Updates on Old and Weary Haematopoiesis


Abstract
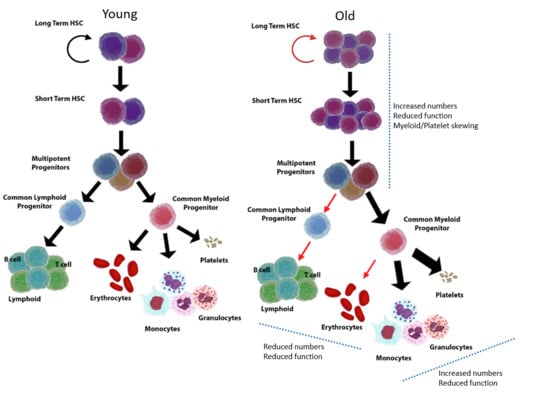
{kind=link}
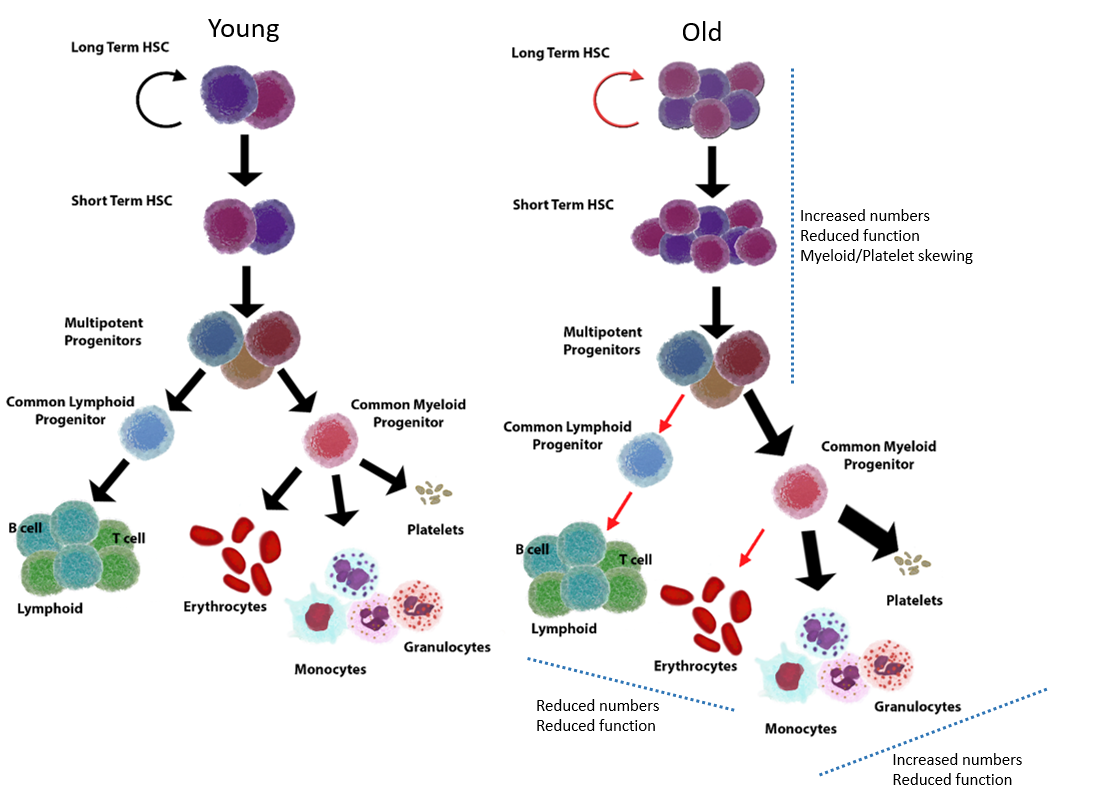
{kind=link}
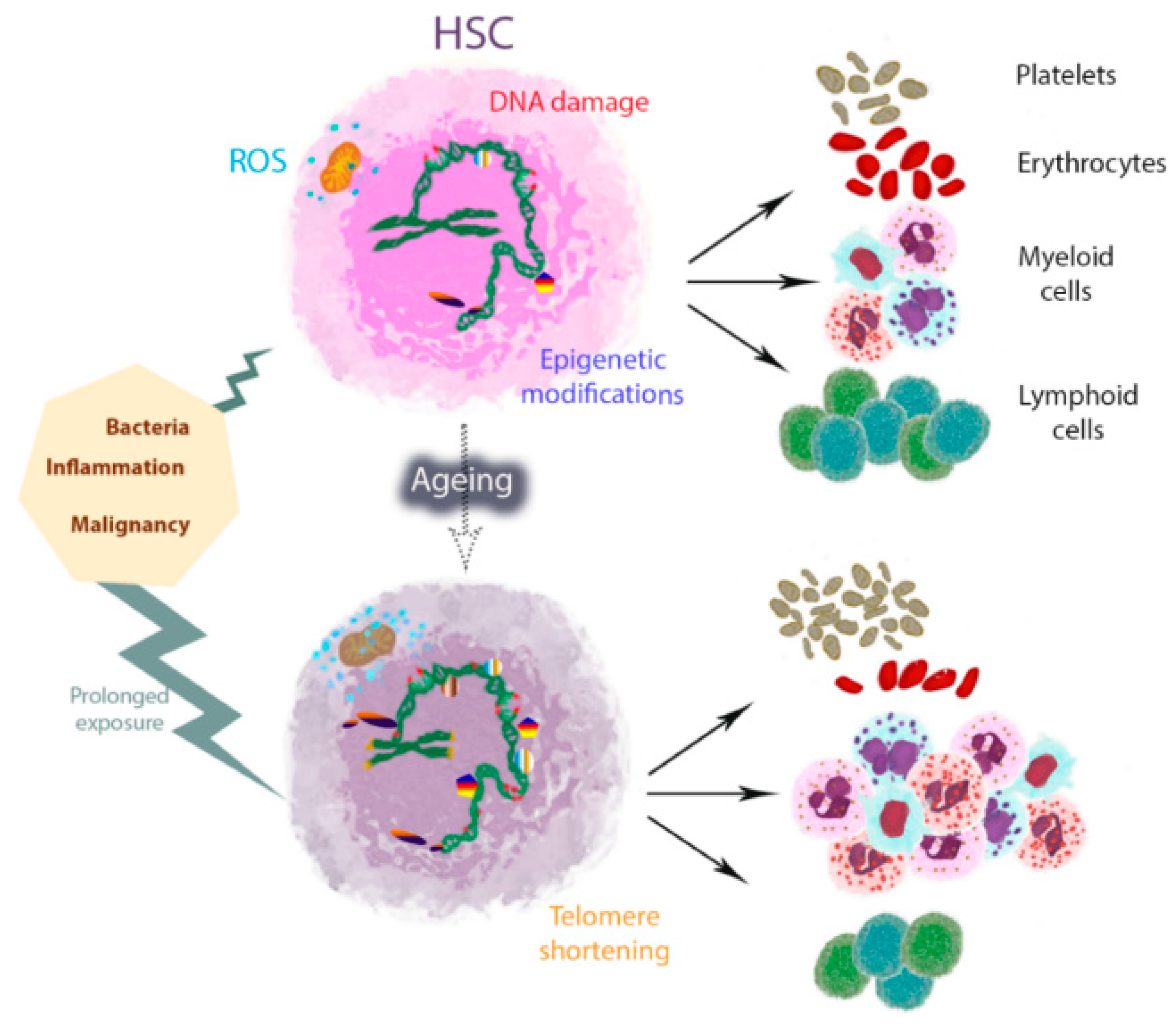
{kind=link}
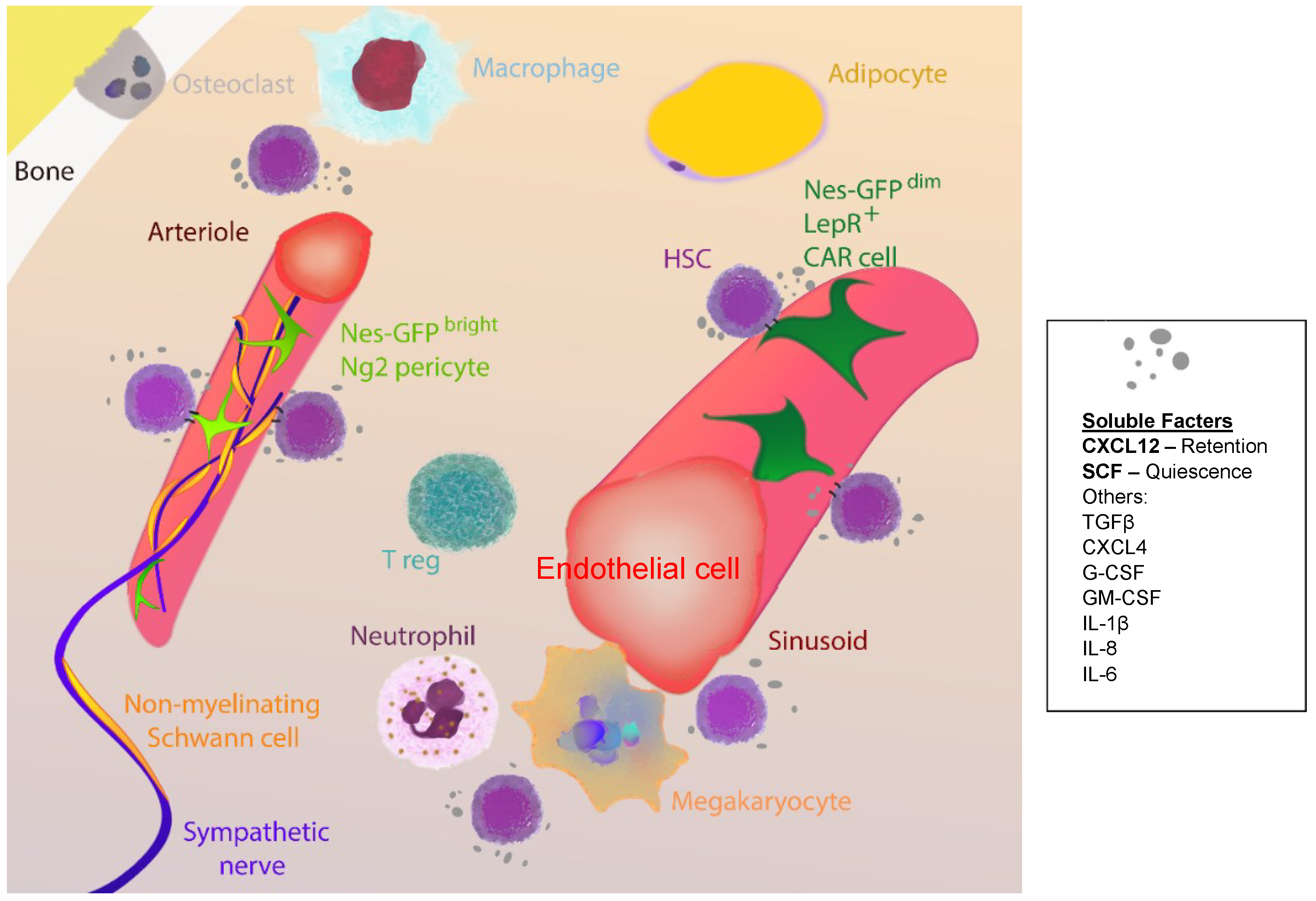
{kind=link}
1. Introduction
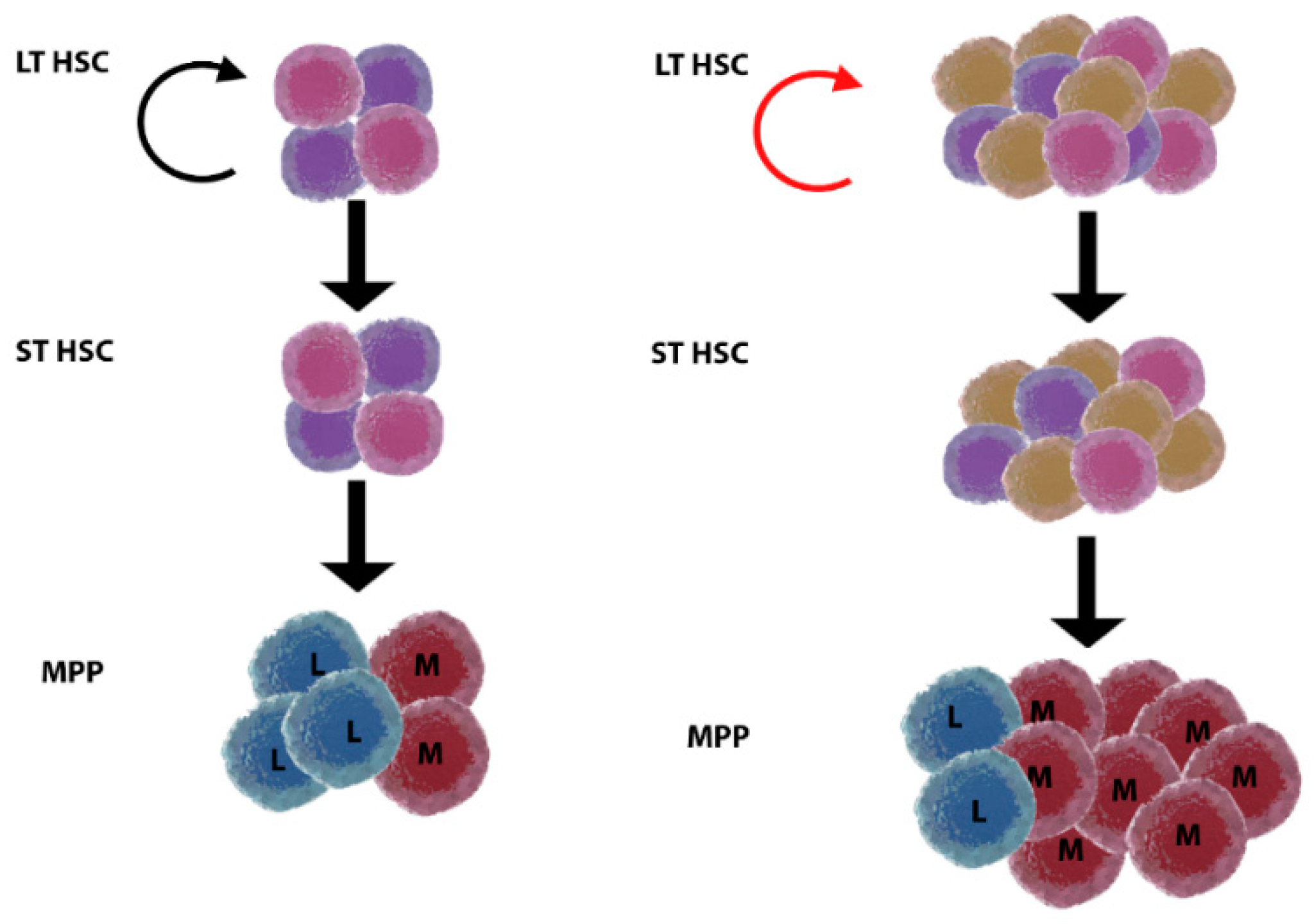
2. HSC Ageing and Myeloid/Platelet Skewing
3. Inflammageing and Its Relation to HSC Ageing
4. Ageing of the HSC Niche
5. Clonal Haematopoiesis and Age-Related Haematological Malignancies
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sanjuan-Pla, A.; Macaulay, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Matsuoka, S.; Bouriez Jones, T.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M.; Reynaud, D.; Kang, Y.A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Gottgens, B.; Passegue, E. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Schofield, R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells 1978, 4, 7–25. [Google Scholar] [PubMed]
- Gomez-Lopez, S.; Lerner, R.G.; Petritsch, C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell. Mol. Life Sci. 2014, 71, 575–597. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef] [PubMed]
- Verovskaya, E.; Broekhuis, M.J.C.; Zwart, E.; Ritsema, M.; van Os, R.; de Haan, G.; Bystrykh, L.V. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood 2013, 122, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-Associated Characteristics of Murine Hematopoietic Stem Cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Denkinger, M.D.; Leins, H.; Schirmbeck, R.; Florian, M.C.; Geiger, H. HSC aging and senescent immune remodeling. Trends Immunol. 2015, 36, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Linton, P.J.; Dorshkind, K. Age-related changes in lymphocyte development and function. Nat. Immunol. 2004, 5, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Montecino-Rodriguez, E.; Dorshkind, K. Reduction in the developmental potential of intrathymic T cell progenitors with age. J. Immunol. 2004, 173, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Young, K.; Borikar, S.; Bell, R.; Kuffler, L.; Philip, V.; Trowbridge, J.J. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J. Exp. Med. 2016, 213, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Montecino-Rodriguez, E.; Dorshkind, K. Effects of aging on the common lymphoid progenitor to pro-B cell transition. J. Immunol. 2006, 176, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- Signer, R.A.; Montecino-Rodriguez, E.; Witte, O.N.; McLaughlin, J.; Dorshkind, K. Age-related defects in B lymphopoiesis underlie the myeloid dominance of adult leukemia. Blood 2007, 110, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Geiger, H.; Van Zant, G. The aging of lympho-hematopoietic stem cells. Nat. Immunol. 2002, 3, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.K.; Arkins, S.; Li, Y.M.; Dantzer, R.; Kelley, K.W. Reduction in superoxide anion secretion and bactericidal activity of neutrophils from aged rats: Reversal by the combination of gamma interferon and growth hormone. Infect. Immun. 1994, 62, 1–8. [Google Scholar] [PubMed]
- Stout, R.D.; Suttles, J. Immunosenescence and macrophage functional plasticity: Dysregulation of macrophage function by age-associated microenvironmental changes. Immunol. Rev. 2005, 205, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Komatsubara, S.; Cinader, B.; Muramatsu, S. Polymorphism of age-related changes in stimulatory capacity of murine dendritic cells. Mech. Ageing Dev. 1986, 37, 163–173. [Google Scholar] [CrossRef]
- Ponnappan, S.; Ponnappan, U. Aging and immune function: Molecular mechanisms to interventions. Antioxid. Redox Signal. 2011, 14, 1551–1585. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Akunuru, S.; Geiger, H. Aging, Clonality; Rejuvenation of Hematopoietic Stem Cells. Trends Mol. Med. 2016, 22, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Muller-Sieburg, C.E.; Sieburg, H.B.; Bernitz, J.M.; Cattarossi, G. Stem cell heterogeneity: Implications for aging and regenerative medicine. Blood 2012, 119, 3900–3907. [Google Scholar] [CrossRef] [PubMed]
- Gekas, C.; Graf, T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 2013, 121, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, Á.M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Moehrle, B.M.; Nattamai, K.; Brown, A.; Florian, M.C.; Ryan, M.; Vogel, M.; Bliederhaeuser, C.; Soller, K.; Prows, D.R.; Abdollahi, A.; et al. Stem cell specific mechanisms ensure genomic fidelity within HSCs and upon aging of HSCs. Cell Rep. 2015, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–790. [Google Scholar] [CrossRef] [PubMed]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dragowska, W.; Allsopp, R.C.; Thomas, T.E.; Harley, C.B.; Lansdorp, P.M. Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age. Proc. Natl. Acad. Sci. USA 1994, 91, 9857–9860. [Google Scholar] [CrossRef] [PubMed]
- Lansdorp, P.M. Role of telomerase in hematopoietic stem cells. Ann. N. Y. Acad. Sci. 2005, 1044, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, K.; MacArthur, B.D.; Ikushima, Y.M.; Toyama, H.; Masuhiro, Y.; Hanazawa, S.; Suda, T.; Arai, F. The telomere binding protein Pot1 maintains haematopoietic stem cell activity with age. Nat. Commun. 2017, 8, 804. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, stress response; cancer in aging telomerase-deficient mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef]
- Raval, A.; Behbehani, G.K.; Nguyen, L.X.T.; Thomas, D.; Kusler, B.; Garbuzov, A.; Ramunas, J.; Holbrook, C.; Park, C.Y.; Blau, H.; et al. Reversibility of Defective Hematopoiesis Caused by Telomere Shortening in Telomerase Knockout Mice. PLoS ONE 2015, 10, e0131722. [Google Scholar] [CrossRef] [PubMed]
- Porto, M.L.; Rodrigues, B.P.; Menezes, T.N.; Ceschim, S.L.; Casarini, D.E.; Gava, A.L.; Pereira, T.M.; Vasquez, E.C.; Campagnaro, B.P.; Meyrelles, S.S. Reactive oxygen species contribute to dysfunction of bone marrow hematopoietic stem cells in aged C57BL/6 J mice. J. Biomed. Sci. 2015, 22, 97. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, D.O.; Byun, J.-E.; Kim, W.S.; Kim, M.J.; Song, H.Y.; Kim, Y.K.; Kang, D.-K.; Park, Y.-J.; Kim, T.-D.; et al. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat. Commun. 2016, 7, 13674. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.L.; Ma, C.; O’Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Lord, J.M.; De la Fuente, M. Preserved ex vivo inflammatory status and cytokine responses in naturally long-lived mice. Age 2010, 32, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Sanchez-Aguilera, A.; Martin-Perez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntion, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447s–455s. [Google Scholar] [CrossRef] [PubMed]
- Dybedal, I.; Bryder, D.; Fossum, A.; Rusten, L.S.; Jacobsen, S.E.W. Tumor necrosis factor (TNF)–mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood 2001, 98, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Kristinsson, S.Y.; Bjorkholm, M.; Hultcrantz, M.; Derolf, A.R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [PubMed]
- Hauer, J.; Martín-Lorenzo, A.; Sánchez-García, I. Infection causes childhood leukemia. Aging 2015, 7, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018, 18, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Smedby, K.E.; Askling, J.; Mariette, X.; Baecklund, E. Autoimmune and inflammatory disorders and risk of malignant lymphomas—An update. J. Intern. Med. 2008, 264, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Farkas, D.K.; Christiansen, C.F.; Hasselbalch, H.C.; Sørensen, H.T. Chronic myeloproliferative neoplasms and subsequent cancer risk: A Danish population-based cohort study. Blood 2011, 118, 6515–6520. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. The platelet-cancer loop in myeloproliferative cancer. Is thrombocythemia an enhancer of cancer invasiveness and metastasis in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk. Res. 2014, 38, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1beta as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Welner, R.S.; Amabile, G.; Bararia, D.; Czibere, A.; Yang, H.; Zhang, H.; Pontes, L.L.; Ye, M.; Levantini, E.; Di Ruscio, A.; et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell 2015, 27, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Williams, D.A.; Watt, F.M. Modulating the stem cell niche for tissue regeneration. Nat. Biotechnol. 2014, 32, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Mayack, S.R.; Shadrach, J.L.; Kim, F.S.; Wagers, A.J. Systemic signals regulate ageing and rejuvenation of blood stem cell niches. Nature 2010, 463, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Itkin, T.; Andaloussi Mäe, M.; Langen, U.H.; Betsholtz, C.; Lapidot, T.; Adams, R.H. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 2016, 532, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Bernal, A.; Arranz, L. Nestin-expressing progenitor cells: Function, identity and therapeutic implications. Cell. Mol. Life Sci. 2018, 75, 2177–2195. [Google Scholar] [CrossRef] [PubMed]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Marchand, T.; Yang, E.; Wei, Q.; Nerlov, C.; Frenette, P.S. Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev. Cell 2018, 44, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Deschler, B.; Lubbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Grisolano, J.L.; O’Neal, J.; Cain, J.; Tomasson, M.H. An activated receptor tyrosine kinase, TEL/PDGFβR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 9506–9511. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.T.; Kutok, J.L.; Williams, I.R.; Cohen, S.; Kelly, L.; Shigematsu, H.; Johnson, L.; Akashi, K.; Tuveson, D.A.; Jacks, T.; et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J. Clin. Investig. 2004, 113, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Heiser, D.; Li, L.; Kaplan, I.; Collector, M.; Huso, D.; Sharkis, S.J.; Civin, C.; Small, D. FLT3-ITD Knock-in Impairs Hematopoietic Stem Cell Quiescence/Homeostasis, Leading to Myeloproliferative Neoplasm. Cell Stem Cell 2012, 11, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Haigis, K.M.; McDaniel, A.; Harding-Theobald, E.; Kogan, S.C.; Akagi, K.; Wong, J.C.; Braun, B.S.; Wolff, L.; Jacks, T.; et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood 2011, 117, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Vera, G.; Claudia, H.; Niroshan, N.; Annette, F.; Sandra, W.; Andreas, R.; Christiane, E.; Elisa, S.; Wolfgang, K.; Torsten, H.; et al. CEBPA double-mutated acute myeloid leukaemia harbours concomitant molecular mutations in 76·8% of cases with TET2 and GATA2 alterations impacting prognosis. Br. J. Haematol. 2013, 161, 649–658. [Google Scholar]
- Zhao, S.; Zhang, Y.; Sha, K.; Tang, Q.; Yang, X.; Yu, C.; Liu, Z.; Sun, W.; Cai, L.; Xu, C.; et al. KRAS (G12D) cooperates with AML1/ETO to initiate a mouse model mimicking human acute myeloid leukemia. Cell. Physiol. Biochem. 2014, 33, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; McArthur, G.A.; Westmoreland, S.V.; Chambon, P.; Scadden, D.T.; Purton, L.E. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007, 129, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.H.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yu, W.M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Asada, N. Regulation of Malignant Hematopoiesis by Bone Marrow Microenvironment. Front. Oncol. 2018, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Itzkovitz, S.; Schuh, A.C. Aging, clonal hematopoiesis and preleukemia: Not just bad luck? Int. J. Hematol. 2015, 102, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukemic hematopoietic stem cells in acute leukemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T. Hematopoietic stem cell heterogeneity: Subtypes, not unpredictable behavior. Cell Stem Cell 2010, 6, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.M.; Simons, B.D. Universal patterns of stem cell fate in cycling adult tissues. Development 2011, 138, 3103–3111. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Ueno, T.; Fukumura, K.; Yamato, A.; Ando, M.; Yamaguchi, H.; Soda, M.; Kawazu, M.; Sai, E.; Yamashita, Y.; et al. Leukemic evolution of donor-derived cells harboring IDH2 and DNMT3A mutations after allogeneic stem cell transplantation. Leukemia 2014, 28, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Vas, V.; Senger, K.; Dorr, K.; Niebel, A.; Geiger, H. Aging of the microenvironment influences clonality in hematopoiesis. PLoS ONE 2012, 7, e42080. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konieczny, J.; Arranz, L. Updates on Old and Weary Haematopoiesis. Int. J. Mol. Sci. 2018, 19, 2567. https://doi.org/10.3390/ijms19092567
Konieczny J, Arranz L. Updates on Old and Weary Haematopoiesis. International Journal of Molecular Sciences. 2018; 19(9):2567. https://doi.org/10.3390/ijms19092567
Chicago/Turabian StyleKonieczny, Joanna, and Lorena Arranz. 2018. "Updates on Old and Weary Haematopoiesis" International Journal of Molecular Sciences 19, no. 9: 2567. https://doi.org/10.3390/ijms19092567
APA StyleKonieczny, J., & Arranz, L. (2018). Updates on Old and Weary Haematopoiesis. International Journal of Molecular Sciences, 19(9), 2567. https://doi.org/10.3390/ijms19092567