inhibitors: Future therapeutics for insulin resistance and type 2 diabetes. Pharmacol. Res. 2016, 113, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Improving insulin sensitivity with HDAC inhibitor. Diabetes 2013, 62, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Dirice, E.; Ng, R.W.S.; Martinez, R.; Hu, J.; Wagner, F.F.; Holson, E.B.; Wagner, B.K.; Kulkarni, R.N. Isoform-selective inhibitor of histone deacetylase 3 (HDAC3) limits pancreatic islet infiltration and protects female nonobese diabetic mice from diabetes. J. Biol. Chem. 2017, 292, 17598–17608. [Google Scholar] [CrossRef] [PubMed]
- Kochar, D.K.; Rawat, N.; Agrawal, R.P.; Vyas, A.; Beniwal, R.; Kochar, S.K.; Garg, P. Sodium valproate for painful diabetic neuropathy: A randomized double-blind placebo-controlled study. QJM 2004, 97, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Moradei, O.; Raeppel, S.; Leit, S.; Frechette, S.; Gaudette, F.; Paquin, I.; Bernstein, N.; Bouchain, G.; Vaisburg, A.; et al. Discovery of n-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide (mgcd0103), an orally active histone deacetylase inhibitor. J. Med. Chem. 2008, 51, 4072–4075. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Pidugu, V.R.; Yarla, N.S.; Bishayee, A.; Kalle, A.M.; Satya, A.K. Novel histone deacetylase 8-selective inhibitor 1,3,4-oxadiazole-alanine hybrid induces apoptosis in breast cancer cells. Apoptosis 2017, 22, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Huber, E.; Kiefer, F.; Gottlicher, M. Specific and redundant functions of histone deacetylases in regulation of cell cycle and apoptosis. Cell Cycle 2004, 3, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Carrer, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Redundant control of adipogenesis by histone deacetylases 1 and 2. J. Biol. Chem. 2010, 285, 14663–14670. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Jurkin, J.; Zupkovitz, G.; Lagger, S.; Grausenburger, R.; Hagelkruys, A.; Kenner, L.; Seiser, C. Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle 2011, 10, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.T.; van Diepen, J.A.; Riksen, N.P.; El-Osta, A. Epigenetics in diabetic nephropathy, immunity and metabolism. Diabetologia 2018, 61, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Prescribing Information from BELEODAQ. Available online: http://www.beleodaq.com/downloads/Beleodaq_PI.pdf (accessed on 10 August 2018).
- Prescribing Information from FARYDAK. Available online: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/farydak.pdf (accessed on 10 August 2018).
- Prescribing Information from ISTODAX. Available online: https://media.celgene.com/content/uploads/sites/23/ISTODAX_Product_Monograph_English_Version.pdf (accessed on 10 August 2018).
- Product Monograph from ZOLINZA. Available online: http://www.merck.ca/static/pdf/ZOLINZA-PM_E.pdf (accessed on 10 August 2018).
- Prescribing Information from DEPAKENE. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/018081s056lbl.pdf (accessed on 10 August 2018).



{kind=link}
{kind=link}
| HDAC Inhibitor | Structural Class | HDAC Class Inhibited | Indication or Latest Phase of Development and Clinicaltrials.gov Identifier for that Condition |
|---|---|---|---|
| Vorinostat | Hydroxamic acid | Broad-spectrum | U.S. FDA approval (2006); cutaneous T cell lymphoma |
| Belinostat | Hydroxamic acid | Broad-spectrum | U.S. FDA approval (2014); peripheral T cell lymphoma |
| Panobinostat | Hydroxamic acid | Broad-spectrum | U.S. FDA approval (2015); multiple myeloma (in combination with bortezomib and dexamethasone) |
| Abexinostat | Hydroxamic acid | Broad-spectrum | Phase 1/2; sarcoma (NCT01027910) |
| Pracinostat | Hydroxamic acid | Broad-spectrum | Phase 3; acute myeloid leukemia (NCT03151408) |
| Resminostat | Hydroxamic acid | Broad-spectrum | Phase 2; advanced stage mycosis fungoides or Sézary syndrome (NCT02953301), Hodkin’s lymphoma (NCT01037478), hepatocellular carcinoma (NCT00943449) |
| Givinostat | Hydroxamic acid | Broad-spectrum | Phase 2/3; Duchenne muscular dystrophy (NCT03373968) |
| Quisinostat | Hydroxamic acid | Broad-spectrum | Phase 2; ovarian cancer (NCT02948075), cutaneous T cell lymphoma (NCT01486277) |
| Ricolinostat | Hydroxamic acid | HDAC6 (with some Class I inhibition) | Phase 2; diabetic neuropathic pain (NCT03176472) |
| Citarinostat | Hydroxamic acid | HDAC6 (with some Class I inhibition) | Phase 1; multiple myeloma (NCT02886065) |
| Dacinostat | Hydroxamic acid | Broad-spectrum | Not in clinical trial |
| Droxinostat | Hydroxamic acid | HDAC3, HDAC6, HDAC8 | Not in clinical trial |
| Trichostatin A | Hydroxamic acid | Broad-spectrum | Not in clinical trial |
| Valproate | Short-chain fatty acid | I, II | U.S. FDA approval for seizures, bipolar disorder and migraine (more recently reported to have HDAC inhibitory effects) |
| Sodium butyrate | Short-chain fatty acid | I, II | Phase 2/3; schizophrenia (NCT02654405; NCT03010865) |
| Romidepsin | Cyclic peptide | I | U.S. FDA approval (2009); cutaneous T cell lymphoma and peripheral T cell lymphoma |
| Tacedinaline | Benzamide | I | Phase 3; lung cancer (NCT00005093) |
| Chidamide | Benzamide | I, IIb | China FDA approval (2014); peripheral T cell lymphoma |
| Mocetinostat | Benzamide | I, IV | Phase 2; urothelial carcinoma (NCT02236195), metastatic leiomyosarcoma (NCT02303262), non-small cell lung cancer (NCT02954991) |
| AR-42 | Benzamide | I, II | Phase 1; renal cell carcinoma or soft tissue sarcoma (NCT02795819), vestibular schwannoma and meningioma (NCT02282917), acute myeloid leukemia (NCT01798901), multiple myeloma (NCT02569320), multiple myeloma, chronic lymphocytic leukemia or lymphoma (NCT01129193) |
| Entinostat | Benzamide | I | Phase 3; breast cancer (NCT03538171, NCT02115282) |
| Tubastatin A | Benzamide | HDAC6 | Not in clinical trial |
| SK-7041 | Hybrid, hydroxamic acid/benzamide | I | Not in clinical trial |
| Citation | HDAC Inhibitor Studied | HDAC Classes Inhibited | Experimental Models | Outcome |
|---|---|---|---|---|
| Noh et al., 2009 [54] | Trichostatin A SK-7041 | Trichostatin A, Class I & II; SK-7041 Class I | STZ-diabetic rats, NRK-52E cells | Trichostatin A decreased proteinuria and extracellular matrix production; SK-7041 decreased matrix protein production in vitro |
| Gilbert et al., 2011 [6] | Vorinostat | Classes I & II | STZ-diabetic rats, NRK-52E cells | Vorinostat downregulated EGFR expression and decreased tubule cell proliferation and diabetes-associated kidney growth |
| Advani et al., 2011 [5] | Vorinostat | Classes I & II | STZ-diabetic wildtype and eNOS−/− mice | Vorinostat downregulated eNOS and reduced oxidative stress, albuminuria and glomerular matrix production in STZ-diabetic wildtype mice but not STZ-diabetic eNOS−/− mice |
| Khan et al., 2015 (1) [63] | Valproate | Classes I & II | STZ-diabetic rats | Valproate decreased tubule injury and renal fibrosis |
| Khan et al., 2015 (2) [64] | Valproate | Classes I & II | STZ-diabetic rats | Valproate decreased proteinuria and normalized NF-κB upregulation and autophagy downregulation |
| Sun et al., 2016 [65] | Valproate | Classes I & II | STZ-diabetic rats | Valproate decreased proteinuria, glomerular matrix deposition, endoplasmic reticulum stress and programmed cell death |
| Khan & Jena, 2014 [66] | Sodium butyrate | Classes I & II | STZ-diabetic rats | Sodium butyrate lowered plasma glucose and NF-κB expression and attenuated kidney injury and matrix deposition |
| Dong et al., 2017 [67] | Sodium butyrate | Classes I & II | STZ-diabetic wildtype and Nrf2−/− mice | Sodium butyrate prevented Nrf2 downregulation and attenuated oxidative damage, inflammation, programmed cell death, fibrosis and albuminuria but was ineffective in STZ-Nrf2−/− mice |
| HDAC Inhibitor | Common Adverse Effects | Serious Adverse Effects |
|---|---|---|
| Belinostat | Nausea (42%), fatigue (37%), pyrexia (35%), anemia (32%), vomiting (29%), constipation (23%), diarrhea (23%), dyspnea (22%), rash (20%), peripheral edema (20%), cough (19%), thrombocytopenia (16%), pruritus (16%), chills (16%), decreased appetite (15%), abdominal pain (11%), hypotension (10%), phlebitis (10%), dizziness (10%) | Pneumonia, pyrexia, infection, anemia, increased creatinine, thrombocytopenia, and multi-organ failure (>2%) |
| Panobinostat (in combination with bortezomib and dexamethasone vs. placebo in combination with bortezomib and dexamethasone) | Arrhythmia (12%), diarrhea (68%), nausea (36%), vomiting (26%), fatigue (60%), peripheral edema (29%), pyrexia (26%), decreased weight (12%), decreased appetite (28%) | Pneumonia (18%), diarrhea (11%), thrombocytopenia (7%), fatigue (6%), sepsis (6%) |
| Romidepsin | Nausea (64%), diarrhea (36%), constipation (30%), hematological disorders (57%) including thrombocytopenia (41%), neutropenia (30%) and anemia (24%), asthenic conditions (55%), including fatigue (41%) and asthenia (16%), infections (55%), pyrexia (35%), anorexia (28%), dysgeusia (21%) | Infection (20%), pyrexia (8%), pneumonia, sepsis, vomiting (5%), cellulitis, deep vein thrombosis (4%), febrile neutropenia, gastrointestinal and abdominal pain (3%), chest pain, neutropenia, pulmonary embolism, dyspnea, dehydration (2%) |
| Valproate | Headache (31%), asthenia (27%), fever (6%), nausea (48%), vomiting (27%), abdominal pain (23%), diarrhea (13%), anorexia (12%), dyspepsia (8%), constipation (5), somnolence (27%), tremor (25%), dizziness (25%), diplopia (16%), amblyopia/blurred vision (12%), ataxia (8%), nystagmus (8%), emotional lability (6%), thinking abnormal (6%), amnesia (5%), flu syndrome (12%), infection (12%), bronchitis (5%), rhinitis (5%), alopecia (6%), weight loss (6%), depression (>5%), dyspnea (>5%), ecchymosis (>5%), increased appetite (>5%), insomnia (>5%), nervousness (>5%), peripheral edema (>5%), pharyngitis (>5%), thrombocytopenia (>5%), tinnitus (>5%), weight gain (>5%) | Hepatotoxicity, birth defects, pancreatitis, suicidal ideation, thromobocytopenia, hyperammonemia and hyperammonemic encephalopathy, hypothermia, multi-organ hypersensitivity reaction |
| Vorinostat | Fatigue (45%), diarrhea (47%), nausea (38%), dysgeusia (23%), thrombocytopenia (26%), anorexia (23%), decreased weight (20%), dry mouth (16%), vomiting (12%), increased blood creatinine (13%), alopecia (16%), decreased appetite (12%), muscle spasms (16%), anemia (13%), constipation (11%), chills (11%), dizziness (11%), abdominal pain (8%), proteinuria (8%), dyspnea (7%), headache (6%) | Pulmonary embolism (4.7%), anemia (2.3%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hadden, M.J.; Advani, A. Histone Deacetylase Inhibitors and Diabetic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 2630. https://doi.org/10.3390/ijms19092630
Hadden MJ, Advani A. Histone Deacetylase Inhibitors and Diabetic Kidney Disease. International Journal of Molecular Sciences. 2018; 19(9):2630. https://doi.org/10.3390/ijms19092630
Chicago/Turabian StyleHadden, Mitchell J., and Andrew Advani. 2018. "Histone Deacetylase Inhibitors and Diabetic Kidney Disease" International Journal of Molecular Sciences 19, no. 9: 2630. https://doi.org/10.3390/ijms19092630
APA StyleHadden, M. J., & Advani, A. (2018). Histone Deacetylase Inhibitors and Diabetic Kidney Disease. International Journal of Molecular Sciences, 19(9), 2630. https://doi.org/10.3390/ijms19092630