Revisiting the Cardioprotective Effects of Acetylcholine Receptor Activation against Myocardial Ischemia/Reperfusion Injury


Abstract
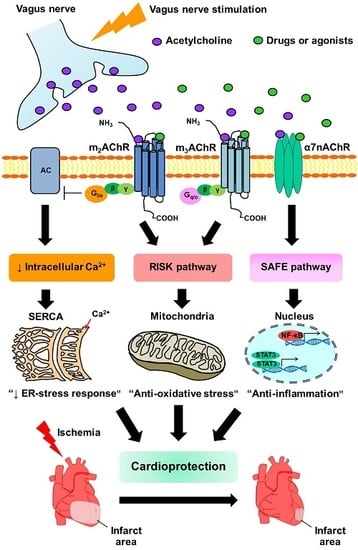
1. Introduction
2. Acute Myocardial Infarction (AMI) and Pathophysiologic Mechanisms of Myocardial Ischemia/Reperfusion (I/R) Injury
3. Parasympathetic Modulation as a Novel Strategy for Attenuating Myocardial I/R Injury
4. The Effects of Muscarinic Receptors (mAChRs) Activation on Myocardial Infarct Size, Hemodynamic and Cardiac Function in the Setting of I/R
5. The Effects of α7 Nicotinic Acetylcholine Receptor (α7nAChR) Activation on Myocardial Infarct Size, Hemodynamic and Cardiac Function in the Setting of I/R
6. Anti-Apoptosis and Anti-Oxidative Stress against I/R-induced Cell Injury through mAChRs Activation
7. Anti-Apoptosis and Anti-Oxidative Stress Against I/R-Induced Cell Injury through α7nAChRs Activation
8. Inflammation and the Cholinergic Anti-Inflammatory Pathway in the Setting of I/R Injury
9. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| ACh | Acetylcholine |
| Akt | Protein kinase B |
| α7nAChR | α7 nicotinic acetylcholine receptor |
| Bcl2 | B-cell lymphoma 2 |
| BK channel | Voltage and Ca2+-activated potassium channel BK |
| cAMP | Cyclic adenosine monophosphate |
| cGMP | Cyclic guanosine monophosphate |
| COX2 | Cyclooxygenase-2 |
| pCx43 | Phosphorylated connexin 43 |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| Fas | TNF superfamily receptor 6 |
| HMGB1 | High mobility group box 1 protein |
| IL-6 | Interleukin 6 |
| IL-1β | Interleukin 1β |
| IP3 | Inositol 1,4,5-triphosphate |
| JAK2 | Janus kinase 2 |
| m2AChR | Muscarinic acetylcholine receptor type 2 |
| m3AChR | Muscarinic acetylcholine receptor type 3 |
| MDA | Malondialdehyde |
| MEK | Mitogen-activated protein kinase/extracellular signal-regulated kinase kinase |
| MPTP | Mitochondrial permeability transition pore |
| NF-κB | Nuclear factor-kappa B |
| NO | Nitric oxide |
| eNOS | Endothelial nitric oxide synthase |
| NOX | Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase |
| pFOXO3a | Phosphorylated forkhead box subfamily O3a |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator-1-alpha |
| pGSK-3β | Phospho glycogen synthase kinase 3 β |
| PKA | Protein kinase A |
| PI3K | Phosphoinositide 3-kinase |
| PLC | Phospholipase C |
| PKC | Protein kinase C |
| p38MAPK | p38 mitogen-activated protein kinases |
| Ras/Raf | Serine/threonine kinase |
| RISK | Reperfusion injury savage kinase |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptors |
| SAFE | Survivor activating factor enhancement |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| SOD | Superoxide dismutase |
| sGC | Soluble guanylyl cyclase |
| STAT3 | Signal transducers and activators of transcription 3 |
| TNF-α | Tumor necrosis factor-α |
| XO | Xanthine oxidase |
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Picatoste, B.; Badimon, J.J. Pathophysiology of acute coronary syndrome. Curr. Atheroscler. Rep. 2014, 16, 401. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T. Heart failure as an autonomic nervous system dysfunction. J. Cardiol. 2012, 59, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Vaseghi, M.; Shivkumar, K. The role of the autonomic nervous system in sudden cardiac death. Prog. Cardiovasc. Dis. 2008, 50, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Shinlapawittayatorn, K.; Chinda, K.; Palee, S.; Surinkaew, S.; Kumfu, S.; Kumphune, S.; Chattipakorn, S.; KenKnight, B.H.; Chattipakorn, N. Vagus nerve stimulation initiated late during ischemia, but not reperfusion, exerts cardioprotection via amelioration of cardiac mitochondrial dysfunction. Heart Rhythm. 2014, 11, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Shinlapawittayatorn, K.; Chinda, K.; Palee, S.; Surinkaew, S.; Thunsiri, K.; Weerateerangkul, P.; Chattipakorn, S.; KenKnight, B.H.; Chattipakorn, N. Low-amplitude, left vagus nerve stimulation significantly attenuates ventricular dysfunction and infarct size through prevention of mitochondrial dysfunction during acute ischemia-reperfusion injury. Heart Rhythm. 2013, 10, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yong, Y.; Li, X.; Hu, Y.; Wang, J.; Wang, Y.Q.; Song, W.; Chen, W.T.; Xie, J.; Chen, X.M.; et al. Vagal modulation of high mobility group box-1 protein mediates electroacupuncture-induced cardioprotection in ischemia-reperfusion injury. Sci. Rep. 2015, 5, 15503. [Google Scholar] [CrossRef] [PubMed]
- Nuntaphum, W.; Pongkan, W.; Wongjaikam, S.; Thummasorn, S.; Tanajak, P.; Khamseekaew, J.; Intachai, K.; Chattipakorn, S.C.; Chattipakorn, N.; Shinlapawittayatorn, K. Vagus nerve stimulation exerts cardioprotection against myocardial ischemia/reperfusion injury predominantly through its efferent vagal fibers. Basic Res. Cardiol. 2018, 113, 22. [Google Scholar] [CrossRef] [PubMed]
- Uitterdijk, A.; Yetgin, T.; te Lintel Hekkert, M.; Sneep, S.; Krabbendam-Peters, I.; van Beusekom, H.M.; Fischer, T.M.; Cornelussen, R.N.; Manintveld, O.C.; Merkus, D.; et al. Vagal nerve stimulation started just prior to reperfusion limits infarct size and no-reflow. Basic Res. Cardiol. 2015, 110, 508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wugeti, N.; Sun, J.; Yan, H.; Guo, Y.; Zhang, L.; Ma, M.; Guo, X.; Jiao, C.; Xu, W.; et al. Effects of vagus nerve stimulation via cholinergic anti-inflammatory pathway activation on myocardial ischemia/reperfusion injury in canine. Int. J. Clin. Exp. Med. 2014, 7, 2615–2623. [Google Scholar] [PubMed]
- Zhao, M.; He, X.; Bi, X.Y.; Yu, X.J.; Gil Wier, W.; Zang, W.J. Vagal stimulation triggers peripheral vascular protection through the cholinergic anti-inflammatory pathway in a rat model of myocardial ischemia/reperfusion. Basic Res. Cardiol. 2013, 108, 345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yu, L.; Wang, S.; Huang, B.; Liao, K.; Saren, G.; Tan, T.; Jiang, H. Chronic intermittent low-level transcutaneous electrical stimulation of auricular branch of vagus nerve improves left ventricular remodeling in conscious dogs with healed myocardial infarction. Circ. Heart Fail. 2014, 7, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Huang, B.; Po, S.S.; Tan, T.; Wang, M.; Zhou, L.; Meng, G.; Yuan, S.; Zhou, X.; Li, X.; et al. Low-level tragus stimulation for the treatment of ischemia and reperfusion injury in patients with ST-segment elevation myocardial infarction: A proof-of-concept study. JACC Cardiovasc. Interv. 2017, 10, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, Y.; Ando, M.; Kuwabara, M.; Katare, R.G.; Okudela, K.; Kobayashi, M.; Sato, T. Acetylcholine from vagal stimulation protects cardiomyocytes against ischemia and hypoxia involving additive non-hypoxic induction of HIF-1α. FEBS Lett. 2005, 579, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Burke, N.; Davidson, S.M.; Yellon, D.M. Intrinsic cardiac ganglia and acetylcholine are important in the mechanism of ischaemic preconditioning. Basic Res. Cardiol. 2017, 112, 11. [Google Scholar] [CrossRef] [PubMed]
- Palee, S.; Apaijai, N.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. Acetylcholine attenuates hydrogen peroxide-induced intracellular calcium dyshomeostasis through both muscarinic and nicotinic receptors in cardiomyocytes. Cell. Physiol. Biochem. 2016, 39, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Liu, B.H.; Sun, L.; Zhao, M.; He, X.; Yu, X.J.; Zang, W.J. Alterations of muscarinic acetylcholine receptors-2, 4 and α7-nicotinic acetylcholine receptor expression after ischaemia/reperfusion in the rat isolated heart. Clin. Exper. Pharmacol. Physiol. 2010, 37, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lin, H.; Xu, C.; Liu, Y.; Wang, H.; Han, H.; Wang, Z. Choline produces cytoprotective effects against ischemic myocardial injuries: Evidence for the role of cardiac m3 subtype muscarinic acetylcholine receptors. Cell. Physiol. Biochem. 2005, 16, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Fields, W.C.; Rocha-Resende, C.; Resende, R.R.; Guatimosim, S.; Prado, V.F.; Gros, R.; Prado, M.A.M. Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. 2013, 27, 5072–5082. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, Y.; Akiyama, T.; Okazaki, K.; Arikawa, M.; Noguchi, T.; Sato, T. A non-neuronal cardiac cholinergic system plays a protective role in myocardium salvage during ischemic insults. PLoS ONE 2012, 7, e50761. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kai, Y.; Tsuda, M.; Ohata, H.; Mano, A.; Mizoguchi, N.; Sugama, S.; Nemoto, T.; Suzuki, K.; Kurabayashi, A.; et al. Non-neuronal cardiac cholinergic system influences cns via the vagus nerve to acquire a stress-refractory propensity. Clin. Sci. 2016, 130, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, M.; Lips, K.S.; Bruggmann, D.; Slavikova, J.; Kuncova, J.; Kummer, W. Developmental changes in the expression of nicotinic acetylcholine receptor α-subunits in the rat heart. Cell Tissue Res. 2005, 319, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, S.; Wang, C.; Song, H.; Han, H.; Hang, P.; Jiang, Y.; Wei, L.; Huo, R.; Sun, L.; et al. Upregulation of m3 muscarinic receptor inhibits cardiac hypertrophy induced by angiotensin ii. J. Transl. Med. 2013, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute myocardial infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Critical issues for the translation of cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Turer, A.T.; Hill, J.A. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 2010, 106, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Roberto, F.; Guardigli, G.; Mele, D.; Percoco, G.F.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischaemia and heart failure. Curr. Pharm. Des. 2004, 10, 1699–1711. [Google Scholar] [CrossRef]
- Avkiran, M.; Marber, M.S. Na+/H+ exchange inhibitors for cardioprotective therapy: Progress, problems and prospects. J. Am. Coll. Cardiol. 2002, 39, 747–753. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Thibault, H.; Gharib, A.; Dumont, J.M.; Vuagniaux, G.; Scalfaro, P.; Derumeaux, G.; Ovize, M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1654–H1661. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair and remodeling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ciarka, A.; Borne, P.v.d.; Pathak, A. Myocardial infarction, heart failure and sympathetic nervous system activity: New pharmacological approaches that affect neurohumoral activation. Expert Opin. Investig. Drugs 2008, 17, 1315–1330. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Ponikowski, P.; Piepoli, M.F.; Banasiak, W.; Anker, S.D.; Poole-Wilson, P.A. Autonomic imbalance and immune activation in chronic heart failure—Pathophysiological links. Cardiovasc. Res. 2006, 70, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Donato, M.; Perez, V.; Deutsch, A.C.; Hocht, C.; del Mauro, J.S.; Rodriguez, M.; Gelpi, R.J. Changes in the loading conditions induced by vagal stimulation modify the myocardial infarct size through sympathetic-parasympathetic interactions. Pflugers Arch. 2015, 467, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, B.; Kelly, J.; Munoz, M.; Bernatene, E.A.; Mendez Diodati, N.; Gonzalez Maglio, D.H.; Dominici, F.P.; Gelpi, R.J. Vagal stimulation mimics preconditioning and postconditioning of ischemic myocardium in mice by activating different protection mechanisms. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1289–H1297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Su, Y.; Zhang, Y.; Pan, Z.; Yang, L.; Chen, X.; Liu, Y.; Lu, Y.; Du, Z.; Yang, B. Activation of cardiac muscarinic m3 receptors induces delayed cardioprotection by preserving phosphorylated connexin43 and up-regulating cyclooxygenase-2 expression. Br. J. Pharmacol. 2010, 159, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Zheng, Y.; Cai, J.; Fan, J.; Wang, J.; Yang, J.; Cui, Q.; Xu, G.; Tang, C.; Geng, B. Catestatin attenuates endoplasmic reticulum induced cell apoptosis by activation type 2 muscarinic acetylcholine receptor in cardiac ischemia/reperfusion. Sci. Rep. 2015, 5, 16590. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.L.; Hussain, A.; Maddock, H.L. Ipratropium bromide-mediated myocardial injury in in vitro models of myocardial ischaemia/reperfusion. Toxicol. Sci. 2014, 138, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.S.; Liu, J.J.; Hwang, T.C.; Yu, X.J.; Zhao, M.; Zhao, M.; Yuan, B.X.; Lu, Y.; Kang, Y.M.; Wang, B.; et al. Optimizing the parameters of vagus nerve stimulation by uniform design in rats with acute myocardial infarction. PLoS ONE 2012, 7, e42799. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.; Buchholz, B.; Rodriguez, M.; Perez, V.; Inserte, J.; Garcia-Dorado, D.; Gelpi, R.J. Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp. Physiol. 2013, 98, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhou, X.; Yu, L.; Liu, Q.; Sheng, X.; Wang, Z.; Wang, S.; Jiang, H.; Zhou, S. Low-level vagus nerve stimulation attenuates myocardial ischemic reperfusion injury by antioxidative stress and antiapoptosis reactions in canines. J. Cardiovasc. Electrophysiol. 2016, 27, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Calvillo, L.; Vanoli, E.; Andreoli, E.; Besana, A.; Omodeo, E.; Gnecchi, M.; Zerbi, P.; Vago, G.; Busca, G.; Schwartz, P.J. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J. Cardiovasc. Pharmacol. 2011, 58, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Tratsiakovich, Y.; Mahdi, A.; Yang, J.; Gonon, A.T.; Podesser, B.K.; Pernow, J. Vagal nerve stimulation reduces infarct size via a mechanism involving the α-7 nicotinic acetylcholine receptor and downregulation of cardiac and vascular arginase. Acta Physiol. 2017, 221, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.Z.; Zhan, J.; He, X.H.; Song, X.M.; Wang, Y.L. Protective effect of PNU-120596, a selective α7 nicotinic acetylcholine receptor–positive allosteric modulator, on myocardial ischemia–reperfusion injury in rats. J. Cardiovasc. Pharmacol. 2012, 59, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Yuan, Y.J.; Xue, F.S.; Wang, Q.; Cheng, Y.; Li, R.P.; Liao, X.; Liu, J.H. Postconditioning with α7nAChR agonist attenuates systemic inflammatory response to myocardial ischemia–reperfusion injury in rats. Inflammation 2012, 35, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Yuan, Y.J.; Xue, F.S.; Wang, Q.; Li, S.; Liao, X.; Liu, J.H.; Chen, Y.; Li, R.P. Combined postconditioning with ischemia and α7nAChR agonist produces an enhanced protection against rat myocardial ischemia reperfusion injury. Chin. Med. J. 2012, 125, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, S.A.; Khan, N.S.; Levy, A.C.J.; Faliks, B.T.; Sison, C.P.; Pavlov, V.A.; Zhang, Y.; Ojamaa, K. Nicotinic acetylcholine receptor-mediated protection of the rat heart exposed to ischemia reperfusion. Mol. Med. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Eefting, F.; Rensing, B.; Wigman, J.; Pannekoek, W.J.; Liu, W.M.; Cramer, M.J.; Lips, D.J.; Doevendans, P.A. Role of apoptosis in reperfusion injury. Cardiovasc. Res. 2004, 61, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A. Cell death pathways in acute ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Vahl, T.P.; Goliasch, G.; Picatoste, B.; Arias, T.; Ishikawa, K.; Njerve, I.U.; Sanz, J.; Narula, J.; Sengupta, P.P.; et al. Sphingosine-1-phosphate receptor agonist fingolimod increases myocardial salvage and decreases adverse postinfarction left ventricular remodeling in a porcine model of ischemia/reperfusion. Circulation 2016, 133, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Liu, J.J.; Liu, B.H.; Hu, H.; Sun, L.; Miao, Y.; Xu, H.F.; Yu, X.J.; Ma, X.; Ren, J.; et al. Acetylcholine inhibits hypoxia-induced tumor necrosis factor-α production via regulation of MAPKs phosphorylation in cardiomyocytes. J. Cell. Physiol. 2011, 226, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, M.; Yang, Y.; Xue, R.Q.; Yu, X.J.; Liu, J.K.; Zang, W.J. Acetylcholine attenuates hypoxia/reoxygenation injury by inducing mitophagy through PINK1/Parkin signal pathway in H9c2 cells. J. Cell. Physiol. 2016, 231, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zang, W.J.; Wang, H.; Zhao, M.; Yu, X.J.; He, X.; Miao, Y.; Zhou, J. Acetylcholine promotes ros detoxification against hypoxia/reoxygenation-induced oxidative stress through FoxO3a/PGC-1α dependent superoxide dismutase. Cell. Physiol. Biochem. 2014, 34, 1614–1625. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, M.; Yu, X.J.; Wang, H.; He, X.; Liu, J.K.; Zang, W.J. Cardioprotection by acetylcholine: A novel mechanism via mitochondrial biogenesis and function involving the PGC-1α pathway. J. Cell. Physiol. 2013, 228, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zhou, J.; Zhao, M.; Liu, J.; Sun, L.; Yu, X.; He, X.; Pan, X.; Zang, W. Acetylcholine attenuates hypoxia/ reoxygenation-induced mitochondrial and cytosolic ROS formation in H9c2 cells via m2 acetylcholine receptor. Cell. Physiol. Biochem. 2013, 31, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Krijnen, P.A.; Nijmeijer, R.; Meijer, C.J.; Visser, C.A.; Hack, C.E.; Niessen, H.W. Apoptosis in myocardial ischaemia and infarction. J. Clin. Pathol. 2002, 55, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia–reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Hammerling, B.C.; Gustafsson, A.B. Mitochondrial quality control in the myocardium: Cooperation between protein degradation and mitophagy. J. Mol. Cell. Cardiol. 2014, 75, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Moore, X.L.; Dart, A.M.; Wang, L.M. Systemic inflammatory response following acute myocardial infarction. J. Geriatr. Cardiol. 2015, 12, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.M.; Tracey, K.J. The pulse of inflammation: Heart rate variability, the cholinergic anti-inflammatory pathway and implications for therapy. J. Intern. Med. 2011, 269, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and immunology of the cholinergic antiinflammatory pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Martelli, D.; McKinley, M.J.; McAllen, R.M. The cholinergic anti-inflammatory pathway: A critical review. Auton. Neurosci. 2014, 182, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Fujii, T.; Moriwaki, Y.; Misawa, H.; Horiguchi, K. Non-neuronal cholinergic system in regulation of immune function with a focus on α7 Nachrs. Int. Immunopharmacol. 2015, 29, 127–134. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, W.J.; Ulloa, L. The α7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex—Linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Abe, C.; Sung, S.S.; Moscalu, S.; Jankowski, J.; Huang, L.; Ye, H.; Rosin, D.L.; Guyenet, P.G.; Okusa, M.D. Vagus nerve stimulation mediates protection from kidney ischemia-reperfusion injury through α7nAChR+ splenocytes. J. Clin. Investig. 2016, 126, 1939–1952. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Fu, H.; Huang, F.; Zhao, T.; Chen, J.K.; Li, D.J.; Shen, F.M. Vagus nerve attenuates hepatocyte apoptosis upon ischemia-reperfusion via α7 nicotinic acetylcholine receptor on kupffer cells in mice. Anesthesiology 2016, 125, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Tong, Y.L.; Li, J.C.; Lu, Z.Q.; Yao, Y.M. The protective effect of α7 nicotinic acetylcholine receptor activation on critical illness and its mechanism. Int. J. Biol. Sci. 2017, 13, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. α7 Nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int. J. Mol. Sci. 2012, 13, 2219–2238. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Tian, J.; Yang, H.; Hou, L.; Wang, Z.; He, Z.; Wang, X. α7 Nicotine acetylcholine receptor agonist PNU-282987 attenuates acute lung injury in a cardiopulmonary bypass model in rats. Shock 2016. [Google Scholar] [CrossRef]
- He, Y.; Ye, Z.Q.; Li, X.; Zhu, G.S.; Liu, Y.; Yao, W.F.; Luo, G.J. α7 Nicotinic acetylcholine receptor activation attenuated intestine-derived acute lung injury. J. Surg. Res. 2016, 201, 258–265. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Model | Study Protocol | Mode of Intervention | Major Finding | Interpretation | Ref. |
|---|---|---|---|---|---|
| Ex Vivo | |||||
| Sprague-Dawley rats | Langendorff perfusion Global ischemia: 30 min Reperfusion: 1 h | Pre-ischemia | CST 100 nM ↓ infarct size: 64.3% ↓ LVEDP ↑ dLVP, LV ±dp/dtmax -Atropine (10 nM) and AF-DX116 (100 nM) abrogated this protection. | m2AChR activation by CST reduces infarct size and attenuates myocardial I/R injury. | [44] |
| Langendorff perfusion LAD ligation Ischemia: 35 min Reperfusion: 1 h | Pre-ischemia | IPC (3 cycle of 5 min-global ischemia/reperfusion) ↓ infarct size: 70.8% ↑ ACh: 88% -Hexamethonium (50 µM) and atropine (100 nM) blocked IPC protection. | IPC involved activation of the intrinsic cardiac nervous system, leading to release of ACh in ventricles via activation of mAChRs. | [16] | |
| Langendorff perfusion LAD ligation Ischemia: 35 min Reperfusion: 2 h | Onset of reperfusion | ACh 10−7 M ↓ infarct size: 17.6% -Ipratropium bromide (10−11–10−4 M) increased infarct size in a dose dependent manner. | ACh treatment reduces infarct size through mAChRs. | [45] | |
| In Vivo | |||||
| New Zealand rabbits | LAD ligation Ischemia: 30 min Reperfusion: 3 h | Pre-ischemia | I-VNS (0.1 ms, 10 Hz, cycles of 10 s ON/50 s OFF) ↓ infarct size: 42.6% C-VNS (0.1 ms, 10 Hz) ↑ infarct size: 36% ↑ LVEDP, AP -Atropine (1.3–2.0 mg/kg) blocked the cardioprotective effects. | VNS performed intermittently antagonizes the sympathetic system and reduces the infarct size through mAChR activation. | [41] |
| Global no-flow Ischemia: 30 min Reperfusion: 3 h | Pre-ischemia | rIPC (three cycles of 5 min of hindlimb ischemia and 5 min of reperfusion) ↓ infarct size: 59.8% ↑ LVDP, ↓ LVEDP -Spinal cord section abolished the reduction of infarct size. VNS (0.1 ms, 10 Hz) ↓ infarct size: 57.8% ↑ LVDP, ↓ LVEDP -Atropine (1.3–2.0 mg/kg) and vagus nerve section abolished the reduction of infarct size. | rIPC and VNS activate a neural afferent pathway and exert cardioprotection through mAChRs activation. | [48] | |
| Wistar rats | LAD ligation Ischemia: 30 min Reperfusion: 3 h | Pre-ischemia | Choline (5 mg/kg) ↓ infarct size: 20.4% ↓ VT, VF ↑ pCx43 | Choline treatment reduces infarct size and preserved pCx43 via m3AChR. | [43] |
| LAD ligation Ischemia: 6 h | Pre-ischemia | Choline (5 mg/kg) ↓ infarct size: 33% ↓ LVEDP, LVSP ↑ ±dP/dt ↓ Arrhythmic scores, VT, VF -4-DAMP (0.5 µg/kg) abolished the protective effect of choline. | Choline treatment reduces infarct size and improves cardiac function via m3AChR. | [19] | |
| FVB mice | LAD ligation Ischemia: 30 min Reperfusion: 2 h | Pre-ischemia | VNS (0.1 ms, 10 Hz) ↓ infarct size: 10% -Atropine (3–5 mg/kg) and wortmannin (1 mg/kg) reverse the reduction of infarct size. | Preischemic vagal stimulation reduces infarct size through mAChRs activation. | [42] |
| Domestic pigs | LAD ligation Ischemia: 1 h Reperfusion: 2 h | During ischemia | I-VNS (3.5 mA, pulse width 0.5 ms, 20 Hz, cycle of 21 s ON/30 s OFF) ↓ infarct size: 89% ↓ number of VT/VF, PVC C-VNS (3.5 mA, 0.5 ms, 20 Hz) ↓ infarct size: 60% ↓ number of PVC -Atropine (1 mg/kg) abolished the beneficial effects of VNS. | Both I-VNS and C-VNS reduce the infarct size and ventricular dysfunction through mAChRs. | [7] |
| LAD ligation Ischemia: 1 h Reperfusion: 2 h | During ischemia | I-VNS (3.5 mA, pulse width 0.5 ms, 20 Hz, cycle of 21 s ON/30 s OFF) ↓ infarct size: 59% ↓ number of VT/VF, PVC ↑ pCx43 -Atropine (1 mg/kg) abolished the beneficial effects of VNS. | I-VNS reduces infarct size and ventricular fibrillation incidence through mAChRs. | [6] | |
| Mongrel dogs | LAD ligation Ischemia: 1 h Reperfusion: 1 h | During ischemia | VNS (0.1 ms, 20 Hz) ↓ infarct size: 47.1% ↓ number of PVC, VT, VF and LF/HF ratio. | VNS reduces infarct size and ventricular arrhythmia. | [49] |
| Model | Study Protocol | Mode of Intervention | Major Finding | Interpretation | Ref. |
|---|---|---|---|---|---|
| In Vivo | |||||
| Sprague-Dawley rats | LAD occlusion Ischemia: 1 h Reperfusion: 2 h | Pre-ischemia | VNS (1 ms, 5 Hz) ↓ infarct size: 43% ↓ LVEDP ↑ LVSP, ±dP/dt ↓ incidence of VF: 81.6% ↓ remote vascular injury -MLA (10 mg/kg) and α7nAChR shRNA inhibited the protective effects for remote vascular injury. | VNS reduces infarct size and remote vascular protection via activating α7nAChR-mediated cholinergic pathway. | [12] |
| LAD ligation Ischemia: 30 min Reperfusion: 2 h | Pre-ischemia | PNU-120596 (1 mg/kg) ↓ infarct size: 27.6% -BGT (1 µg/kg) abolished the effect of PNU-120596. | α7nAChR activation by PNU-120596 reduces infarct size. | [52] | |
| LAD ligation Ischemia: 30 min Reperfusion: 24 h | During ischemia | VNS (2.5 V, pulse width 0.5 ms, 8–10 Hz) ↓ infarct size: 87.7% -MEC (2.1 mg/kg) reduced the protective effect of VNS. | VNS decreases infarct size through α7nAChR. | [50] | |
| LAD ligation Ischemia: 30 min Reperfusion: 24 h | During ischemia | VNS (0.5 ms, 0.1–1 mA,15 Hz, cycle of 40 s ON/20 s OFF) ↓ infarct size: 38.8% -MLA (10 mg/kg) partially abolished the protective effect of VNS. | VNS decreases infarct size through α7nAChR. | [51] | |
| LAD ligation Ischemia: 30 min Reperfusion: 3 h | Onset of reperfusion | PNU-282987 (2.4 mg/kg) ↓ infarct size: 23% | α7nAChR activation by PNU-282987 protects against myocardial I/R injury. | [53] | |
| LAD ligation Ischemia: 30 min Reperfusion: 3 h | Onset of reperfusion | Combined ischemia postconditioning and PNU-282987 (2.4 mg/kg) ↓ infarct size: 45% | Combined ischemia postconditioning and α7nAChR activation by PNU-282987 protect against myocardial I/R injury. | [54] | |
| LAD ligation Ischemia: 30 min Reperfusion: 1 h | Onset of reperfusion | GTS21 (0.06–1.0 mg/kg) ↓ infarct size: 42% ↑ LVDP, ±dP/dt | α7nAChR activation by GTS21 at initial of reperfusion reduces infarct size and improved LV function. | [55] | |
| C57BL6 mice | LAD ligation Ischemia: 30 min Reperfusion: 24 h | Pre-ischemia | Electroacupuncture (1 mA: 2 Hz and 100 Hz) at Neiguan acupoint (PC6) ↓ infarct size: 33.9% ↓ serum cardiac troponin 1 -MEC (1 mg/kg) and MLA (1 mg/kg) reversed the cardioprotective effect of electroacupuncture. | Electroacupuncture at Neiguan acupoint reduces infarct size through α7nAChR. | [8] |
| FVB mice | LAD ligation Ischemia: 30 min Reperfusion: 2 h | Onset of reperfusion | VNS (0.1 ms, 10 Hz) ↓ infarct size: 9% -MLA (5 mg/kg) and AG490 (4 mg/kg) reverse the reduction of infarct size. | Vagal stimulation during the first 10 min of reperfusion reduces infarct size through α7nAChR activation. | [42] |
| Model | Study Protocol | Mode of Intervention | Major Finding | Interpretation | Ref. | |
|---|---|---|---|---|---|---|
| Anti-Apoptosis | Anti-Oxidative Stress | |||||
| In Vitro | ||||||
| Neonatal rat cardiomyocytes | Hypoxia: 12 h Reoxygenation: 24 h | Pre-hypoxia | CST (100 nM) ↓ cleaved caspase-3, -9, and -7, PARP ↓ apoptotic cell, cAMP ↓ p-ERK1/2, pAkt -PD98059 (20 µM) and wortmannin (10 nM) blocked the CST protection on cell apoptosis. -AF-DX116 (100 nM) blocked the effect of CST. | - | m2AChR activation by CST activates ERK1/2 and PI3K/Akt pathways to inhibit ER stress-induced cell apoptosis. | [44] |
| H9c2 cells | Hypoxia: 24 h | During hypoxia | ACh (10−6 M) and SB203580 (10−5 M) ↓ TNFα, cleaved caspase-3, ↓ % cell death ↓ p-p38MAPK, p-JNK ↓ p-ERK -Atropine (10−4 M) and Methoctramine (10−4 M) abrogated the effect of ACh treatment. | - | ACh treatment inhibits hypoxia-induced TNFα production via MAPK phosphorylation through m2AChR. | [59] |
| Hypoxia: 12 h Reoxygenation: 2 h | During hypoxia | - | ACh (10−5 M) ↓ mtROS, ↓ XO, NOX activity-Atropine (10−4 M) and m2AChR siRNA abolished the antioxidant and cardioprotective effect of ACh. | ACh treatment inhibits mitochondrial and cytosolic ROS production via m2AChR. | [63] | |
| Hypoxia: 8 h Reoxygenation: 4 h | Onset of reoxygenation | ACh (10−6 M) ↓ Cleaved caspase-3, cytC ↓ mtROS and mitochondria swelling ↑ ATP content, preserved mitochondrial membrane potential -Methoctramine (10−6 M) and m2AChR siRNA reversed the effect of ACh. | - | ACh treatment promotes cytoprotective mitophagy and preserved cardiac homeostasis via m2AChR. | [60] | |
| Hypoxia: 8 h Reoxygenation: 3 h | Onset of reoxygenation | ACh (10−3 M) ↓ Apoptotic cell ↑ ATP synthesis, ↑ mtDNA copy -Atropine (10−3 M), PGC-1α siRNA and AMPK siRNA blocked the effect of ACh on mitochondria function. | ACh treatment reduces H/R injury through promoting mitochondria biogenesis and function through AMPK/PGC-1α pathway via mAChRs. | [62] | ||
| Hypoxia: 8 h Reoxygenation: 2 h | Onset of reoxygenation | ACh (10−5 M) ↓ Apoptotic cell ↑ ATP synthesis -Atropine (10−4 M), PGC-1α siRNA block the effect of ACh. | ACh (10−5 M) ↑ SOD1, SOD2 ↓ ROS, ∆Ψm -Atropine (10−4 M) reversed the effect of ACh. -FoxO3a siRNA blocked the effect of SOD activities. | ACh treatment reduces H/R injury through promoting mitochondria function and ROS detoxification through FoxO3a/PGC-1α pathway via mAChRs. | [61] | |
| Adult isolated rat ventricular cardio- myocytes | Hypoxia: 4 h Reoxygenation: 2 h | Onset of reoxygenation | ACh (10−7 M) ↓ cleaved caspase-3 ↑ cell viability -Atropine (10−7 M) reversed the effect of ACh on cell apoptosis. | ACh (10−7 M) ↑ time of myocardial depolarization and hypercontraction | ACh treatment reduces apoptosis and oxidative stress via muscarinic receptors. | [45] |
| In Vivo | ||||||
| Wistar rats | LAD ligation Ischemia: 30 min Reperfusion: 3 h | Pre-hypoxia | - | Choline (5 mg/kg) ↑ Hsp70, COX-2 ↓ dephosphorylated Cx43 | Choline treatment exerts cytoprotective effect against ischemic myocardial injury via m3AChR. | [43] |
| LAD ligation Ischemia: 6 h | Pre-hypoxia | Choline (5 mg/kg) ↓ Fas, p38MAPK, apoptotic cells ↑ ERK1, ERK2, Bcl-2 -4-DAMP reversed the effect of choline. | Choline (5 mg/kg) ↑ SOD ↓ MDA -4-DAMP abolished the protective effects of choline. | Choline treatment exerts cytoprotective effect against ischemic myocardial injury via m3AChR. | [19] | |
| Domestic pigs | LAD ligation Ischemia: 1 h Reperfusion: 2 h | During hypoxia | - | C-VNS and I-VNS (3.5 mA, pulse width 0.5 ms, 20 Hz, cycle of 21 s ON/30 s OFF) ↓ mitochondria ROS production, swelling preserved mitochondrial membrane potential -Atropine (1 mg/kg) abolished the beneficial effects of VNS. | VNS decreases mitochondrial ROS production and swelling and prevents mitochondrial membrane depolarization via mAChRs. | [7] |
| Mongrel dogs | LAD ligation Ischemia: 1 h Reperfusion: 1 h | During hypoxia | VNS ↓ apoptotic cells, MPO, Bax protein ↑ Bcl-2 protein | VNS ↓ serum MDA, myocardial MDA ↑ serum SOD and myocardial SOD | VNS suppresses apoptosis and oxidative stress. | [49] |
| Model | Study Protocol | Mode of Intervention | Major Finding | Interpretation | Ref. | ||
|---|---|---|---|---|---|---|---|
| Anti- Apoptosis | Anti-Oxidative Stress | Anti- Inflammation | |||||
| Ex Vivo | |||||||
| Sprague-Dawley rats | Global ischemia Ischemia: 30 min Reperfusion: 40 min | Pre-ischemia | GTS21 (1.6 × 10−8 M) ↓ ROS production maintenance of ∆Ψ -MLA (2.33 × 10−7 M) blocked the effect of GTS21. | GTS21 treatment reduces I/R injury by preserving mitochondrial membrane potential, maintaining intracellular ATP and reducing ROS production via α7nAChR. | [55] | ||
| In Vivo | |||||||
| Sprague- Dawley rats | LAD ligation Ischemia: 30 min Reperfusion: 2 h | Pre-ischemia | - | PNU-120596 (1 mg/kg) ↑ SOD ↓ MDA, MPO -BGT (1 µg/kg) abolished the effect of PNU-120596. | PNU-120596 (1 mg/kg) ↓ serum TNF-α, IL-6 ↓ NF-κB p65 protein expression -BGT (1 µg/kg) abolished the effect of PNU-120596. | PNU-120596 treatment increases SOD activities, attenuated MPO activities and MDA contents in myocardium and decreased serum pro-inflammatory cytokine production via α7nAChR. | [52] |
| LAD ligation Ischemia: 30 min Reperfusion: 24 h | During ischemia | VNS (2.5 V, pulse width 0.5 ms, 8–10 Hz) ↓ Apoptotic cell -MEC (2.1 mg/kg) reduced the protective effect of VNS. | - | VNS (2.5 V, pulse width 0.5 ms, 8–10 Hz) ↓ macrophage infiltration ↓ PMN infiltration -MEC (2.1 mg/kg) reduced the protective effect of VNS. | VNS decreases apoptotic cell, macrophage and PMN infiltration through nAChRs. | [50] | |
| LAD ligation Ischemia: 30 min Reperfusion: 3 h | Onset of reperfusion | PNU-282987 (2.4 mg/kg) ↓ serum TNFα, IL-6, HMGB1 | Postconditioning with PNU-282987 attenuates systemic inflammatory response to myocardial I/R injury. | [53] | |||
| LAD ligation Ischemia: 30 min Reperfusion: 3 h | Onset of reperfusion | PNU-282987 (2.4 mg/kg) ↓ serum TnI, TNFα and HMGB1 | Combined Postconditioning with PNU-282987 and ischemia postconditioning attenuate systemic inflammatory response to myocardial I/R injury. | [54] | |||
| Mongrel dog | LAD ligation Ischemia: 1 h Reperfusion: 6 h | During ischemia | - | - | VNS (pulse width 0.5 ms, 10 Hz, 1.5–3 V) ↓ serum TNF-α ↓ serum IL-6 ↓ neutrophil infiltration ↑ α7nAChR protein | VNS activates anti-inflammatory pathway and inhibits the systemic and local inflammatory reaction. | [11] |
| C57BL6 mice | LAD ligation Ischemia: 30 min Reperfusion: 24 h | Pre-ischemia | Electroacupuncture (1 mA: 2 Hz and 100 Hz) at Neiguan acupoint (PC6) ↓ HMGB1 ↓ neutrophil infiltration -MEC (1 mg/kg) and MLA (1 mg/kg) reversed the cardioprotective effect of electroacupuncture. | Electroacupuncture attenuates pro-inflammatory responses and I/R injury via α7nAChR. | [8] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Intachai, K.; C. Chattipakorn, S.; Chattipakorn, N.; Shinlapawittayatorn, K. Revisiting the Cardioprotective Effects of Acetylcholine Receptor Activation against Myocardial Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 2466. https://doi.org/10.3390/ijms19092466
Intachai K, C. Chattipakorn S, Chattipakorn N, Shinlapawittayatorn K. Revisiting the Cardioprotective Effects of Acetylcholine Receptor Activation against Myocardial Ischemia/Reperfusion Injury. International Journal of Molecular Sciences. 2018; 19(9):2466. https://doi.org/10.3390/ijms19092466
Chicago/Turabian StyleIntachai, Kannaporn, Siriporn C. Chattipakorn, Nipon Chattipakorn, and Krekwit Shinlapawittayatorn. 2018. "Revisiting the Cardioprotective Effects of Acetylcholine Receptor Activation against Myocardial Ischemia/Reperfusion Injury" International Journal of Molecular Sciences 19, no. 9: 2466. https://doi.org/10.3390/ijms19092466
APA StyleIntachai, K., C. Chattipakorn, S., Chattipakorn, N., & Shinlapawittayatorn, K. (2018). Revisiting the Cardioprotective Effects of Acetylcholine Receptor Activation against Myocardial Ischemia/Reperfusion Injury. International Journal of Molecular Sciences, 19(9), 2466. https://doi.org/10.3390/ijms19092466