Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist

 , and
, and 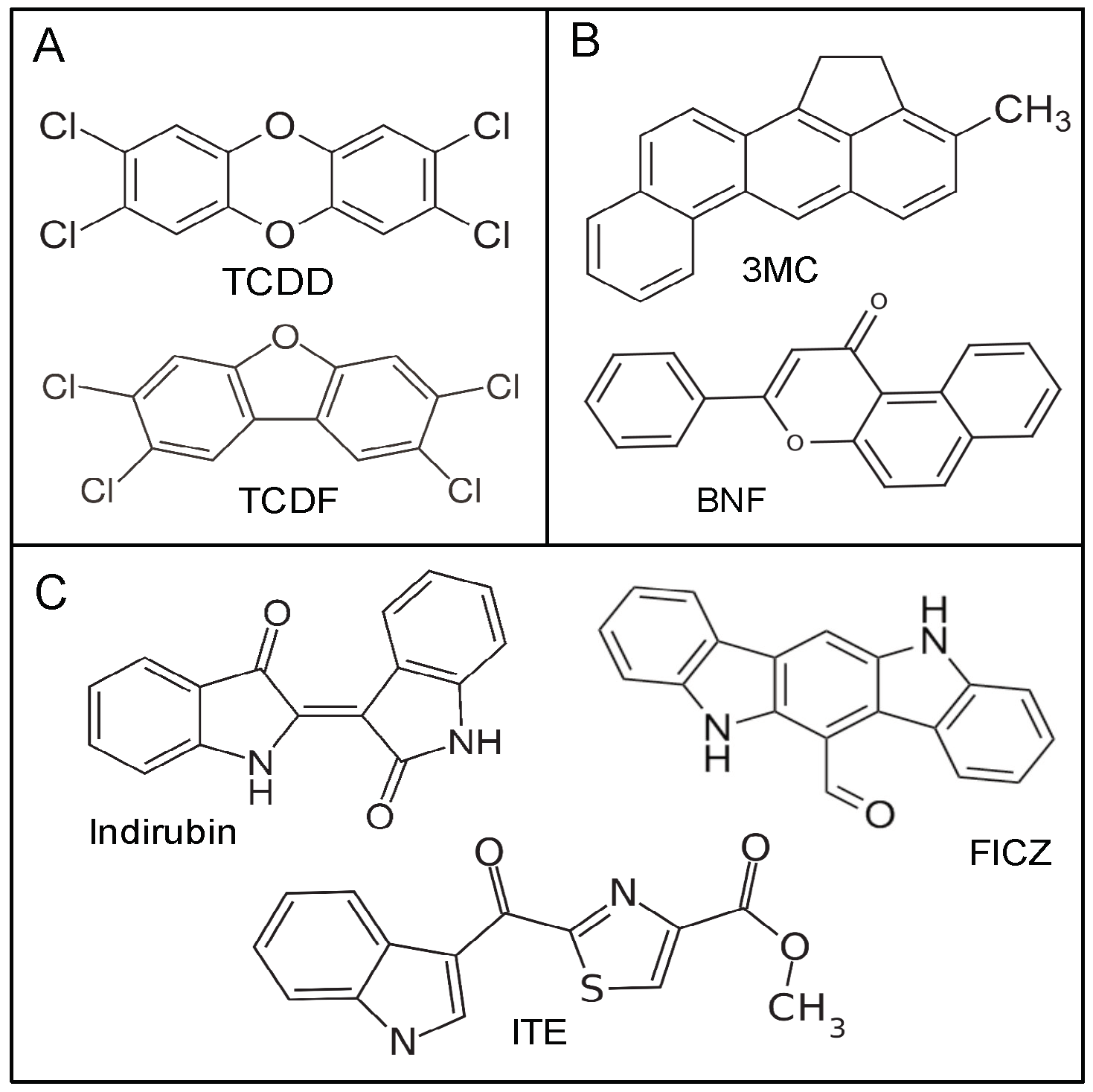{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
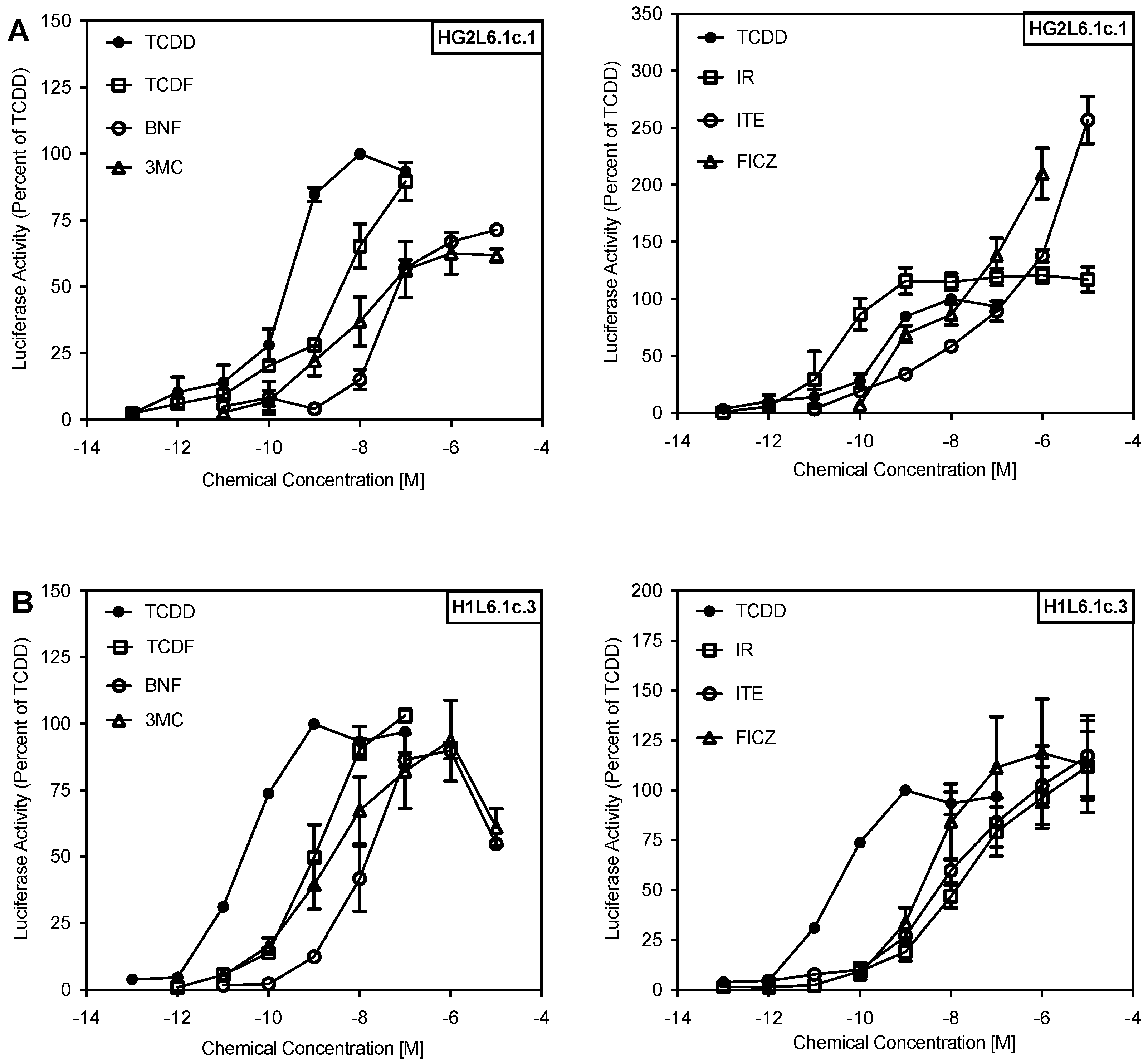
2.1. Comparison of the Relative Potency and Efficacy of AhR Agonists in Human and Mouse Cells
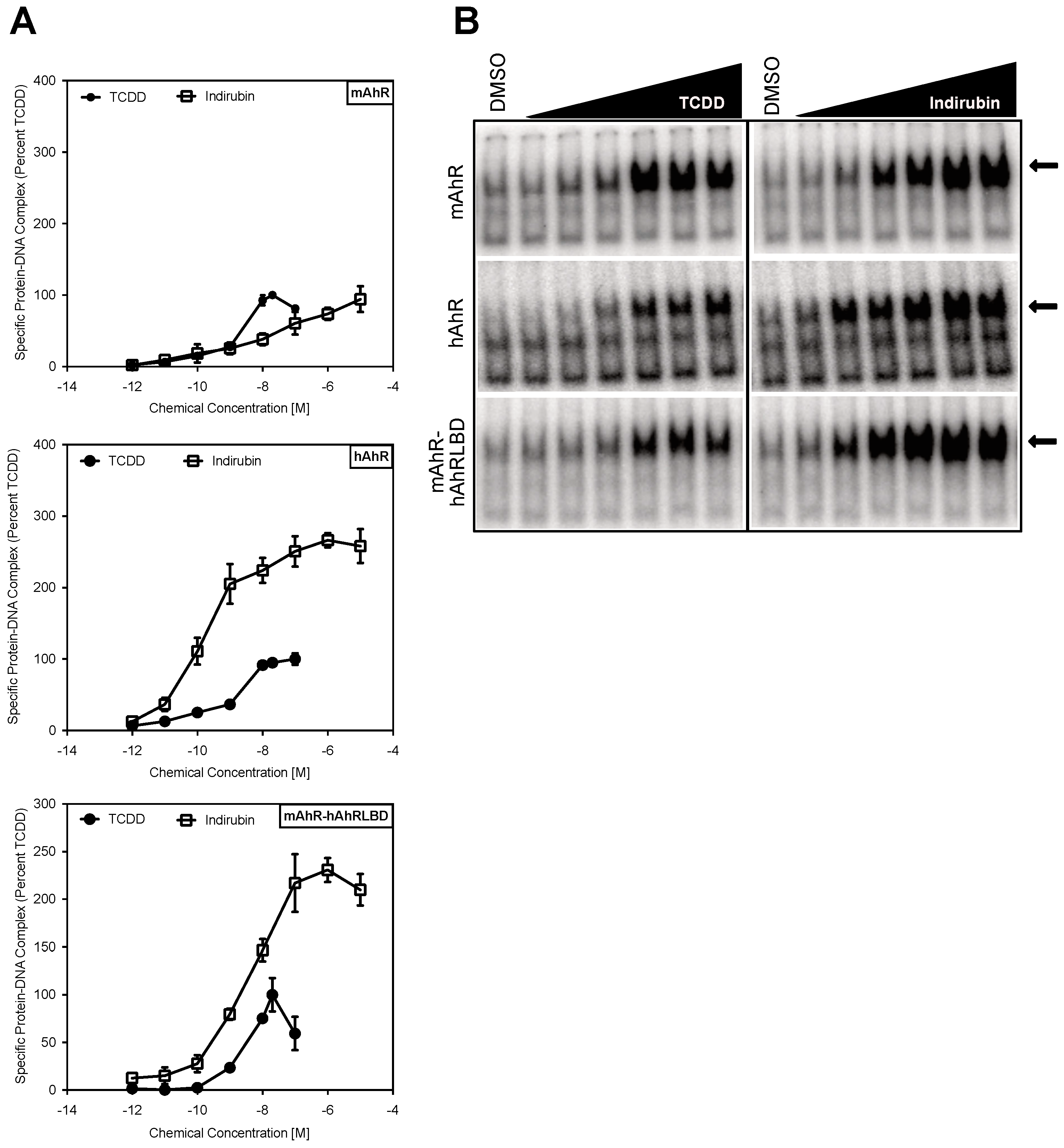
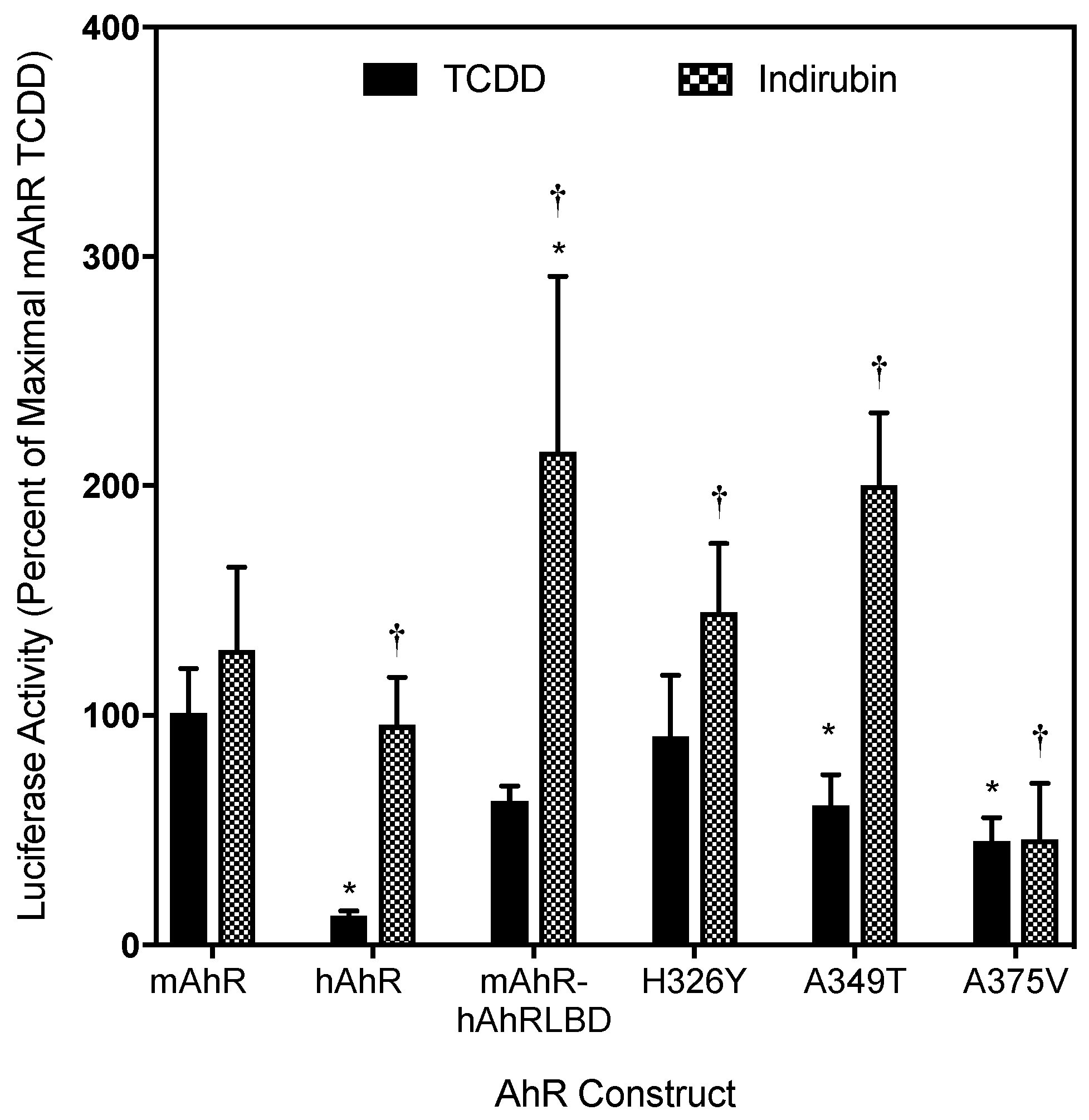
2.2. The Human AhR Ligand Binding Domain Is Essential for Ligand-Selective Activation by IR
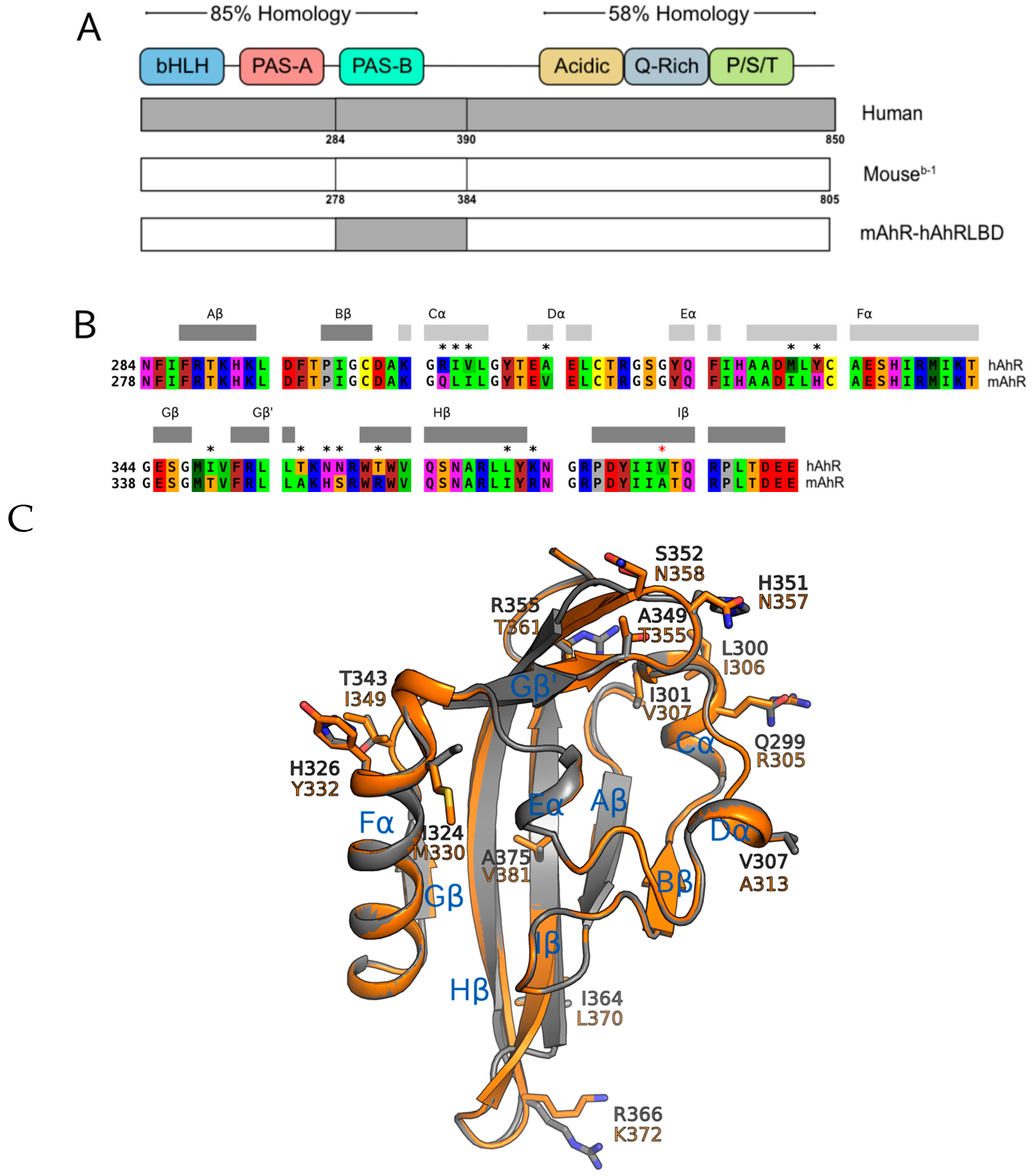
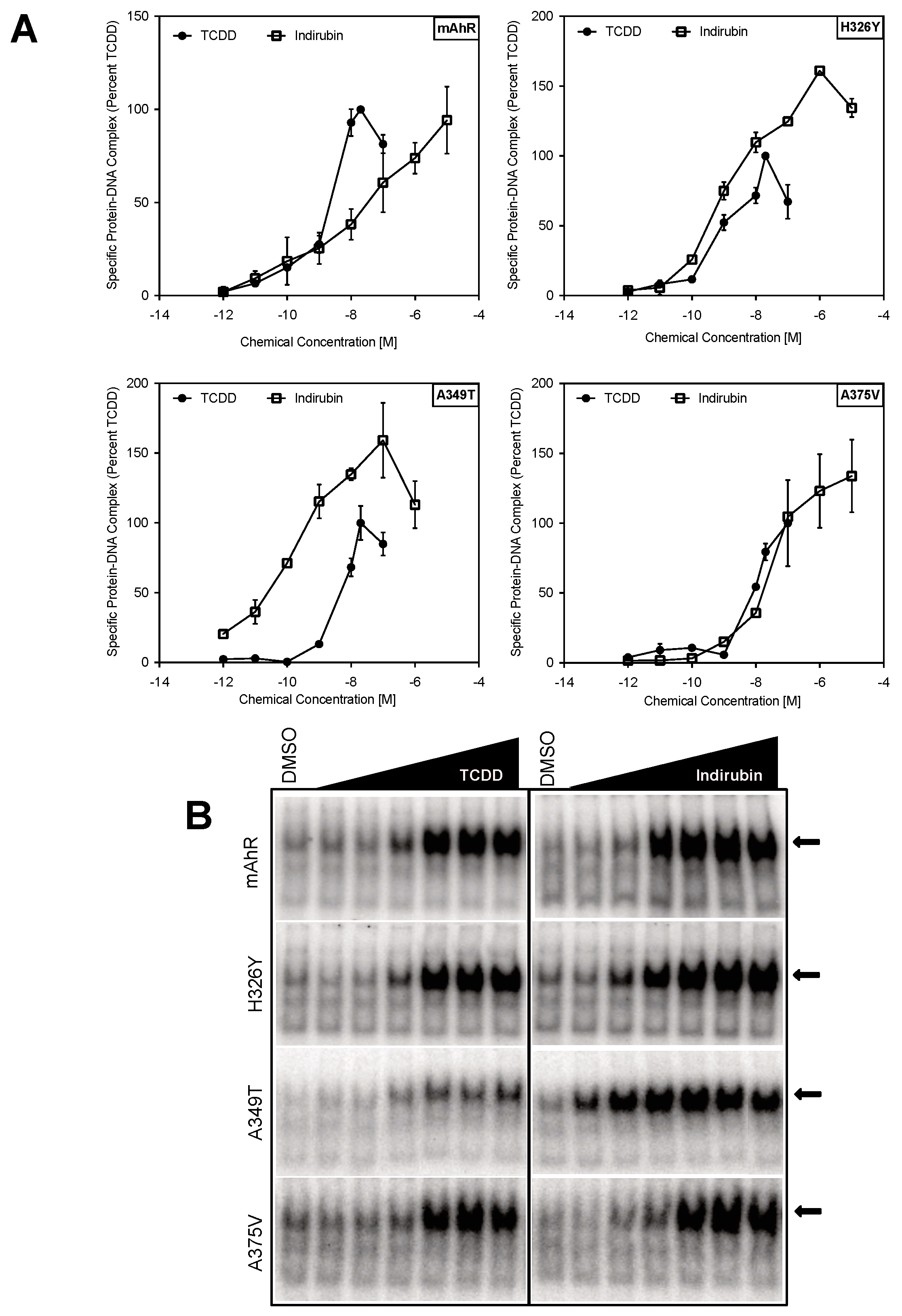
2.3. Point Mutations in the mAhR LBD Produce a hAhR-Like Response with IR
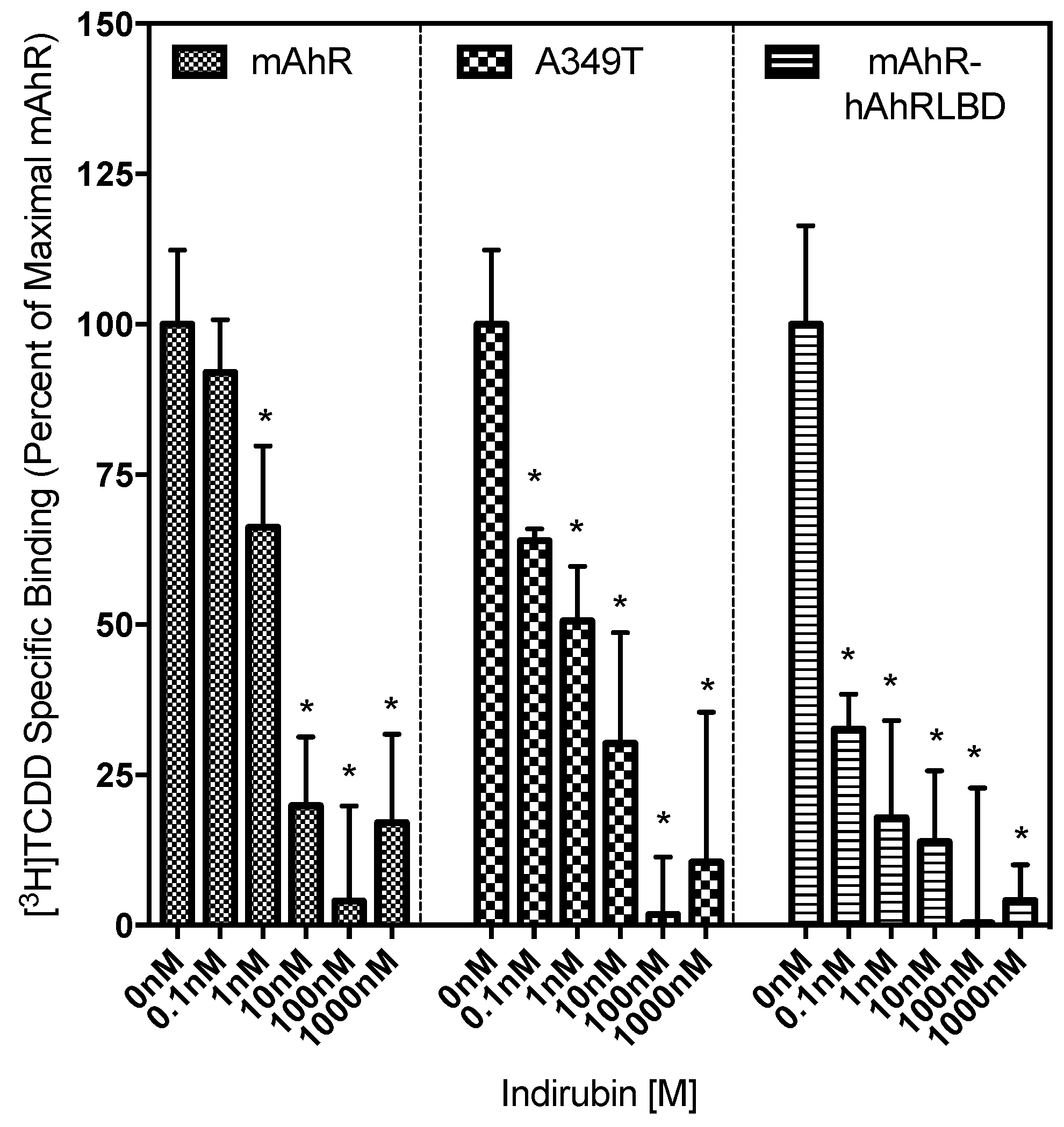
2.4. Ligand Binding Analysis Reveals That the A349T Mutation Differentially Affects TCDD and IR
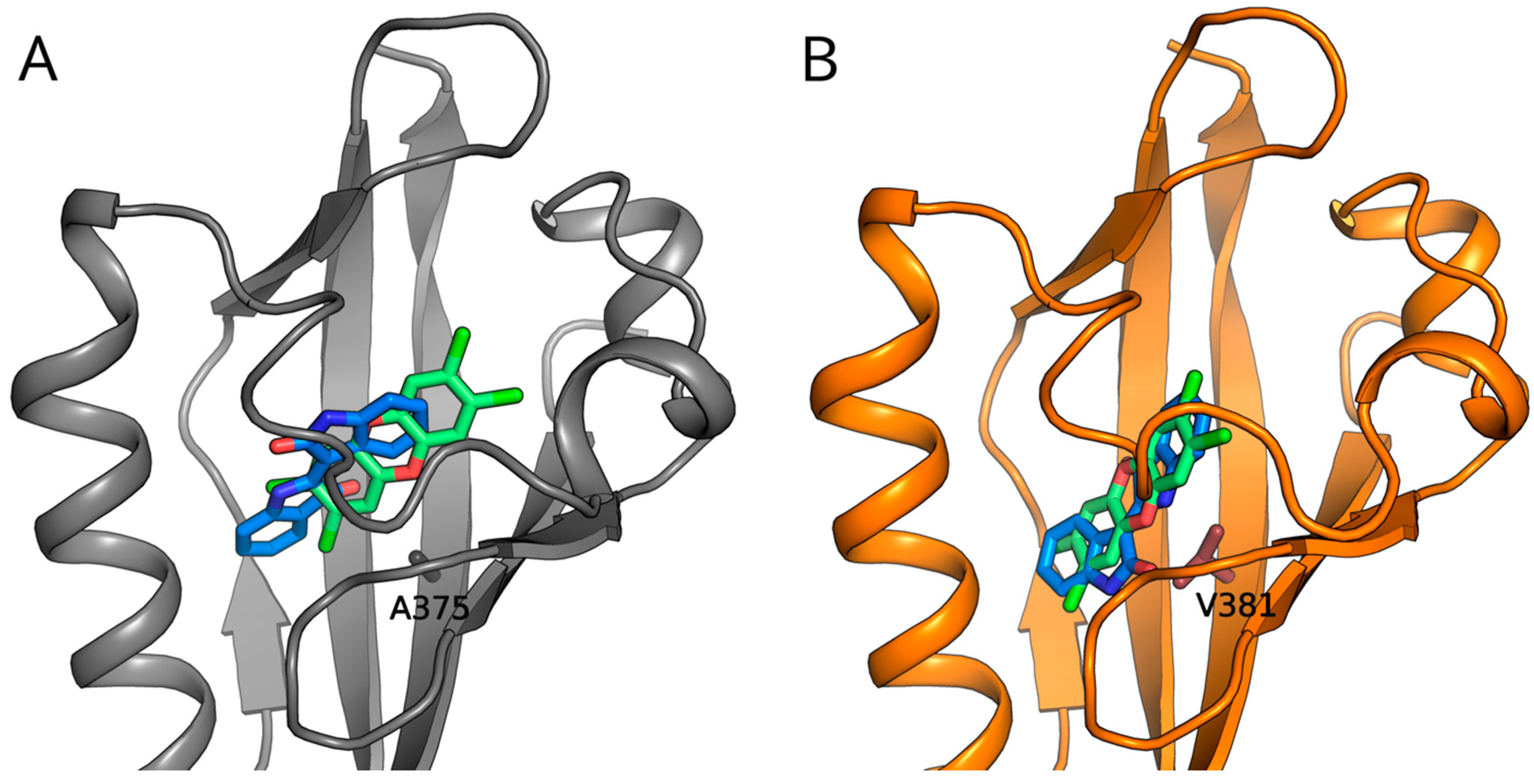
2.5. Molecular Docking Predicts Differences in TCDD and IR Binding within the mAhR and hAhR LBDs
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Plasmids
4.3. Hydroxyapatite [3H]TCDD Ligand Binding Assay
4.4. AhR DNA Binding (Gel Retardation) Assay
4.5. Reporter Gene Assays
4.6. Transient Transfection Assays
4.7. Statistical Analysis
4.8. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, D.E.; He, G.; Fraccalvieri, D.; Bonati, L.; Pandini, A.; Denison, M.S. AhR ligands: Promiscuity in binding and diversity in response. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 63–79. ISBN 9780470601822. [Google Scholar]
- Chen, H.S.; Singh, S.S.; Perdew, G.H. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch. Biochem. Biophys. 1997, 348, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Henry, E.C.; Gasiewicz, T.A. Transformation of the aryl hydrocarbon receptor to a DNA-binding form is accompanied by release of the 90 kDa heat-shock protein and increased affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. J. 1993, 294, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Pongratz, I.; Mason, G.G.; Poellinger, L. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Evidence that the dioxin receptor functionally belongs to a subclass of nuclear receptors which require hsp90 both for ligand binding activity and repression of intrinsic DNA binding activity. J. Biol. Chem. 1992, 267, 13728–13734. [Google Scholar] [PubMed]
- Soshilov, A.; Denison, M.S. Ligand displaces heat shock protein 90 from overlapping binding sites within the aryl hydrocarbon receptor ligand binding domain. J. Biol. Chem. 2011, 286, 35275–35282. [Google Scholar] [CrossRef] [PubMed]
- Probst, M.R.; Reisz-Porszasz, S.; Agbunag, R.V.; Ong, M.S.; Hankinson, O. Role of the aryl hydrocarbon receptor nuclear translocator protein in aryl hydrocarbon (dioxin) receptor action. Mol. Pharmacol. 1993, 44, 511–518. [Google Scholar] [PubMed]
- Bunger, M.K.; Glover, E.; Moran, S.M.; Walisser, J.A.; Lahvis, G.P.; Hsu, E.L.; Bradfield, C.A. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 2008, 106, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Fisher, J.M.; Whitlock, J.P., Jr. The DNA recognition site for the dioxin-Ah receptor complex. Proc. Natl. Acad. Sci. USA 1988, 263, 17221–17224. [Google Scholar]
- Denison, M.S.; Faber, S.C. And now for something completely different: Diversity in ligand-dependent activation of Ah receptor responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; Joshi, A.D.; Elferink, C.J. The tumor suppressor Kruppel-like factor 6 is a novel aryl hydrocarbon receptor DNA binding partner. J. Pharmacol. Exp. Ther. 2013, 345, 419–429. [Google Scholar] [CrossRef] [PubMed]
- White, S.S.; Birnbaum, L.S. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.E.; Noble, S.M.; Redinbo, M.R. Structural insights into the promiscuity and function of the human pregnane X receptor. Curr. Opin. Drug Discov. Dev. 2002, 5, 150–158. [Google Scholar]
- Fraccalvieri, D.; Soshilov, A.A.; Karchner, S.I.; Franks, D.G.; Pandini, A.; Bonati, L.; Hahn, M.E.; Denison, M.S. Comparative analysis of homology models of the Ah receptor ligand binding domain: Verification of structure-function predictions by site-directed mutagenesis of a nonfunctional receptor. Biochemistry 2013, 52, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.A.; Denison, M.S. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell. Biol. 2014, 34, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Whelan, F.; Hao, N.; Furness, S.G.; Whitelaw, M.L.; Chapman-Smith, A. Amino acid substitutions in the aryl hydrocarbon receptor ligand binding domain reveal YH439 as an atypical AhR activator. Mol. Pharmacol. 2010, 77, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Goodale, B.C.; Tilton, S.C.; Corvi, M.M.; Wilson, G.R.; Janszen, D.B.; Anderson, K.A.; Waters, K.M.; Tanguay, R.L. Structurally distinct polycyclic aromatic hydrocarbons induce differential transcriptional responses in developing zebrafish. Toxicol. Appl. Pharmacol. 2013, 272, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Aarts, J.M.M.J.G.; Denison, M.S.; Cox, M.A.; Schalk, M.A.C.; Garrison, P.M.; Tullis, K.; de Haan, L.H.J.; Brouwer, A. Species-specific antagonism of Ah receptor action by 2,2′,5,5′-tetrachloro- and 2,2′,3,3′,4,4′-hexachlorobiphenyl. Eur. J. Pharmacol. Environ. Toxicol. 1995, 293, 463–474. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; DeGroot, D.; Hayashi, A.; He, G.; Denison, M.S. CH223191 is a ligand-selective antagonist of the Ah (dioxin) receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Murray, I.A.; Chiaro, C.R.; Perdew, G.H. Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol. Pharmacol. 2009, 75, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Murray, I.A.; Perdew, G.H. Differential gene regulation by the human and mouse aryl hydrocarbon receptor. Toxicol. Sci. 2010, 114, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Ohe, N.; Suzuki, M.; Mimura, J.; Sogawa, K.; Ikawa, S.; Fujii-Kuriyama, Y. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J. Biol. Chem. 1994, 269, 27337–27343. [Google Scholar] [PubMed]
- Moriguchi, T.; Motohashi, H.; Hosoya, T.; Nakajima, O.; Takahashi, S.; Ohsako, S.; Aoki, Y.; Nishimura, N.; Tohyama, C.; Fujii-Kuriyama, Y.; et al. Distinct response to dioxin in an arylhydrocarbon receptor (AHR)-humanized mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Pandini, A.; Denison, M.S.; Song, Y.; Soshilov, A.; Bonati, L. Structural and functional characterization of the aryl hydrocarbon receptor ligand binding domain by homology modeling and mutational analysis. Biochemistry 2007, 46, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Pandini, A.; Soshilov, A.A.; Song, Y.; Zhao, J.; Bonati, L.; Denison, M.S. Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry 2009, 48, 5972–5983. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and tryptophan metabolism: Endogenous and dietary routes to ah receptor activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Mori, Y.; Matsui, S.; Takigami, H.; Fujino, J.; Kitagawa, H.; Miller, C.A., 3rd; Kato, T.; Saeki, K.; Matsuda, T. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 2001, 276, 31475–31478. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Mori, Y.; Matsui, S.; Matsuda, T. Comparison of gene expression patterns between 2,37,8-tetrachlorodibenzo-p-dioxin and a natural arylhydrocarbon receptor ligand, indirubin. Toxicol. Sci. 2004, 80, 161–169. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Baston, D.S.; Denison, M.S. Considerations for potency equivalent calculations in the Ah receptor-based CALUX bioassay: Normalization of superinduction results for improved sample potency estimation. Talanta 2011, 83, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Motto, I.; Bordogna, A.; Soshilov, A.; Denison, M.S.; Bonati, L. New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J. Chem. Inf. Model. 2011, 51, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Pandini, A.; Nagy, S.R.; Baldwin, E.P.; Bonati, L. Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 2002, 141, 3–24. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Carter, D.E.; Harper, T.A., Jr.; Elferink, C.J. Aryl hydrocarbon receptor–dependent stanniocalcin 2 induction by cinnabarinic acid provides cytoprotection against endoplasmic reticulum and oxidative stress. J. Pharmacol. Exp. Ther. 2015, 353, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.M.; Mold, J.E.; Kanwar, B.; Huang, Y.; Louie, A.; Pollastri, M.P.; Wang, C.; Patel, G.; Franks, D.G.; Schlezinger, J.; et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE 2014, 9, e87877. [Google Scholar] [CrossRef] [PubMed]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. BHLH-PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Möglich, A.; Ayers, R.A.; Moffat, K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Corrada, D.; Soshilov, A.A.; Denison, M.S.; Bonati, L. Deciphering dimerization modes of PAS domains: Computational and rxperimental analyses of the AhR:ARNT complex reveal new insights into the mechanisms of AhR transformation. PLoS Comput. Biol. 2016, 12, e1004981. [Google Scholar] [CrossRef] [PubMed]
- Corrada, D.; Denison, M.S.; Bonati, L. Structural modeling of the AhR:ARNT complex in the bHLH-PASA-PASB region elucidates the key determinants of dimerization. Mol. Biosyst. 2017, 13, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Bonati, L.; Corrada, D.; Tagliabue, S.G.; Motta, S. Molecular modeling of the AhR structure and interactions can shed light on ligand-dependent activation and transformation mechanisms. Curr. Opin. Toxicol. 2016, 1, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.W.; Green, E.; Wilz, A.; Platten, M.; Daumke, O. Structural basis for aryl hydrocarbon receptor-mediated gene activation. Structure 2017, 25, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.-H.; Lee, W.; Jiang, L.; Molugu, K.; Zheng, A.; Li, Y.; Park, S.; Bradfield, C.A.; Xing, Y. Structural hierarchy controlling dimerization and target DNA recognition in the AHR transcriptional complex. Proc. Natl. Acad. Sci. USA 2017, 114, 5431–5436. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.A.; Denison, M.S. DNA binding (gel retardation assay) analysis for identification of aryl hydrocarbon (Ah) receptor agonists and antagonists. In Optimization of Drug Discovery: In Vitro Methods, 2nd ed.; Yan, A., Caldwell, G.W., Eds.; Humana Press: New York, NY, USA, 2014; pp. 207–219. ISBN 978-1-62703-742-6. [Google Scholar]
- Soshilov, A.; Denison, M.S. Role of the Per/Arnt/Sim domains in ligand-dependent transformation of the aryl hydrocarbon receptor. J. Biol. Chem. 2008, 283, 32995–33005. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhao, J.; Brennan, J.C.; Affatato, A.A.; Zhao, B.; Rice, R.H.; Denison, M.S. Cell-based assays for identification of aryl hydrocarbon (Ah) receptor activators. In Optimization of Drug Discovery: In Vitro Methods, 2nd ed.; Yan, A., Caldwell, G.W., Eds.; Humana Press: New York, NY, USA, 2014; pp. 221–235. ISBN 978-1-62703-742-6. [Google Scholar]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016. [Google Scholar] [CrossRef]
- MacroModel, Schrödinger Release 2015-4, Schrödinger, LLC: New York, NY, USA, 2015.
- LigPrep, Schrödinger Release 2015-4, Schrödinger, LLC: New York, NY, USA, 2015.
- Glide, Schrödinger Release 2015-4, Schrödinger, LLC: New York, NY, USA, 2015.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free energy calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Prime, Schrödinger Release 2015-4, Schrödinger, LLC: New York, NY, USA, 2015.
- PyMOL. The PyMOL Molecular Graphics System, version 1.6; Schrödinger LLC: New York, NY, USA, 2010. [Google Scholar]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faber, S.C.; Soshilov, A.A.; Giani Tagliabue, S.; Bonati, L.; Denison, M.S. Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist. Int. J. Mol. Sci. 2018, 19, 2692. https://doi.org/10.3390/ijms19092692
Faber SC, Soshilov AA, Giani Tagliabue S, Bonati L, Denison MS. Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist. International Journal of Molecular Sciences. 2018; 19(9):2692. https://doi.org/10.3390/ijms19092692
Chicago/Turabian StyleFaber, Samantha C., Anatoly A. Soshilov, Sara Giani Tagliabue, Laura Bonati, and Michael S. Denison. 2018. "Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist" International Journal of Molecular Sciences 19, no. 9: 2692. https://doi.org/10.3390/ijms19092692
APA StyleFaber, S. C., Soshilov, A. A., Giani Tagliabue, S., Bonati, L., & Denison, M. S. (2018). Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist. International Journal of Molecular Sciences, 19(9), 2692. https://doi.org/10.3390/ijms19092692