The Aryl Hydrocarbon Receptor and the Nervous System


Abstract
{kind=link}
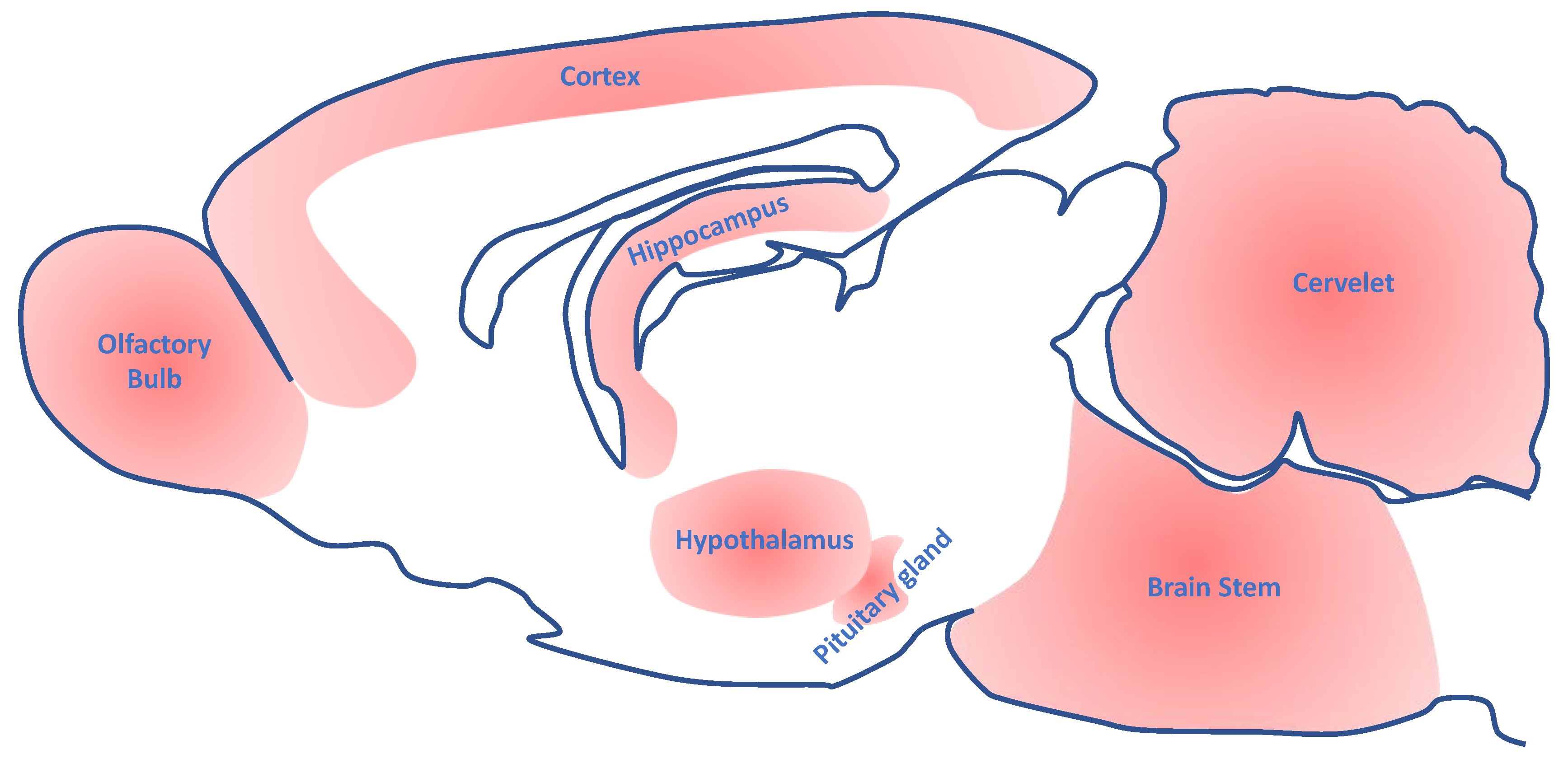{kind=link}
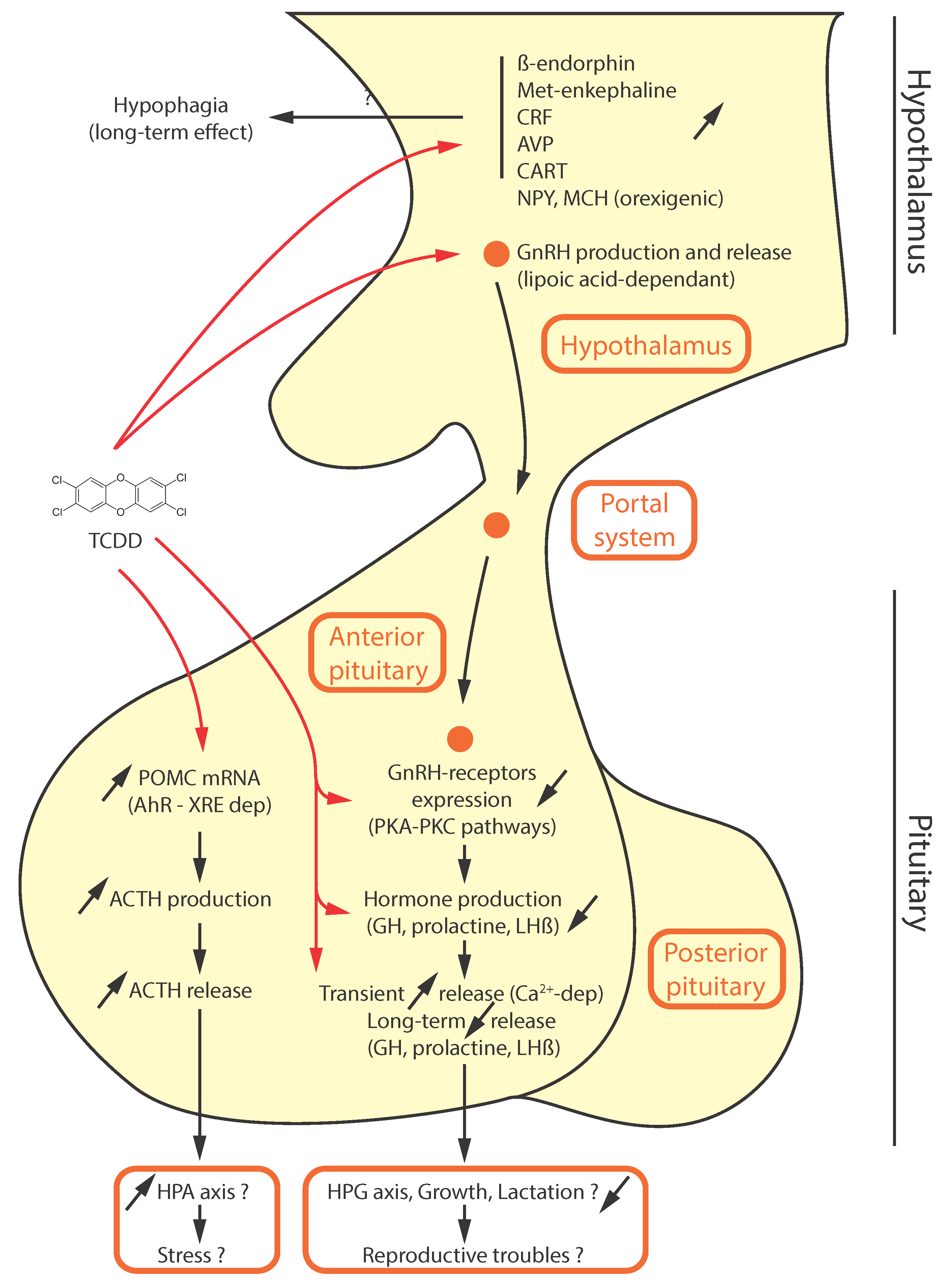{kind=link}
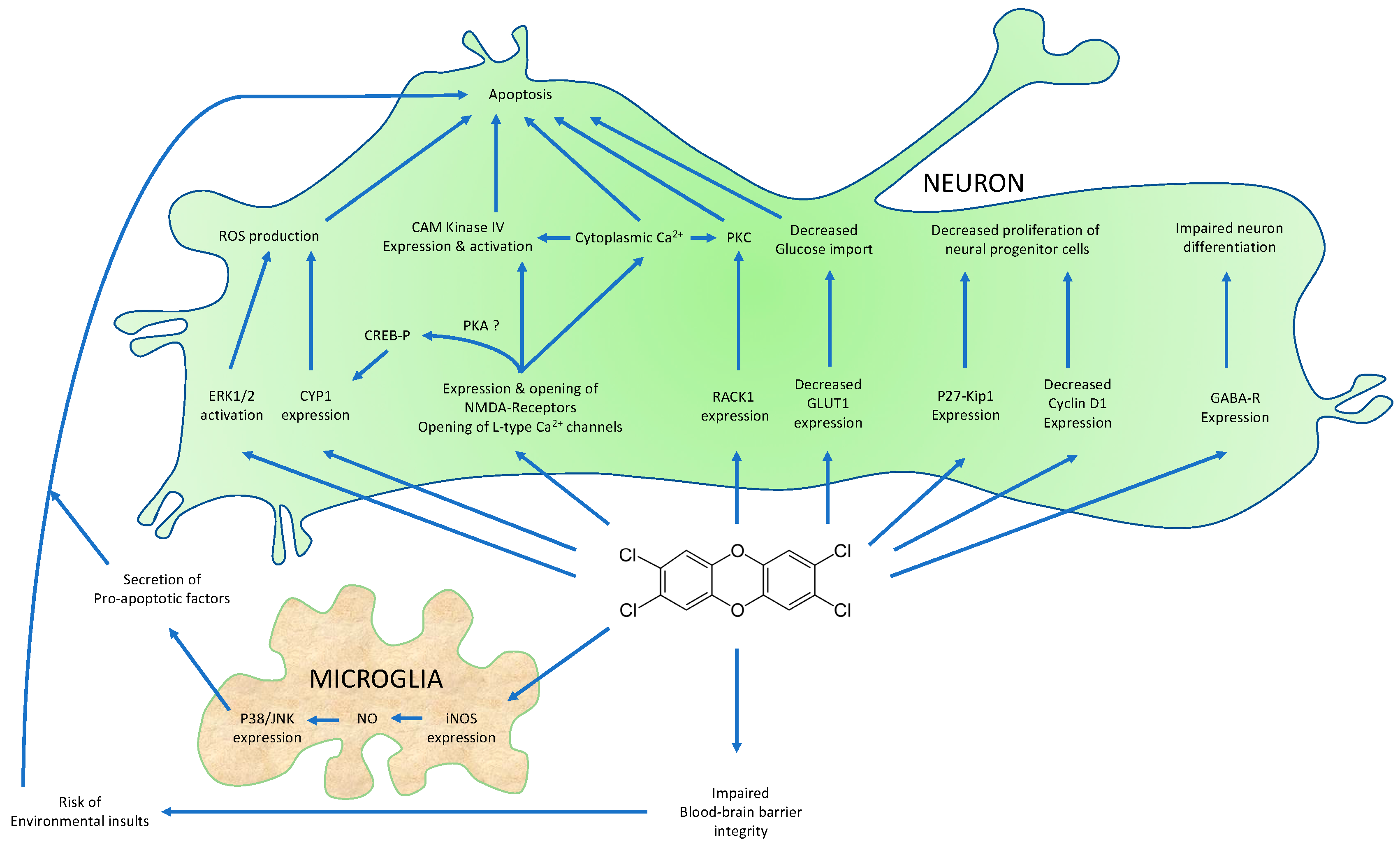{kind=link}
1. Introduction
2. Expression and Functions of the AhR in Invertebrates
3. Expression of the AhR in the Nervous System of Vertebrates
4. Disruption of Neuroendocrine Functions by AhR Ligands
5. Impact of AhR Ligands on Neurogenesis, Cell Proliferation, Differentiation, and Survival in the Nervous System of Vertebrates and Possible Mechanisms
5.1. Neurogenesis, Cell Proliferation, Differentiation and Migration
5.2. Cell Survival
5.3. Indirect Effects of AhR Ligands on Neuron Survival
6. Influence of AhR Ligands on Rodents’ Behavior and Neurotransmitter Levels
7. The AhR Regulates the Function of the Blood Brain Barrier (BBB)
8. The Physiological Functions of the AhR in the Central Nervous System
9. Conclusions
Conflicts of Interest
References
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Barouki, R.; Aggerbeck, M.; Aggerbeck, L.; Coumoul, X. The aryl hydrocarbon receptor system. Drug Metabol. Drug Interact. 2012, 27, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Barouki, R.; Coumoul, X.; Fernandez-Salguero, P.M. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 2007, 581, 3608–3615. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Powell-Coffman, J.A.; Jin, Y. The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Dev. Camb. Engl. 2004, 131, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Powell-Coffman, J.A. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev. Biol. 2004, 270, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhai, Z.; Powell-Coffman, J.A. The Caenorhabditis elegans AHR-1 transcription complex controls expression of soluble guanylate cyclase genes in the URX neurons and regulates aggregation behavior. Dev. Biol. 2006, 298, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Jevince, A.R.; Guan, L.; Wang, J.; Hall, D.H.; Huang, X.; Ding, M. Neuronal target identification requires AHA-1-mediated fine-tuning of Wnt signaling in C. elegans. PLoS Genet. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed]
- Schantz, S.L.; Gasior, D.M.; Polverejan, E.; McCaffrey, R.J.; Sweeney, A.M.; Humphrey, H.E.; Gardiner, J.C. Impairments of memory and learning in older adults exposed to polychlorinated biphenyls via consumption of Great Lakes fish. Environ. Health Perspect. 2001, 109, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Schantz, S.L.; Bowman, R.E. Learning in monkeys exposed perinatally to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Neurotoxicol. Teratol. 1989, 11, 13–19. [Google Scholar] [CrossRef]
- Negishi, T.; Shimomura, H.; Koyama, T.; Kawasaki, K.; Ishii, Y.; Kyuwa, S.; Yasuda, M.; Kuroda, Y.; Yoshikawa, Y. Gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin affects social behaviors between developing rhesus monkeys (Macaca mulatta). Toxicol. Lett. 2006, 160, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.D.; Jan, L.Y.; Jan, Y.N. The bHLH-PAS protein Spineless is necessary for the diversification of dendrite morphology of Drosophila dendritic arborization neurons. Genes Dev. 2006, 20, 2806–2819. [Google Scholar] [CrossRef] [PubMed]
- Wernet, M.F.; Mazzoni, E.O.; Celik, A.; Duncan, D.M.; Duncan, I.; Desplan, C. Stochastic spineless expression creates the retinal mosaic for colour vision. Nature 2006, 440, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Bats, A.-S.; Chevallier, A.; Bui, L.-C.; Ambolet-Camoit, A.; Garlatti, M.; Aggerbeck, M.; Barouki, R.; Coumoul, X. Induction of the Ras activator Son of Sevenless 1 by environmental pollutants mediates their effects on cellular proliferation. Biochem. Pharmacol. 2011, 81, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.A.; Kelley, M.L.; Powell, W.H.; Hahn, M.E.; Van Beneden, R.J. An aryl hydrocarbon receptor (AHR) homologue from the soft-shell clam, Mya arenaria: Evidence that invertebrate AHR homologues lack 2,3,7,8-tetrachlorodibenzo-p-dioxin and beta-naphthoflavone binding. Gene 2001, 278, 223–234. [Google Scholar] [CrossRef]
- Jain, S.; Maltepe, E.; Lu, M.M.; Simon, C.; Bradfield, C.A. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech. Dev. 1998, 73, 117–123. [Google Scholar] [CrossRef]
- Kimura, E.; Tohyama, C. Embryonic and postnatal expression of aryl hydrocarbon receptor mRNA in mouse brain. Front. Neuroanat. 2017, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Moran, T.B.; Brannick, K.E.; Raetzman, L.T. Aryl-hydrocarbon receptor activity modulates prolactin expression in the pituitary. Toxicol. Appl. Pharmacol. 2012, 265, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Pravettoni, A.; Colciago, A.; Negri-Cesi, P.; Villa, S.; Celotti, F. Ontogenetic development, sexual differentiation, and effects of Aroclor 1254 exposure on expression of the arylhydrocarbon receptor and of the arylhydrocarbon receptor nuclear translocator in the rat hypothalamus. Reprod. Toxicol. Elmsford N. Y. 2005, 20, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Latchney, S.E.; Hein, A.M.; O’Banion, M.K.; DiCicco-Bloom, E.; Opanashuk, L.A. Deletion or activation of the aryl hydrocarbon receptor alters adult hippocampal neurogenesis and contextual fear memory. J. Neurochem. 2013, 125, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.A.; Gasiewicz, T.A.; Opanashuk, L.A. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: Implications for development and dioxin neurotoxicity. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 83, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.L.; Williamson, M.A.; Thompson, B.D.; Dever, D.P.; Gasiewicz, T.A.; Opanashuk, L.A. 2,3,7,8-Tetracholorodibenzo-p-dioxin exposure disrupts granule neuron precursor maturation in the developing mouse cerebellum. Toxicol. Sci. Off. J. Soc. Toxicol. 2008, 103, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, A.; Mialot, A.; Petit, J.-M.; Fernandez-Salguero, P.; Barouki, R.; Coumoul, X.; Beraneck, M. Oculomotor deficits in aryl hydrocarbon receptor null mouse. PLoS ONE 2013, 8, e53520. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, J.M.; Stockton, P.S.; Sieber, S.; Foley, J.; Portier, C.J. AhR-mediated gene expression in the developing mouse telencephalon. Reprod. Toxicol. Elmsford N. Y. 2009, 28, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Filbrandt, C.R.; Wu, Z.; Zlokovic, B.; Opanashuk, L.; Gasiewicz, T.A. Presence and functional activity of the aryl hydrocarbon receptor in isolated murine cerebral vascular endothelial cells and astrocytes. Neurotoxicology 2004, 25, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Stegeman, J.J.; Woodin, B.R.; Iwanaga, T.; Harano, R.; Peterson, R.E.; Hiraga, T.; Teraoka, H. Role of zebrafish cytochrome P450 CYP1C genes in the reduced mesencephalic vein blood flow caused by activation of AHR2. Toxicol. Appl. Pharmacol. 2011, 253, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, G.; Zhao, J.; Nie, X.; Wan, C.; Liu, J.; Duan, Z.; Xu, G. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces microglial nitric oxide production and subsequent rat primary cortical neuron apoptosis through p38/JNK MAPK pathway. Toxicology 2013, 312, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.L.; Curran, M.A.; Marconi, S.A.; Carpenter, C.D.; Lubbers, L.S.; McAbee, M.D. Distribution of mRNAs encoding the arylhydrocarbon receptor, arylhydrocarbon receptor nuclear translocator, and arylhydrocarbon receptor nuclear translocator-2 in the rat brain and brainstem. J. Comp. Neurol. 2000, 427, 428–439. [Google Scholar] [CrossRef]
- Mukai, M.; Lin, T.-M.; Peterson, R.E.; Cooke, P.S.; Tischkau, S.A. Behavioral rhythmicity of mice lacking AhR and attenuation of light-induced phase shift by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Rhythms 2008, 23, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Yang, Z.; Shi, R.; Luo, C.; Zhang, Z. Expression of aryl hydrocarbon receptor in rat brain lesions following traumatic brain injury. Diagn. Pathol. 2016, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Wójtowicz, A.K.; Szychowski, K.A.; Wnuk, A.; Kajta, M. Dibutyl Phthalate (DBP)-Induced Apoptosis and Neurotoxicity are Mediated via the Aryl Hydrocarbon Receptor (AhR) but not by Estrogen Receptor Alpha (ERα), Estrogen Receptor Beta (ERβ), or Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) in Mouse Cortical Neurons. Neurotox. Res. 2017, 31, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Imanishi, S.; Manabe, N. Effects of exposure in utero to bisphenol a on the expression of aryl hydrocarbon receptor, related factors, and xenobiotic metabolizing enzymes in murine embryos. J. Reprod. Dev. 2005, 51, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Takata, K.; Kakimura, J.; Umeki, M.; Azukawa, S.; Suzuki, S.; Taniguchi, T. Aryl hydrocarbon receptor nuclear translocator is induced by kainic acid in rat hippocampal glial cells. Neurosci. Lett. 2000, 291, 117–120. [Google Scholar] [CrossRef]
- Elovaara, E.; Savolainen, H.; Parkki, M.G.; Aitio, A.; Vainio, H. Neurochemical effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Wistar and Gunn rats. Res. Commun. Chem. Pathol. Pharmacol. 1977, 18, 487–494. [Google Scholar] [PubMed]
- Henshel, D.S.; Martin, J.W.; Norstrom, R.; Whitehead, P.; Steeves, J.D.; Cheng, K.M. Morphometric abnormalities in brains of great blue heron hatchlings exposed in the wild to PCDDs. Environ. Health Perspect. 1995, 103, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Henshel, D.S.; Martin, J.W.; DeWitt, J.C. Brain asymmetry as a potential biomarker for developmental TCDD intoxication: A dose-response study. Environ. Health Perspect. 1997, 105, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Patisaul, H.B.; Petersen, S.L. Aryl hydrocarbon receptor activation in lactotropes and gonadotropes interferes with estradiol-dependent and -independent preprolactin, glycoprotein alpha and luteinizing hormone beta gene expression. Mol. Cell. Endocrinol. 2011, 333, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Aluru, N.; Vijayan, M.M. Brain transcriptomics in response to beta-naphthoflavone treatment in rainbow trout: The role of aryl hydrocarbon receptor signaling. Aquat. Toxicol. Amst. Neth. 2008, 87, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gouédard, C.; Barouki, R.; Morel, Y. Dietary polyphenols increase paraoxonase 1 gene expression by an aryl hydrocarbon receptor-dependent mechanism. Mol. Cell. Biol. 2004, 24, 5209–5222. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, D.; Xiao, G.-H.; Leingartner, K.; Chu, I.; Musicki, B.; Tsang, B.K. Comparisons of brain, uterus, and liver mRNA expression for cytochrome p450s, DNA methyltransferase-1, and catechol-o-methyltransferase in prepubertal female Sprague-Dawley rats exposed to a mixture of aryl hydrocarbon receptor agonists. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 86, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mizuyachi, K.; Terranova, P.F.; Rozman, K.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin decreases responsiveness of the hypothalamus to estradiol as a feedback inducer of preovulatory gonadotropin secretion in the immature gonadotropin-primed rat. Toxicol. Appl. Pharmacol. 2001, 170, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Chaffin, C.L.; Peterson, R.E.; Hutz, R.J. In utero and lactational exposure of female Holtzman rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin: Modulation of the estrogen signal. Biol. Reprod. 1996, 55, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lindén, J.; Korkalainen, M.; Lensu, S.; Tuomisto, J.; Pohjanvirta, R. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and leptin on hypothalamic mRNA expression of factors participating in food intake regulation in a TCDD-sensitive and a TCDD-resistant rat strain. J. Biochem. Mol. Toxicol. 2005, 19, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bestervelt, L.L.; Nolan, C.J.; Cai, Y.; Maimansomsuk, P.; Mousigian, C.A.; Piper, W.N. Tetrachlorodibenzo-p-dioxin alters rat hypothalamic endorphin and mu opioid receptors. Neurotoxicol. Teratol. 1991, 13, 495–497. [Google Scholar] [CrossRef]
- Cheng, S.B.; Kuchiiwa, S.; Kawachi, A.; Gao, H.Z.; Gohshi, A.; Kozako, T.; Kuchiiwa, T.; Nakagawa, S. Up-regulation of methionine-enkephalin-like immunoreactivity by 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment in the forebrain of the Long-Evans rat. J. Chem. Neuroanat. 2003, 25, 73–82. [Google Scholar] [CrossRef]
- Moon, B.H.; Hong, C.G.; Kim, S.Y.; Kim, H.J.; Shin, S.K.; Kang, S.; Lee, K.J.; Kim, Y.K.; Lee, M.S.; Shin, K.H. A single administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin that produces reduced food and water intake induces long-lasting expression of corticotropin-releasing factor, arginine vasopressin, and proopiomelanocortin in rat brain. Toxicol. Appl. Pharmacol. 2008, 233, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Shridhar, S.; Farley, A.; Reid, R.L.; Foster, W.G.; Van Vugt, D.A. The effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on corticotrophin-releasing hormone, arginine vasopressin, and pro-opiomelanocortin mRNA levels in the hypothalamus of the cynomolgus monkey. Toxicol. Sci. Off. J. Soc. Toxicol. 2001, 63, 181–188. [Google Scholar] [CrossRef]
- Fetissov, S.O.; Huang, P.; Zhang, Q.; Mimura, J.; Fujii-Kuriyama, Y.; Rannug, A.; Hökfelt, T.; Ceccatelli, S. Expression of hypothalamic neuropeptides after acute TCDD treatment and distribution of Ah receptor repressor. Regul. Pept. 2004, 119, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Solak, K.A.; Wijnolts, F.M.J.; Pralong, F.P.; Blaauboer, B.J.; van den Berg, M.; Westerink, R.H.; van Duursen, M.B.M. In vitro neuroendocrine effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the AhR-expressing hypothalamic rat GnV-3 cell line. Toxicology 2013, 311, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Pohjanvirta, R.; Rahko, T.; Tuomisto, J. TCDD decreases rapidly and persistently serum melatonin concentration without morphologically affecting the pineal gland in TCDD-resistant Han/Wistar rats. Pharmacol. Toxicol. 1991, 69, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Ishida, T.; Takeda, T.; Koga, T.; Fujii, M.; Ishii, Y.; Fujimura, Y.; Miura, D.; Wariishi, H.; Yamada, H. Maternal exposure to dioxin reduces hypothalamic but not pituitary metabolome in fetal rats: A possible mechanism for a fetus-specific reduction in steroidogenesis. J. Toxicol. Sci. 2010, 35, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Kakeyama, M.; Sone, H.; Tohyama, C. Perinatal exposure of female rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin induces central precocious puberty in the offspring. J. Endocrinol. 2008, 197, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.H.; Buckley, A.R.; Shah, G.N.; Sipes, I.G.; Blask, D.E.; Benson, B. Hypothalamic site of action of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Appl. Pharmacol. 1988, 94, 496–502. [Google Scholar] [CrossRef]
- Chaffin, C.L.; Trewin, A.L.; Watanabe, G.; Taya, K.; Hutz, R.J. Alterations to the pituitary-gonadal axis in the peripubertal female rat exposed in utero and through lactation to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol. Reprod. 1997, 56, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Johnson, D.C.; Rozman, K.K. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) increases release of luteinizing hormone and follicle-stimulating hormone from the pituitary of immature female rats in vivo and in vitro. Toxicol. Appl. Pharmacol. 1997, 142, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, J.; Taketoh, J.; Okamura, K.; Kagawa, T.; Ishida, T.; Ishii, Y.; Yamada, H. Fetal pituitary gonadotropin as an initial target of dioxin in its impairment of cholesterol transportation and steroidogenesis in rats. Endocrinology 2006, 147, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Taketoh, J.; Mutoh, J.; Takeda, T.; Ogishima, T.; Takeda, S.; Ishii, Y.; Ishida, T.; Yamada, H. Suppression of fetal testicular cytochrome P450 17 by maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin: A mechanism involving an initial effect on gonadotropin synthesis in the pituitary. Life Sci. 2007, 80, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Ishida, T.; Takeda, T.; Ishii, Y.; Uchi, H.; Tsukimori, K.; Yamamoto, M.; Himeno, M.; Furue, M.; Yamada, H. Restoration of dioxin-induced damage to fetal steroidogenesis and gonadotropin formation by maternal co-treatment with α-lipoic acid. PLoS ONE 2012, 7, e40322. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Matsumoto, Y.; Koga, T.; Mutoh, J.; Nishimura, Y.; Shimazoe, T.; Ishii, Y.; Ishida, T.; Yamada, H. Maternal exposure to dioxin disrupts gonadotropin production in fetal rats and imprints defects in sexual behavior. J. Pharmacol. Exp. Ther. 2009, 329, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Bookstaff, R.C.; Kamel, F.; Moore, R.W.; Bjerke, D.L.; Peterson, R.E. Altered regulation of pituitary gonadotropin-releasing hormone (GnRH) receptor number and pituitary responsiveness to GnRH in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated male rats. Toxicol. Appl. Pharmacol. 1990, 105, 78–92. [Google Scholar] [CrossRef]
- Bookstaff, R.C.; Moore, R.W.; Peterson, R.E. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases the potency of androgens and estrogens as feedback inhibitors of luteinizing hormone secretion in male rats. Toxicol. Appl. Pharmacol. 1990, 104, 212–224. [Google Scholar] [CrossRef]
- Clements, R.J.; Lawrence, R.C.; Blank, J.L. Effects of intrauterine 2,3,7,8-tetrachlorodibenzo-p-dioxin on the development and function of the gonadotrophin releasing hormone neuronal system in the male rat. Reprod. Toxicol. Elmsford N. Y. 2009, 28, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Fujii, M.; Hattori, Y.; Yamamoto, M.; Shimazoe, T.; Ishii, Y.; Himeno, M.; Yamada, H. Maternal exposure to dioxin imprints sexual immaturity of the pups through fixing the status of the reduced expression of hypothalamic gonadotropin-releasing hormone. Mol. Pharmacol. 2014, 85, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Trewin, A.L.; Woller, M.J.; Wimpee, B.A.B.; Conley, L.K.; Baldridge, M.G.; Hutz, R.J. Short-term hormone release from adult female rat hypothalamic and pituitary explants is not altered by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Reprod. Dev. 2007, 53, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Yamamoto, M.; Himeno, M.; Takechi, S.; Yamaguchi, T.; Ishida, T.; Ishii, Y.; Yamada, H. 2,3,7,8-tetrachlorodibenzo-p-dioxin potentially attenuates the gene expression of pituitary gonadotropin β-subunits in a fetal age-specific fashion: A comparative study using cultured pituitaries. J. Toxicol. Sci. 2011, 36, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Elango, A.; Shepherd, B.; Chen, T.T. Effects of endocrine disrupters on the expression of growth hormone and prolactin mRNA in the rainbow trout pituitary. Gen. Comp. Endocrinol. 2006, 145, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Ceccatelli, S.; Håkansson, H.; Grandison, L.; Rannug, A. Constitutive and TCDD-induced expression of Ah receptor-responsive genes in the pituitary. Neurotoxicology 2002, 23, 783–793. [Google Scholar] [CrossRef]
- Bestervelt, L.L.; Pitt, J.A.; Nolan, C.J.; Cai, Y.; Piper, D.W.; Dybowski, J.A.; Dayharsh, G.A.; Piper, W.N. In vitro 2,3,7,8-tetrachlorodibenzo-p-dioxin interference with the anterior pituitary hormone adrenocorticortropin. Toxicol. Sci. Off. J. Soc. Toxicol. 1998, 44, 107–115. [Google Scholar] [CrossRef]
- Bestervelt, L.L.; Pitt, J.A.; Piper, W.N. Evidence for Ah receptor mediation of increased ACTH concentrations in primary cultures of rat anterior pituitary cells exposed to TCDD. Toxicol. Sci. Off. J. Soc. Toxicol. 1998, 46, 294–299. [Google Scholar] [CrossRef]
- Ahmed, R.G. Perinatal TCDD exposure alters developmental neuroendocrine system. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2011, 49, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Miyabara, Y.; Sato, M.; Yonemoto, J.; Tohyama, C. Immunohistochemical localization of thyroid stimulating hormone induced by a low oral dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin in female Sprague-Dawley rats. Toxicology 2002, 171, 73–82. [Google Scholar] [CrossRef]
- Latchney, S.E.; Lioy, D.T.; Henry, E.C.; Gasiewicz, T.A.; Strathmann, F.G.; Mayer-Pröschel, M.; Opanashuk, L.A. Neural precursor cell proliferation is disrupted through activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Stem Cells Dev. 2011, 20, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Q.; Jung, J.W.; Lee, Y.S.; Kim, J.A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits cell proliferation through arylhydrocarbon receptor-mediated G1 arrest in SK-N-SH human neuronal cells. Neurosci. Lett. 2004, 363, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Paradisi, M.; D’Intino, G.; Del Vecchio, G.; Sivilia, S.; Giardino, L.; Calzà, L. A single prenatal exposure to the endocrine disruptor 2,3,7,8-tetrachlorodibenzo-p-dioxin alters developmental myelination and remyelination potential in the rat brain. J. Neurochem. 2010, 115, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Hirano, T.; Omotehara, T.; Hashimoto, R.; Umemura, Y.; Yuasa, H.; Masuda, N.; Kubota, N.; Minami, K.; Yanai, S.; et al. Immunohistochemical analysis of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) toxicity on the developmental dentate gyrus and hippocampal fimbria in fetal mice. J. Vet. Med. Sci. Jpn. Soc. Vet. Sci. 2015, 77, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kubo, K.I.; Endo, T.; Ling, W.; Nakajima, K.; Kakeyama, M.; Tohyama, C. Impaired dendritic growth and positioning of cortical pyramidal neurons by activation of aryl hydrocarbon receptor signaling in the developing mouse. PLoS ONE 2017, 12, e0183497. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.Q.; Xu, H.M.; Fu, H.L.; Hu, Q.; Tian, W.J.; Pei, X.H.; Zhao, B. AhR-mediated effects of dioxin on neuronal acetylcholinesterase expression in vitro. Environ. Health Perspect. 2013, 121, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Takanaga, H.; Yoshitake, T.; Yatabe, E.; Hara, S.; Kunimoto, M. Beta-naphthoflavone disturbs astrocytic differentiation of C6 glioma cells by inhibiting autocrine interleukin-6. J. Neurochem. 2004, 90, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Liang, L.; Xi, H.; Jiang, S.; Jiang, J.; Tang, C.; Liu, X.; Liu, S.; Wan, C.; Zhao, J.; et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces premature senescence of astrocytes via WNT/β-catenin signaling and ROS production. J. Appl. Toxicol. JAT 2015, 35, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Li, Y.; Yoshimoto, K.; Wu, Q.; Chen, G.; Iwata, T.; Mizusawa, N.; Wan, C.; Nie, X. 2,3,7,8-Tetrachlorodibenzo-p-dioxin stimulates proliferation of HAPI microglia by affecting the Akt/GSK-3β/cyclin D1 signaling pathway. Toxicol. Lett. 2014, 224, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Legare, M.E.; Hanneman, W.H.; Barhoumi, R.; Burghardt, R.C.; Tiffany-Castiglioni, E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters hippocampal astroglia-neuronal gap junctional communication. Neurotoxicology 2000, 21, 1109–1116. [Google Scholar] [PubMed]
- Mitsui, T.; Taniguchi, N.; Kawasaki, N.; Kagami, Y.; Arita, J. Fetal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin induces expression of the chemokine genes Cxcl4 and Cxcl7 in the perinatal mouse brain. J. Appl. Toxicol. JAT 2011, 31, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Rothhammer, V.; Quintana, F.J. Control of immune-mediated pathology via the aryl hydrocarbon receptor. J. Biol. Chem. 2017, 292, 12383–12389. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Quintana, F.J. Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Pantazis, G.; Schittenhelm, J.; Tabatabai, G.; Köhle, C.; Wick, W.; Schwarz, M.; Weller, M.; Tritschler, I. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 2009, 28, 2593–2605. [Google Scholar] [CrossRef] [PubMed]
- Silginer, M.; Burghardt, I.; Gramatzki, D.; Bunse, L.; Leske, H.; Rushing, E.J.; Hao, N.; Platten, M.; Weller, M.; Roth, P. The aryl hydrocarbon receptor links integrin signaling to the TGF-β pathway. Oncogene 2016, 35, 3260–3271. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kubo, K.; Matsuyoshi, C.; Benner, S.; Hosokawa, M.; Endo, T.; Ling, W.; Kohda, M.; Yokoyama, K.; Nakajima, K.; et al. Developmental origin of abnormal dendritic growth in the mouse brain induced by in utero disruption of aryl hydrocarbon receptor signaling. Neurotoxicol. Teratol. 2015, 52, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Kubo, K.-I.; Endo, T.; Nakajima, K.; Kakeyama, M.; Tohyama, C. Excessive activation of AhR signaling disrupts neuronal migration in the hippocampal CA1 region in the developing mouse. J. Toxicol. Sci. 2017, 42, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Gassmann, K.; Abel, J.; Bothe, H.; Haarmann-Stemmann, T.; Merk, H.F.; Quasthoff, K.N.; Rockel, T.D.; Schreiber, T.; Fritsche, E. Species-specific differential AhR expression protects human neural progenitor cells against developmental neurotoxicity of PAHs. Environ. Health Perspect. 2010, 118, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Imran, S.; Ferretti, P.; Vrzal, R. Different regulation of aryl hydrocarbon receptor-regulated genes in response to dioxin in undifferentiated and neuronally differentiated human neuroblastoma SH-SY5Y cells. Toxicol. Mech. Methods 2015, 25, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Morales-Hernández, A.; Corrales-Redondo, M.; Marcos-Merino, J.M.; González-Rico, F.J.; Sánchez-Martín, F.J.; Merino, J.M. AhR-dependent 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in human neuronal cell line SHSY5Y. NeuroToxicology 2016, 56, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, M.C.; Beggiato, S.; Ferraro, L.; Tanganelli, S.; Marani, L.; Lorenzini, L.; Antonelli, T. Prenatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin produces alterations in cortical neuron development and a long-term dysfunction of glutamate transmission in rat cerebral cortex. Neurochem. Int. 2012, 61, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Howard, C.V.; Strahle, U.; Cossins, A. Neurodevelopmental defects in zebrafish (Danio rerio) at environmentally relevant dioxin (TCDD) concentrations. Toxicol. Sci. Off. J. Soc. Toxicol. 2003, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Szychowski, K.A.; Wnuk, A.; Kajta, M.; Wójtowicz, A.K. Triclosan activates aryl hydrocarbon receptor (AhR)-dependent apoptosis and affects Cyp1a1 and Cyp1b1 expression in mouse neocortical neurons. Environ. Res. 2016, 151, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.P.; Hegde, V.L.; Hofseth, L.J.; Nagarkatti, M.; Nagarkatti, P. Resveratrol (trans-3,5,4′-trihydroxystilbene) ameliorates experimental allergic encephalomyelitis, primarily via induction of apoptosis in T cells involving activation of aryl hydrocarbon receptor and estrogen receptor. Mol. Pharmacol. 2007, 72, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, F.J.; Fernández-Salguero, P.M.; Merino, J.M. Aryl hydrocarbon receptor-dependent induction of apoptosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells from mouse. J. Neurochem. 2011, 118, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, F.J.; Fernández-Salguero, P.M.; Merino, J.M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptosis in neural growth factor (NGF)-differentiated pheochromocytoma PC12 cells. Neurotoxicology 2010, 31, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Rzemieniec, J.; Litwa, E.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. Selective Aryl Hydrocarbon Receptor Modulator 3,3′-Diindolylmethane Impairs AhR and ARNT Signaling and Protects Mouse Neuronal Cells Against Hypoxia. Mol. Neurobiol. 2016, 53, 5591–5606. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.F.; Sun, Y.Y.; Yang, L.Y.; Hu, S.Y.; Tsai, S.Y.; Lee, W.S.; Lee, Y.H. Bcl-2 gene family expression in the brain of rat offspring after gestational and lactational dioxin exposure. Ann. N. Y. Acad. Sci. 2005, 1042, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Jung, J.S.; Jin, I.; Jung, Y.W.; Ko, B.H.; Nam, K.S.; Park, I.K.; Moon, I.S. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the expression of synaptic proteins in dissociated rat cortical cells. Mol. Cells 2002, 14, 238–244. [Google Scholar] [PubMed]
- Dong, W.; Teraoka, H.; Kondo, S.; Hiraga, T. 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin induces apoptosis in the dorsal midbrain of zebrafish embryos by activation of arylhydrocarbon receptor. Neurosci. Lett. 2001, 303, 169–172. [Google Scholar] [CrossRef]
- Huang, P.; Tofighi, R.; Emgard, M.; Ceccatelli, S. Cell death induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (2,3,7,8-TCDD) in AtT-20 pituitary cells. Toxicology 2005, 207, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Hanneman, W.H.; Legare, M.E.; Tiffany-Castiglioni, E.; Safe, S.H. The need for cellular, biochemical, and mechanistic studies. Neurotoxicol. Teratol. 1996, 18, 247–250. [Google Scholar] [CrossRef]
- Hanneman, W.H.; Legare, M.E.; Barhoumi, R.; Burghardt, R.C.; Safe, S.; Tiffany-Castiglioni, E. Stimulation of calcium uptake in cultured rat hippocampal neurons by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology 1996, 112, 19–28. [Google Scholar] [CrossRef]
- Morales-Hernández, A.; Sánchez-Martín, F.J.; Hortigón-Vinagre, M.P.; Henao, F.; Merino, J.M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptosis by disruption of intracellular calcium homeostasis in human neuronal cell line SHSY5Y. Apoptosis Int. J. Program. Cell. Death 2012, 17, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Zhang, Y.; Jiang, J.; Jiang, S.; Nie, X.; Li, A.; Guo, A.; Wu, Q. Critical Role of TAK1-Dependent Nuclear Factor-κB Signaling in 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced Astrocyte Activation and Subsequent Neuronal Death. Neurochem. Res. 2015, 40, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Kim, S.Y.; Choi, E.J.; Park, K.Y.; Yang, J.H. Translocation of PKC-betaII is mediated via RACK-1 in the neuronal cells following dioxin exposure. Neurotoxicology 2007, 28, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, H.G.; Choi, E.J.; Park, K.Y.; Yang, J.H. TCDD alters PKC signaling pathways in developing neuronal cells in culture. Chemosphere 2007, 67, S421–S427. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Grover, C.A.; Safe, S.H.; Tiffany-Castiglioni, E.; Frye, G.D. Halogenated aromatic hydrocarbons suppress CA1 field excitatory postsynaptic potentials in rat hippocampal slices. Toxicol. Appl. Pharmacol. 1998, 148, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yang, J.H. Neurotoxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells. Exp. Mol. Med. 2005, 37, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhou, Q.; Wan, C.; Wang, Y.; Liu, J.; Li, Y.; Nie, X.; Cheng, C.; Chen, G. 2,3,7,8-TCDD induces neurotoxicity and neuronal apoptosis in the rat brain cortex and PC12 cell line through the down-regulation of the Wnt/β-catenin signaling pathway. Neurotoxicology 2013, 37, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, E.A.; Li, F.; Abushaban, A.; Stohs, S.J. The relative abilities of TCDD and its congeners to induce oxidative stress in the hepatic and brain tissues of rats after subchronic exposure. Toxicology 2000, 145, 103–113. [Google Scholar] [CrossRef]
- Hassoun, E.A.; Li, F.; Abushaban, A.; Stohs, S.J. Production of superoxide anion, lipid peroxidation and DNA damage in the hepatic and brain tissues of rats after subchronic exposure to mixtures of TCDD and its congeners. J. Appl. Toxicol. JAT 2001, 21, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, E.A.; Wang, H.; Abushaban, A.; Stohs, S.J. Induction of oxidative stress in the tissues of rats after chronic exposure to TCDD, 2,3,4,7,8-pentachlorodibenzofuran, and 3,3’,4,4’,5-pentachlorobiphenyl. J. Toxicol. Environ. Health A 2002, 65, 825–842. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, E.A.; Al-Ghafri, M.; Abushaban, A. The role of antioxidant enzymes in TCDD-induced oxidative stress in various brain regions of rats after subchronic exposure. Free Radic. Biol. Med. 2003, 35, 1028–1036. [Google Scholar] [CrossRef]
- Lin, C.H.; Chen, C.C.; Chou, C.M.; Wang, C.Y.; Hung, C.C.; Chen, J.Y.; Chang, H.W.; Chen, Y.C.; Yeh, G.C.; Lee, Y.H. Knockdown of the aryl hydrocarbon receptor attenuates excitotoxicity and enhances NMDA-induced BDNF expression in cortical neurons. J. Neurochem. 2009, 111, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Liu, J.; Yoshimoto, K.; Chen, G.; Iwata, T.; Mizusawa, N.; Duan, Z.; Wan, C.; Jiang, J. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces expression of p27(kip1) and FoxO3a in female rat cerebral cortex and PC12 cells. Toxicol. Lett. 2014, 226, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.B.; Woods, L.; Brown, L.; Johnson, S.; Ebner, F.F. Gestational 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure effects on sensory cortex function. Neurotoxicology 2006, 27, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Kakeyama, M.; Sone, H.; Tohyama, C. Changes in expression of NMDA receptor subunit mRNA by perinatal exposure to dioxin. Neuroreport 2001, 12, 4009–4012. [Google Scholar] [CrossRef] [PubMed]
- Nayyar, T.; Wu, J.; Hood, D.B. Downregulation of hippocampal NMDA receptor expression by prenatal exposure to dioxin. Cell. Mol. Biol. Noisy-le-Grand Fr. 2003, 49, 1357–1362. [Google Scholar]
- Monteiro, P.; Gilot, D.; Langouet, S.; Fardel, O. Activation of the Aryl Hydrocarbon Receptor by the Calcium/Calmodulin-Dependent Protein Kinase Kinase Inhibitor 7-Oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic Acid (STO-609). Drug Metab. Dispos. 2008, 36, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Juan, S.H.; Wang, C.Y.; Sun, Y.Y.; Chou, C.M.; Chang, S.F.; Hu, S.Y.; Lee, W.S.; Lee, Y.H. Neuronal activity enhances aryl hydrocarbon receptor-mediated gene expression and dioxin neurotoxicity in cortical neurons. J. Neurochem. 2008, 104, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, T.; Yonemoto, J.; Sone, H.; Kosuge, Y.; Kosaki, K.; Takahashi, T. In utero exposure to dioxin causes neocortical dysgenesis through the actions of p27Kip1. Proc. Natl. Acad. Sci. USA 2010, 107, 16331–16335. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Domin, H.; Grynkiewicz, G.; Lason, W. Genistein inhibits glutamate-induced apoptotic processes in primary neuronal cell cultures: An involvement of aryl hydrocarbon receptor and estrogen receptor/glycogen synthase kinase-3beta intracellular signaling pathway. Neuroscience 2007, 145, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wójtowicz, A.K.; Maćkowiak, M.; Lasoń, W. Aryl hydrocarbon receptor-mediated apoptosis of neuronal cells: A possible interaction with estrogen receptor signaling. Neuroscience 2009, 158, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Duan, Z.; Nie, X.; Xi, H.; Li, A.; Guo, A.; Wu, Q.; Jiang, S.; Zhao, J.; Chen, G. Activation of neuronal nitric oxide synthase (nNOS) signaling pathway in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced neurotoxicity. Environ. Toxicol. Pharmacol. 2014, 38, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Stanford, E.A.; Al Abdulatif, A.; Ramirez-Cardenas, A.; Ballance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.; DeWitt, J.; Henshel, D.; Watkins, S.; Lasley, B. Fatty acid metabolism in neonatal chickens (Gallus domesticus) treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) or 3,3′,4,4′,5-pentachlorobiphenyl (PCB-126) in ovo. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2003, 136, 73–84. [Google Scholar] [CrossRef]
- Liu, P.C.; Matsumura, F. Differential effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the “adipose- type” and “brain-type” glucose transporters in mice. Mol. Pharmacol. 1995, 47, 65–73. [Google Scholar] [PubMed]
- Khanna, A.; Guo, M.; Mehra, M.; Royal, W. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J. Neuroimmunol. 2013, 254, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y.; et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nie, X.; Tao, T.; Qian, W.; Jiang, S.; Jiang, J.; Li, A.; Guo, A.; Xu, G.; Wu, Q. 2,3,7,8-Tetrachlorodibenzo-p-dioxin promotes astrocyte activation and the secretion of tumor necrosis factor-α via PKC/SSeCKS-dependent mechanisms. J. Neurochem. 2014, 129, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, D.; Balmoori, J.; Bagchi, M.; Ye, X.; Williams, C.B.; Stohs, S.J. Role of p53 tumor suppressor gene in the toxicity of TCDD, endrin, naphthalene, and chromium (VI) in liver and brain tissues of mice. Free Radic. Biol. Med. 2000, 28, 895–903. [Google Scholar] [CrossRef]
- Mukai, M.; Tischkau, S.A. Effects of tryptophan photoproducts in the circadian timing system: Searching for a physiological role for aryl hydrocarbon receptor. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 95, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Kakeyama, M.; Uemura, Y.; Haijima, A.; Okuno, H.; Bito, H.; Tohyama, C. Executive function deficits and social-behavioral abnormality in mice exposed to a low dose of dioxin in utero and via lactation. PLoS ONE 2012, 7, e50741. [Google Scholar] [CrossRef] [PubMed]
- Haijima, A.; Endo, T.; Zhang, Y.; Miyazaki, W.; Kakeyama, M.; Tohyama, C. In utero and lactational exposure to low doses of chlorinated and brominated dioxins induces deficits in the fear memory of male mice. Neurotoxicology 2010, 31, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Powers, B.E.; Lin, T.M.; Vanka, A.; Peterson, R.E.; Juraska, J.M.; Schantz, S.L. Tetrachlorodibenzo-p-dioxin exposure alters radial arm maze performance and hippocampal morphology in female AhR mice. Genes Brain Behav. 2005, 4, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, M.; Kuriwaki, J.I.; Hori, E.; Tawara, K.; Nakagawa, H.; Nishijo, H. Effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on fetal brain growth and motor and behavioral development in offspring rats. Toxicol. Lett. 2007, 173, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, M.; Tawara, K.; Nakagawa, H.; Honda, R.; Kido, T.; Nishijo, H.; Saito, S. 2,3,7,8-Tetrachlorodibenzo-p-dioxin in maternal breast milk and newborn head circumference. J. Expo. Sci. Environ. Epidemiol. 2008, 18, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Hojo, R.; Zareba, G.; Kai, J.W.; Baggs, R.B.; Weiss, B. Sex-specific alterations of cerebral cortical cell size in rats exposed prenatally to dioxin. J. Appl. Toxicol. JAT 2006, 26, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, T.; Sugiyama, N.; Maeda, S.; Tohyama, C.; Arita, J. Perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin suppresses contextual fear conditioning-accompanied activation of cyclic AMP response element-binding protein in the hippocampal CA1 region of male rats. Neurosci. Lett. 2006, 398, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Piedrafita, B.; Erceg, S.; Cauli, O.; Monfort, P.; Felipo, V. Developmental exposure to polychlorinated biphenyls PCB153 or PCB126 impairs learning ability in young but not in adult rats. Eur. J. Neurosci. 2008, 27, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Byers, J.P.; Masters, K.; Sarver, J.G.; Hassoun, E.A. Association between the levels of biogenic amines and superoxide anion production in brain regions of rats after subchronic exposure to TCDD. Toxicology 2006, 228, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Tanida, T.; Warita, K.; Ishihara, K.; Fukui, S.; Mitsuhashi, T.; Sugawara, T.; Tabuchi, Y.; Nanmori, T.; Qi, W.-M.; Inamoto, T.; et al. Fetal and neonatal exposure to three typical environmental chemicals with different mechanisms of action: Mixed exposure to phenol, phthalate, and dioxin cancels the effects of sole exposure on mouse midbrain dopaminergic nuclei. Toxicol. Lett. 2009, 189, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Tanida, T.; Tasaka, K.; Akahoshi, E.; Ishihara-Sugano, M.; Saito, M.; Kawata, S.; Danjo, M.; Tokumoto, J.; Mantani, Y.; Nagahara, D.; et al. Fetal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin transactivates aryl hydrocarbon receptor-responsive element III in the tyrosine hydroxylase immunoreactive neurons of the mouse midbrain. J. Appl. Toxicol. JAT 2014, 34, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Akahoshi, E.; Yoshimura, S.; Uruno, S.; Ishihara-Sugano, M. Effect of dioxins on regulation of tyrosine hydroxylase gene expression by aryl hydrocarbon receptor: A neurotoxicology study. Environ. Health Glob. Access Sci. Source 2009, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kuchiiwa, S.; Cheng, S.B.; Nagatomo, I.; Akasaki, Y.; Uchida, M.; Tominaga, M.; Hashiguchi, W.; Kuchiiwa, T. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin decreases serotonin-immunoreactive neurons in raphe nuclei of male mouse offspring. Neurosci. Lett. 2002, 317, 73–76. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsui, T.; Setani, K.; Tamura, M.; Kakeyama, M.; Sone, H.; Tohyama, C.; Tomita, T. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats disrupts brain sexual differentiation. Toxicol. Appl. Pharmacol. 2005, 205, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kakeyama, M.; Sone, H.; Miyabara, Y.; Tohyama, C. Perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters activity-dependent expression of BDNF mRNA in the neocortex and male rat sexual behavior in adulthood. Neurotoxicology 2003, 24, 207–217. [Google Scholar] [CrossRef]
- Hays, L.E.; Carpenter, C.D.; Petersen, S.L. Evidence that GABAergic neurons in the preoptic area of the rat brain are targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin during development. Environ. Health Perspect. 2002, 110, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.N.; Nishijo, M.; Nguyen, A.T.N.; Bor, A.; Nakamura, T.; Hori, E.; Nakagawa, H.; Ono, T.; Nishijo, H. Effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on parvalbumin- and calbindin-immunoreactive neurons in the limbic system and superior colliculus in rat offspring. Toxicology 2013, 314, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, D.L.; Brown, T.J.; MacLusky, N.J.; Hochberg, R.B.; Peterson, R.E. Partial demasculinization and feminization of sex behavior in male rats by in utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin is not associated with alterations in estrogen receptor binding or volumes of sexually differentiated brain nuclei. Toxicol. Appl. Pharmacol. 1994, 127, 258–267. [Google Scholar] [PubMed]
- Yang, E.J.; Stokes, J.V.; Kummari, E.; Eells, J.; Kaplan, B.L.F. Immunomodulation By Subchronic Low Dose 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Experimental Autoimmune Encephalomyelitis in the Absence of Pertussis Toxin. Toxicol. Sci. 2016, 151, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Potin, S.; Chapy, H.; Crete, D.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O.; Scherrmann, J.-M.; Declèves, X. Aryl hydrocarbon receptor regulates CYP1B1 but not ABCB1 and ABCG2 in hCMEC/D3 human cerebral microvascular endothelial cells after TCDD exposure. Brain Res. 2015, 1613, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Nannelli, A.; Rossignolo, F.; Tolando, R.; Rossato, P.; Longo, V.; Gervasi, P.G. Effect of beta-naphthoflavone on AhR-regulated genes (CYP1A1, 1A2, 1B1, 2S1, Nrf2, and GST) and antioxidant enzymes in various brain regions of pig. Toxicology 2009, 265, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Dauchy, S.; Miller, F.; Couraud, P.-O.; Weaver, R.J.; Weksler, B.; Romero, I.-A.; Scherrmann, J.-M.; De Waziers, I.; Declèves, X. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem. Pharmacol. 2009, 77, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Matsumoto, Y.; Takeda, T.; Koga, T.; Ishii, Y.; Yamada, H. Distribution of 14C-2,3,7,8-tetrachlorodibenzo-p-dioxin to the brain and peripheral tissues of fetal rats and its comparison with adults. J. Toxicol. Sci. 2010, 35, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lee, P.-S.; Chou, Y.; Hwang, L.-L.; Juan, S.-H. Mediating effects of aryl-hydrocarbon receptor and RhoA in altering brain vascular integrity: The therapeutic potential of statins. Am. J. Pathol. 2012, 181, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.J.; Branam, A.M.; Peterson, R.E. Intersection of AHR and Wnt signaling in development, health, and disease. Int. J. Mol. Sci. 2014, 15, 17852–17885. [Google Scholar] [CrossRef] [PubMed]
- Procházková, J.; Kabátková, M.; Bryja, V.; Umannová, L.; Bernatík, O.; Kozubík, A.; Machala, M.; Vondrácek, J. The interplay of the aryl hydrocarbon receptor and β-catenin alters both AhR-dependent transcription and Wnt/β-catenin signaling in liver progenitors. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 122, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Teraoka, H.; Tsujimoto, Y.; Stegeman, J.J.; Hiraga, T. Role of aryl hydrocarbon receptor in mesencephalic circulation failure and apoptosis in zebrafish embryos exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 77, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Teraoka, H.; Yamazaki, K.; Tsukiyama, S.; Imani, S.; Imagawa, T.; Stegeman, J.J.; Peterson, R.E.; Hiraga, T. 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in the zebrafish embryo: Local circulation failure in the dorsal midbrain is associated with increased apoptosis. Toxicol. Sci. Off. J. Soc. Toxicol. 2002, 69, 191–201. [Google Scholar] [CrossRef]
- Teraoka, H.; Dong, W.; Hiraga, T. Zebrafish as a novel experimental model for developmental toxicology. Congenit. Anom. 2003, 43, 123–132. [Google Scholar] [CrossRef]
- Teraoka, H.; Kubota, A.; Dong, W.; Kawai, Y.; Yamazaki, K.; Mori, C.; Harada, Y.; Peterson, R.E.; Hiraga, T. Role of the cyclooxygenase 2-thromboxane pathway in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced decrease in mesencephalic vein blood flow in the zebrafish embryo. Toxicol. Appl. Pharmacol. 2009, 234, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Dever, D.P.; Adham, Z.O.; Thompson, B.; Genestine, M.; Cherry, J.; Olschowka, J.A.; DiCicco-Bloom, E.; Opanashuk, L.A. Aryl hydrocarbon receptor deletion in cerebellar granule neuron precursors impairs neurogenesis. Dev. Neurobiol. 2016, 76, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Carvajal-González, J.M.; Riolobos, A.S.; Sevilla, M.A.; Menacho-Márquez, M.; Román, A.C.; Abad, A.; Montero, M.J.; Fernández-Salguero, P.; Bustelo, X.R. Transcriptional factor aryl hydrocarbon receptor (Ahr) controls cardiovascular and respiratory functions by regulating the expression of the Vav3 proto-oncogene. J. Biol. Chem. 2011, 286, 2896–2909. [Google Scholar] [CrossRef] [PubMed]
- Juricek, L.; Carcaud, J.; Pelhaitre, A.; Riday, T.T.; Chevallier, A.; Lanzini, J.; Auzeil, N.; Laprévote, O.; Dumont, F.; Jacques, S.; et al. AhR-deficiency as a cause of demyelinating disease and inflammation. Sci. Rep. 2017, 7, 9794. [Google Scholar] [CrossRef] [PubMed]
- Shackleford, G.; Sampathkumar, N.K.; Hichor, M.; Weill, L.; Meffre, D.; Juricek, L.; Laurendeau, I.; Chevallier, A.; Ortonne, N.; Larousserie, F.; et al. Involvement of Aryl hydrocarbon receptor in myelination and in human nerve sheath tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E1319–E1328. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.L.; Jeong, K.W. Regulation of aryl hydrocarbon receptor-mediated transcription in human retinal pigmented epithelial cells. Biochem. Biophys. Res. Commun. 2016, 472, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Kazmin, D.; Hu, P.; Thomas, R.S.; McDonnell, D.P.; Malek, G. Aryl hydrocarbon receptor knock-out exacerbates choroidal neovascularization via multiple pathogenic pathways. J. Pathol. 2015, 235, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Yang, H.-J.; Chang, Y.-S.; Kim, J.-W.; Brooks, M.; Chew, E.Y.; Wong, W.T.; Fariss, R.N.; Rachel, R.A.; Cogliati, T.; et al. Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Invest. Ophthalmol. Vis. Sci. 2014, 55, 6031–6040. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.A.; Davis, S.S.; Rosko, A.; Nguyen, S.M.; Mitchell, K.P.; Mateen, S.; Neves, J.; Garcia, T.Y.; Mooney, S.; Perdew, G.H.; et al. A novel AhR ligand, 2AI, protects the retina from environmental stress. Sci. Rep. 2016, 6, 29025. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.S.M.; Tillitt, D.E. 2,3,7,8-TCDD effects on visual structure and function in swim-up rainbow trout. Environ. Sci. Technol. 2004, 38, 6300–6306. [Google Scholar] [CrossRef] [PubMed]
- Aluru, N.; Jenny, M.J.; Hahn, M.E. Knockdown of a zebrafish aryl hydrocarbon receptor repressor (AHRRa) affects expression of genes related to photoreceptor development and hematopoiesis. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 139, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.G.; Mouchiroud, L.; Frochaux, M.; Pandey, A.; Andreux, P.A.; Deplancke, B.; Auwerx, J. An evolutionarily conserved role for the aryl hydrocarbon receptor in the regulation of movement. PLoS Genet. 2014, 10, e1004673. [Google Scholar] [CrossRef] [PubMed]
- García-Lara, L.; Pérez-Severiano, F.; González-Esquivel, D.; Elizondo, G.; Segovia, J. Absence of aryl hydrocarbon receptors increases endogenous kynurenic acid levels and protects mouse brain against excitotoxic insult and oxidative stress. J. Neurosci. Res. 2015, 93, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ray, B.; Neavin, D.R.; Zhang, J.; Athreya, A.P.; Biernacka, J.M.; Bobo, W.V.; Hall-Flavin, D.K.; Skime, M.K.; Zhu, H.; et al. Beta-defensin 1, aryl hydrocarbon receptor and plasma kynurenine in major depressive disorder: Metabolomics-informed genomics. Transl. Psychiatry 2018, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, M.I.; Ballesteros, I.; de la Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernandez-Salguero, P.; Corbi, A.L.; Lizasoain, I.; Moro, M.A. L-Kynurenine/Aryl Hydrocarbon Receptor Pathway Mediates Brain Damage After Experimental Stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Zheng, X.; Hou, Y.; Hu, M.; Wang, H.; Bao, X.; Zhou, F.; Wang, G.; Hao, H. Regulation of proinflammatory monocyte activation by the kynurenine–AhR axis underlies immunometabolic control of depressive behavior in mice. FASEB J. 2018, 32, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.O.; Föcking, M.; Cotter, D.R. Proteomic pathway analysis of the hippocampus in schizophrenia and bipolar affective disorder implicates 14-3-3 signaling, aryl hydrocarbon receptor signaling, and glucose metabolism: Potential roles in GABAergic interneuron pathology. Schizophr. Res. 2015, 167, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.E.J.; Ostby, J.; Wolf, C.; Miller, D.B.; Kelce, W.R.; Gordon, C.J.; Birnbaum, L.S. Functional developmental toxicity of low doses of 2,3,7,8-tetrachlorodibenzo-p-dioxin and a dioxin-like PCB (169) in Long Evans rats and Syrian hamsters: Reproductive, behavioral and thermoregulatory alterations. Organohalogen Compd. 1995, 25, 33–38. [Google Scholar]
- Mably, T.A.; Moore, R.W.; Goy, R.W.; Peterson, R.E. In utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin: 2. Effects on sexual behavior and the regulation of luteinizing hormone secretion in adulthood. Toxicol. Appl. Pharmacol. 1992, 114, 108–117. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juricek, L.; Coumoul, X. The Aryl Hydrocarbon Receptor and the Nervous System. Int. J. Mol. Sci. 2018, 19, 2504. https://doi.org/10.3390/ijms19092504
Juricek L, Coumoul X. The Aryl Hydrocarbon Receptor and the Nervous System. International Journal of Molecular Sciences. 2018; 19(9):2504. https://doi.org/10.3390/ijms19092504
Chicago/Turabian StyleJuricek, Ludmila, and Xavier Coumoul. 2018. "The Aryl Hydrocarbon Receptor and the Nervous System" International Journal of Molecular Sciences 19, no. 9: 2504. https://doi.org/10.3390/ijms19092504
APA StyleJuricek, L., & Coumoul, X. (2018). The Aryl Hydrocarbon Receptor and the Nervous System. International Journal of Molecular Sciences, 19(9), 2504. https://doi.org/10.3390/ijms19092504