Machine Learning Based Toxicity Prediction: From Chemical Structural Description to Transcriptome Analysis

Abstract
1. Introduction
2. Machine Learning
2.1. Shallow Architectures
2.2. Deep Learning
3. Chemical Structure Descriptors
3.1. Traditional Chemical Descriptors
3.2. Deep-Minded Chemical Descriptor
3.3. Chemical Properties
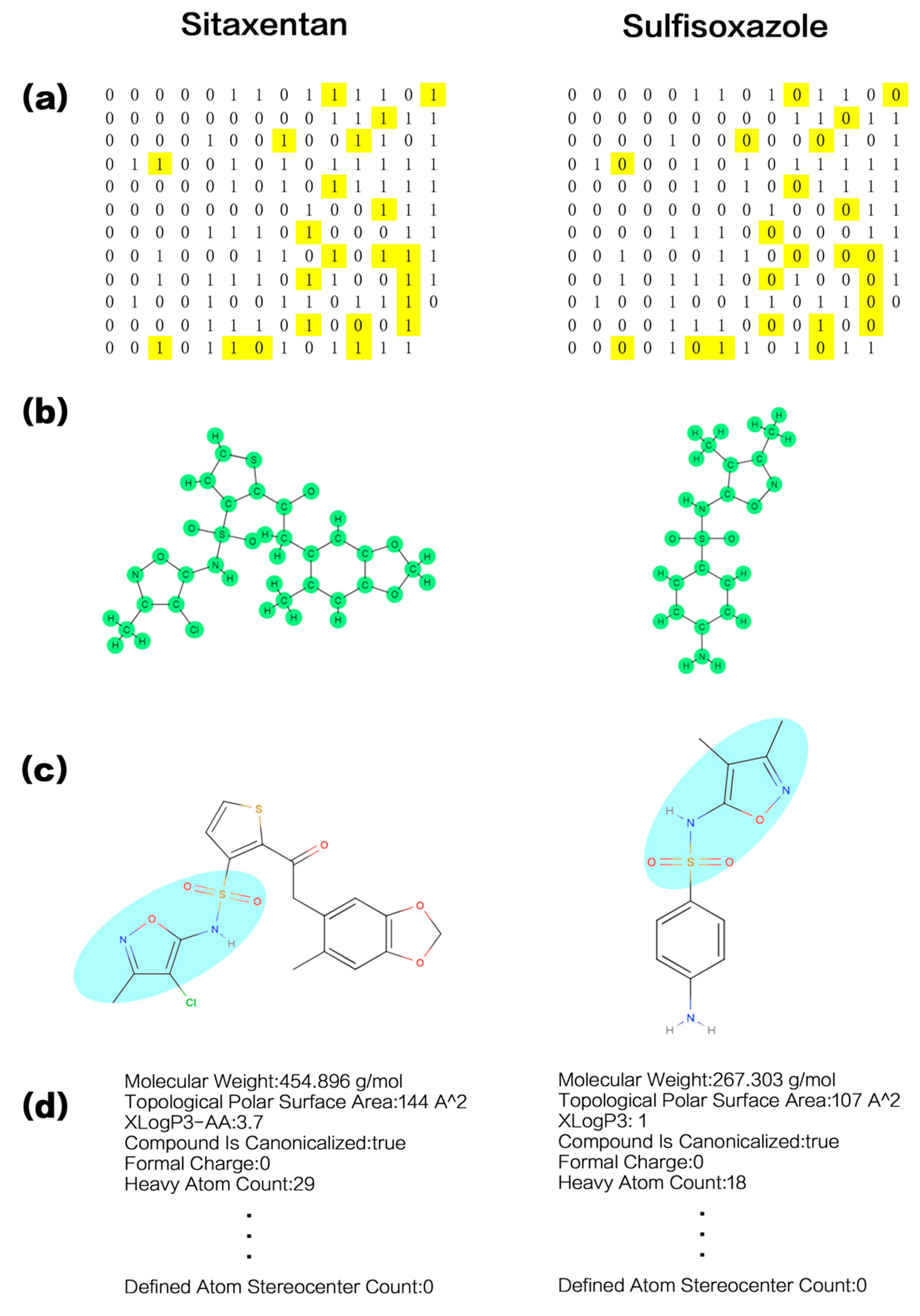
3.4. Examples of Chemical Structural Description
4. Chemical Structure Based Toxicity Prediction by Machine Learning
4.1. Data Collection
4.2. Performance
5. Acute (Immediate) Toxicity Prediction
6. Chronic (Delayed) Toxicity Prediction
6.1. Prediction Based on Chemical Structure
6.2. Prediction with Cellular Transcriptome Information
7. An in Silico Platform of Deep Learning Based Toxicity Prediction
7.1. Collection of Gene Expression Data
7.2. Representation of Gene Expression Data
7.3. Toxicity Prediction
8. Summary
Acknowledgments
Conflicts of Interest
References
- Ting, N. (Ed.) Introduction and New Drug Development Process. In Dose Finding in Drug Development; Springer: New York, NY, USA, 2006; pp. 1–17. [Google Scholar]
- Janodia, M.D.; Sreedhar, D.; Virendra, L.; Ajay, P.; Udupa, N. Drug Development Process: A review. Pharm. Rev. 2007, 5, 2214–2221. [Google Scholar]
- Hwang, T.J.; Carpenter, D.; Lauffenburger, J.C.; Wang, B.; Franklin, J.M.; Kesselheim, A.S. Failure of Investigational Drugs in Late-Stage Clinical Development and Publication of Trial Results. JAMA Intern. Med. 2016, 176, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Erve, J.C.; Gauby, S.; Maynard, M.J., Jr.; Svensson, M.A.; Tonn, G.; Quinn, K.P. Bioactivation of sitaxentan in liver microsomes, hepatocytes, and expressed human P450s with characterization of the glutathione conjugate by liquid chromatography tandem mass spectrometry. Chem. Res. Toxicol. 2013, 26, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Hoeper, M.M.; Simon, J.; Gibbs, R.; Simonneau, G. Liver toxicity of sitaxentan in pulmonary arterial hypertension. Eur. Heart J. 2011, 32, 386–387. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E. Fusion of nonclinical and clinical data to predict human drug safety. Expert Rev. Clin. Pharmacol. 2013, 6, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A. The Flaws and Human Harms of Animal Experimentation. Camb. Q. Healthc. Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.; Cross, D.M.; Derzi, M.; Horsley, E.; Stavros, F.L. An overview of the preclinical toxicity and potential carcinogenicity of sitaxentan (Thelin®), a potent endothelin receptor antagonist developed for pulmonary arterial hypertension. Regul. Toxicol. Pharmacol. 2012, 64, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.S.; Paules, R.S.; Simeonov, A.; Fitzpatrick, S.C.; Crofton, K.M.; Casey, W.M.; Mendrick, D.L. The US Federal Tox21 Program: A strategic and operational plan for continued leadership. Altex 2018, 35, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR Modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Das, R.N. Chapter 7—Validation of QSAR Models. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Roy, K., Kar, S., Das, R.N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 231–289. [Google Scholar]
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar] [CrossRef]
- Free, S.M.; Wilson, J.W. A Mathematical Contribution to Structure-Activity Studies. J. Med. Chem. 1964, 7, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Quinn, F.R.; Neiman, Z.; Beisler, J.A. Toxicity and quantitative structure-activity relationships of colchicines. J. Med. Chem. 1981, 24, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Cain, B.F.; Atwell, G.J.; Hansch, C.; Panthananickal, A.; Leo, A. Potential antitumor agents. 36. Quantitative relationships between experimental antitumor activity, toxicity, and structure for the general class of 9-anilinoacridine antitumor agents. J. Med. Chem. 1982, 25, 276–315. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A.; Atwell, G.J.; Cain, B.F. Potential antitumor agents. 32. Role of agent base strength in the quantitative structure-antitumor relationships for 4′-(9-acridinylamino) methanesulfonanilide analogs. J. Med. Chem. 1979, 22, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Barratt, M.D. Prediction of toxicity from chemical structure. Cell Biol. Toxicol. 2000, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Compton, P.; Preston, P.; Edwards, G.; Kang, B. Knowledge Based Systems That Have Some Idea of Their Limits. CIO 2000, 15, 57–63. [Google Scholar]
- Mitchell, T.M. Machine Learning; McGraw Hill: Ridge, IL, USA, 1997; Volume 45, pp. 870–877. [Google Scholar]
- Bishop, C.M. Pattern Recognition and Machine Learning, 1st ed.; Springer: New York, NY, USA, 2006; p. 738. [Google Scholar]
- Fürnkranz, J.; Gamberger, D.; Lavrač, N. Machine Learning and Data Mining. Comput. Study 2010, 42, 110–114. [Google Scholar]
- Yang, H.; Sun, L.; Li, W.; Liu, G.; Tang, Y. Corrigendum: In Silico Prediction of Chemical Toxicity for Drug Design Using Machine Learning Methods and Structural Alerts. Front. Chem. 2018, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Hemmateenejad, B.; Akhond, M.; Miri, R.; Shamsipur, M. Genetic algorithm applied to the selection of factors in principal component-artificial neural networks: Application to QSAR study of calcium channel antagonist activity of 1,4-dihydropyridines (nifedipine analogous). Cheminform 2003, 34, 1328–1334. [Google Scholar] [CrossRef]
- Hoffman, B.T.; Kopajtic, T.; And, J.L.K.; Newman, A.H. 2D QSAR Modeling and Preliminary Database Searching for Dopamine Transporter Inhibitors Using Genetic Algorithm Variable Selection of Molconn Z Descriptors. J. Med. Chem. 2000, 43, 4151–4159. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, P.G.; Muratov, E.N.; Artemenko, A.G.; Kolumbin, O.G.; Muratov, N.N.; Kuz’Min, V.E. Application of random forest approach to QSAR prediction of aquatic toxicity. J. Chem. Inform. Model. 2009, 49, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inform. Comput. Sci. 2015, 43, 1947. [Google Scholar] [CrossRef] [PubMed]
- Svetnik, V.; Liaw, A.; Tong, C.; Wang, T. Application of Breiman’s Random Forest to Modeling Structure-Activity Relationships of Pharmaceutical Molecules. In Proceedings of the Multiple Classifier Systems, International Workshop, MCS 2004, Cagliari, Italy, 9–11 June 2004; Roli, F., Kittler, J., Windeatt, T., Eds.; Springer: Berlin/Heidelberg, Germany; Cagliari, Italy, 2004; pp. 334–343. [Google Scholar]
- Agrafiotis, D.K.; Cedeño, W.; Lobanov, V.S. On the use of neural network ensembles in QSAR and QSPR. J. Chem. Inform. Comput. Sci. 2002, 42, 903–911. [Google Scholar] [CrossRef]
- Wikel, J.H.; Dow, E.R. The use of neural networks for variable selection in QSAR. Bioorgan. Med. Chem. Lett. 1993, 3, 645–651. [Google Scholar] [CrossRef]
- Lu, X.; Ball, J.W.; Dixon, S.L.; Jurs, P.C. Quantitative structure-activity relationships for toxicity of phenols using regression analysis and computational neural networks. Environ. Toxicol. Chem. 1998, 13, 841–851. [Google Scholar]
- Lu, J.; Peng, J.; Wang, J.; Shen, Q.; Bi, Y.; Gong, L.; Zheng, M.; Luo, X.; Zhu, W.; Jiang, H. Estimation of acute oral toxicity in rat using local lazy learning. J. Cheminform. 2014, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Mazzatorta, P.; Cronin, M.T.D.; Benfenati, E. A QSAR Study of Avian Oral Toxicity using Support Vector Machines and Genetic Algorithms. Qsar Comb. Sci. 2010, 25, 616–628. [Google Scholar] [CrossRef]
- Srinivasan, A.; King, R.D. Using Inductive Logic Programming to construct Structure-Activity Relationshipsp; AAAI: Menlo Park, CA, USA, 1999; pp. 64–73. [Google Scholar]
- Rosenblatt, F. The Perceptron: A Probabilistic Model for Information Storage and Organization in the Brain; MIT Press: Cambridge, MA, USA, 1988; pp. 386–408. [Google Scholar]
- Widrow, B.; Hoff, M.E. Adaptive Switching Circuits. In Neurocomputing: Foundations of Research; Ire Wescon Conv. Rec; MIT Press: Cambridge, MA, USA, 1966. [Google Scholar]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 2002, 13, 21–27. [Google Scholar] [CrossRef]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V.; Cortes, C.; Vapnik, V.; Llorens, C.; Vapnik, V.N.; Cortes, C.; Côrtes, M.V.C.B. Support-vector networks. Mach. Learn. 1995, 20, 27–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Tin Kam, H. Random Decision Forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning representations by back-propagating errors. Read. Cognit. Sci. 1986, 323, 399–421. [Google Scholar] [CrossRef]
- Hochreiter, S. The Vanishing Gradient Problem during Learning Recurrent Neural Nets and Problem Solutions. Int. J. Uncertain. Fuzz. Knowl.-Based Syst. 1998, 6, 107–116. [Google Scholar] [CrossRef]
- Glorot, X.; Bordes, A.; Bengio, Y. Deep Sparse Rectifier Neural Networks. In Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics, Fort Lauderdale, FL, USA, 11–13 April 2011; Geoffrey, G., David, D., Miroslav, D., Eds.; PMLR Proceedings of Machine Learning Research, 2011; Volume 15, pp. 315–323. [Google Scholar]
- Zahangir Alom, M.; Taha, T.M.; Yakopcic, C.; Westberg, S.; Hasan, M.; Van Esesn, B.C.; Awwal, A.A.S.; Asari, V.K. The History Began from AlexNet: A Comprehensive Survey on Deep Learning Approaches. arXiv 2018, arXiv:1803.01164. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. In Proceedings of the 25th International Conference on Neural Information Processing Systems, Lake Tahoe, NV, USA, 3–6 December 2012; Curran Associates Inc.: Lake Tahoe, NV, USA, 2012; Volume 1, pp. 1097–1105. [Google Scholar]
- Deng, J.; Dong, W.; Socher, R.; Li, L.J.; Kai, L.; Li, F.-F. ImageNet: A large-scale hierarchical image database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar]
- Hinton, G.; Deng, L.; Yu, D.; Dahl, G.E.; Mohamed, A.R.; Jaitly, N.; Senior, A.; Vanhoucke, V.; Nguyen, P.; Sainath, T.N. Deep Neural Networks for Acoustic Modeling in Speech Recognition: The Shared Views of Four Research Groups. IEEE Signal Process. Mag. 2012, 29, 82–97. [Google Scholar] [CrossRef]
- Dahl, G.E.; Acero, A. Context-Dependent Pre-Trained Deep Neural Networks for Large-Vocabulary Speech Recognition. IEEE Trans. Audio Speech Lang. Process. 2012, 20, 30–42. [Google Scholar] [CrossRef]
- Luong, T.; Socher, R.; Manning, C.D. Better Word Representations with Recursive Neural Networks for Morphology. In Proceedings of the Seventeenth Conference on Computational Natural Language Learning, Sofia, Bulgaria, 8–9 August 2013; pp. 104–113. [Google Scholar]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to Sequence Learning with Neural Networks. In Proceedings of the Advances in Neural Information Processing Systems 27: 28th Annual Conference on Neural Information Processing Systems 2014, Montreal, QC, Canada, 8–13 December 2014; Volume 4, pp. 3104–3112. [Google Scholar]
- Zeiler, M.D.; Fergus, R. Visualizing and Understanding Convolutional Networks. In Proceedings of the European Conference on Computer Vision, Zurich, Switzerland, 6–12 September 2014; Volume 8689, pp. 818–833. [Google Scholar]
- Donahue, J.; Hendricks, L.A.; Guadarrama, S.; Rohrbach, M.; Venugopalan, S.; Darrell, T.; Saenko, K. Long-term recurrent convolutional networks for visual recognition and description. In Proceedings of the Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; p. 677. [Google Scholar]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Webb, S. Deep learning for biology. Nature 2018, 554, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Hopfield, J.J. Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. USA 1982, 79, 2554–2558. [Google Scholar] [CrossRef] [PubMed]
- Pineda, F.J. Recurrent Backpropagation and the Dynamical Approach to Adaptive Neural Computation. Neural Comput. 1989, 1, 161–172. [Google Scholar] [CrossRef]
- Lawrence, S.; Giles, C.L.; Tsoi, A.C.; Back, A.D. Face recognition: A convolutional neural-network approach. IEEE Trans. Neural Netw. 1997, 8, 98–113. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y. Convolutional networks for images, speech, and time series. In The Handbook of Brain Theory and Neural Networks; Michael, A.A., Ed.; MIT Press: Cambridge, MA, USA, 1998; pp. 255–258. [Google Scholar]
- Cho, K.; van Merrienboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning Phrase Representations using RNN Encoder-Decoder for Statistical Machine Translation. arXiv 2014, arXiv:1406.1078. [Google Scholar]
- Che, Z.; Purushotham, S.; Cho, K.; Sontag, D.; Liu, Y. Recurrent Neural Networks for Multivariate Time Series with Missing Values. Sci. Rep. 2018, 8, 6085. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, P.G. Recurrent neural network for time series prediction. In Proceedings of the 15th Annual International Conference of the IEEE Engineering in Medicine and Biology Societ, San Diego, CA, USA, 31 October 1993; pp. 250–251. [Google Scholar]
- Jaderberg, M.; Simonyan, K.; Zisserman, A.; Kavukcuoglu, K. Spatial Transformer Networks. arXiv 2015, arXiv:1506.02025. [Google Scholar]
- Dean, J.; Corrado, G.S.; Monga, R.; Chen, K.; Devin, M.; Le, Q.V.; Mao, M.Z.; Ranzato, M.A.; Senior, A.; Tucker, P.; et al. Large scale distributed deep networks. In Proceedings of the 25th International Conference on Neural Information Processing Systems, Lake Tahoe, NV, USA, 3–6 December 2012; Curran Associates Inc.: Lake Tahoe, NV, USA, 2012; Volume 1, pp. 1223–1231. [Google Scholar]
- Raina, R.; Madhavan, A.; Ng, A.Y. Large-scale deep unsupervised learning using graphics processors. In Proceedings of the 26th Annual International Conference on Machine Learning, Montreal, QC, Canada, 14–18 June 2009; ACM: Montreal, QC, Canada, 2009; pp. 873–880. [Google Scholar]
- Bottou, L. Large-Scale Machine Learning with Stochastic Gradient Descent; Physica-Verlag HD: Heidelberg, Germany, 2010; pp. 177–186. [Google Scholar]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2016, 18, 851–869. [Google Scholar] [CrossRef] [PubMed]
- Kuzminykh, D.; Polykovskiy, D.; Kadurin, A.; Zhebrak, A.; Baskov, I.; Nikolenko, S.; Shayakhmetov, R.; Zhavoronkov, A. 3D Molecular Representations Based on the Wave Transform for Convolutional Neural Networks. Mol. Pharm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lusci, A.; Pollastri, G.; Baldi, P. Deep architectures and deep learning in chemoinformatics: The prediction of aqueous solubility for drug-like molecules. J. Chem. Inform. Model. 2013, 53, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-W.; Oh, J.M. Deep learning: From chemoinformatics to precision medicine. J. Pharm. Investig. 2017, 47, 317–323. [Google Scholar] [CrossRef]
- Cammarata, A.; Menon, G.K. Pattern recognition. Classification of therapeutic agents according to pharmacophores. J. Med. Chem. 1976, 19, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Menon, G.K.; Cammarata, A. Pattern recognition II: Investigation of structure—Activity relationships. J. Pharm. Sci. 1977, 66, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Henry, D.R.; Block, J.H. Classification of drugs by discriminant analysis using fragment molecular connectivity values. J. Med. Chem. 1979, 22, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1044. [Google Scholar] [CrossRef] [PubMed]
- Devillers, J.; Balaban, A.T. Topological Indices and Related Descriptors in QSAR and QSPAR; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Consonni, V.; Todeschini, R.; Pavan, M.; Gramatica, P. Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 2. Application of the novel 3D molecular descriptors to QSAR/QSPR studies. J. Chem. Inform. Comput. Sci. 2002, 42, 693–705. [Google Scholar] [CrossRef]
- Kiss, L.E.; Kövesdi, I.; Rábai, J. An improved design of fluorophilic molecules: Prediction of the ln P fluorous partition coefficient, fluorophilicity, using 3D QSAR descriptors and neural networks. J. Fluor. Chem. 2001, 108, 95–109. [Google Scholar] [CrossRef]
- Ataide Martins, J.P.; Ma, R.D.O.; Ms, O.D.Q. Web-4D-QSAR: A web-based application to generate 4D-QSAR descriptors. J. Comput. Chem. 2018, 39, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Das, R.N. Chapter 2—Chemical Information and Descriptors. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Roy, K., Kar, S., Das, R.N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 47–80. [Google Scholar]
- Koutsoukas, A.; Paricharak, S.; Galloway, W.R.J.D.; Spring, D.R.; Ijzerman, A.P.; Glen, R.C.; Marcus, D.; Bender, A. How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space. J. Chem. Inform. Model. 2014, 54, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. Cheminform 2003, 34, 1273–1280. [Google Scholar] [CrossRef]
- Greg Landrum. Source Code for Module rdkit.Chem.MACCSkeys; Greg Landrum: Basel, Switzerland, 2011. [Google Scholar]
- Banerjee, P.; Siramshetty, V.B.; Drwal, M.N.; Preissner, R. Computational methods for prediction of in vitro effects of new chemical structures. J. Cheminform. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Yang, H.; Li, F.; Sun, L.; Di, P.; Li, W.; Tang, Y.; Liu, G. In silico prediction of chemical genotoxicity using machine learning methods and structural alerts. Toxicol. Res. 2018, 7, 211–220. [Google Scholar] [CrossRef]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. Acs Cent. Sci. 2016, 3, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. Deep Learning for Drug-Induced Liver Injury. J. Chem. Inform. Model. 2015, 55, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.R.; Milne, G.W.A. Chemical Applications of Graph Theory. J. Chem. Inform. Model. 1976, 32, 210–242. [Google Scholar] [CrossRef]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Hirzel, T.; Adams, R.P. Convolutional networks on graphs for learning molecular fingerprints. In Proceedings of the International Conference on Neural Information Processing Systems, Istanbul, Turkey, 9–12 November 2015; pp. 2224–2232. [Google Scholar]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Marvuglia, A.; Kanevski, M.; Benetto, E. Machine learning for toxicity characterization of organic chemical emissions using USEtox database: Learning the structure of the input space. Environ. Int. 2015, 83, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Srivastava, G.N.; Roy, A.; Sharma, V.K. ToxiM: A Toxicity Prediction Tool for Small Molecules Developed Using Machine Learning and Chemoinformatics Approaches. Front. Pharmacol. 2017, 8, 880. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A. Inductive QSAR Descriptors. Distinguishing Compounds with Antibacterial Activity by Artificial Neural Networks. Int. J. Mol. Sci. 2005, 6, 63–86. [Google Scholar] [CrossRef]
- Chavan, S.; Friedman, R.; Nicholls, I.A. Acute Toxicity-Supported Chronic Toxicity Prediction: A k-Nearest Neighbor Coupled Read-Across Strategy. Int. J. Mol. Sci. 2015, 16, 11659–11677. [Google Scholar] [CrossRef] [PubMed]
- Sunghwan, K.; Thiessen, P.A.; Bolton, E.E.; Jie, C.; Gang, F.; Asta, G.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar]
- Fonger, G.C. Hazardous substances data bank (HSDB) as a source of environmental fate information on chemicals. Toxicology 1995, 103, 137–145. [Google Scholar] [CrossRef]
- Fonger, G.C.; Hakkinen, P.; Jordan, S.; Publicker, S. The National Library of Medicine’s (NLM) Hazardous Substances Data Bank (HSDB): Background, Recent Enhancements and Future Plans. Toxicology 2014, 325, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Fonger, G.C.; Stroup, D.; Thomas, P.L.; Wexler, P. TOXNET: A computerized collection of toxicological and environmental health information. Toxicol. Ind. Health 2000, 16, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Kavlock, R.; Chandler, K.; Houck, K.; Hunter, S.; Judson, R.; Kleinstreuer, N.; Knudsen, T.; Martin, M.; Padilla, S.; Reif, D. Update on EPA’s ToxCast Program: Providing High Throughput Decision Support Tools for Chemical Risk Management. Chem. Res. Toxicol. 2012, 25, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Tice, R.R.; Austin, C.P.; Kavlock, R.J.; Bucher, J.R. Improving the Human Hazard Characterization of Chemicals: A Tox21 Update. Environ. Health Perspect. 2013, 121, 756–765. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. A National Toxicology Program for the 21st Century: A Roadmap for the Future; National Toxicology Program: Research Triangle Park, NC, USA, 2004.
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Kohonen, P.; Benfenati, E.; Bower, D.; Ceder, R.; Crump, M.; Cross, K.; Grafstrom, R.C.; Healy, L.; Helma, C.; Jeliazkova, N.; et al. The ToxBank Data Warehouse: Supporting the Replacement of In Vivo Repeated Dose Systemic Toxicity Testing. Mol. Inform. 2013, 32, 47–63. [Google Scholar] [CrossRef] [PubMed]
- U.S. Environmental Protection Agency. ECOTOX User Guide: ECOTOXicology Knowledgebase System, version 4.0; U.S. Environmental Protection Agency: Washington, DC, USA, 2018.
- Schmidt, U.; Struck, S.; Gruening, B.; Hossbach, J.; Jaeger, I.S.; Parol, R.; Lindequist, U.; Teuscher, E.; Preissner, R. SuperToxic: A comprehensive database of toxic compounds. Nucleic Acids Res 2009, 37, D295–D299. [Google Scholar] [CrossRef] [PubMed]
- Attene-Ramos, M.S.; Miller, N.; Huang, R.; Michael, S.; Itkin, M.; Kavlock, R.J.; Austin, C.P.; Shinn, P.; Simeonov, A.; Tice, R.R. The Tox21 robotic platform for the assessment of environmental chemicals—From vision to reality. Drug Discov. Today 2013, 18, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C. Quantitative approach to biochemical structure-activity relationships. Acc. Chem. Res. 1969, 2, 232–239. [Google Scholar] [CrossRef]
- Bradbury, S.P. Predicting modes of toxic action from chemical structure: An overview. SAR QSAR Environ. Res. 1994, 2, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.T.D.; Dearden, J.C. QSAR in Toxicology. 1. Prediction of Aquatic Toxicity. QSAR Comb. Sci. 2010, 14, 1–7. [Google Scholar] [CrossRef]
- Dunn, W.J., III. QSAR approaches to predicting toxicity. Toxicol. Lett. 1988, 43, 277–283. [Google Scholar]
- Kumar, R.S.; Anitha, Y. An Efficient Approach for Asymmetric Data Classification. Int. J. Innov. Res. Adv. Eng. 2014, 1, 157–161. [Google Scholar]
- Yi, L.M.; Hong, G.; Feldkamp, L.A. Neural Learning from Unbalanced Data. Appl. Intell. 2004, 21, 117–128. [Google Scholar]
- Chen, C.; Breiman, L. Using Random Forest to Learn Imbalanced Data; University of California: Berkeley, CA, USA, 2004. [Google Scholar]
- Wang, S.; Liu, W.; Wu, J.; Cao, L.; Meng, Q.; Kennedy, P.J. Training deep neural networks on imbalanced data sets. In Proceedings of the 2016 International Joint Conference on Neural Networks (IJCNN), Vancouver, BC, Canada, 24–29 July 2016; pp. 4368–4374. [Google Scholar]
- Myint, K.Z.; Wang, L.; Tong, Q.; Xie, X.Q. Molecular fingerprint-based artificial neural networks QSAR for ligand biological activity predictions. Mol. Pharm. 2012, 9, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.Z.; Xie, X.Q. Ligand biological activity predictions using fingerprint-based artificial neural networks (FANN-QSAR). Methods Mol. Biol. 2015, 1260, 149–164. [Google Scholar] [PubMed]
- Dahl, G.E.; Jaitly, N.; Salakhutdinov, R. Multi-task Neural Networks for QSAR Predictions. arXiv 2014, arXiv:1406.1231. [Google Scholar]
- Lee, K.; Lee, M.; Kim, D. Utilizing random Forest QSAR models with optimized parameters for target identification and its application to target-fishing server. BMC Bioinform. 2017, 18 (Suppl. 16), 567. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Wei, G.W. Quantitative toxicity prediction using topology based multi-task deep neural networks. J. Chem. Inform. Model. 2018, 58, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Politi, R.; Isayev, O.; Farag, S.; Tropsha, A. QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front. Environ. Sci. 2016. [Google Scholar] [CrossRef]
- Kearnes, S.; Mccloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Binetti, R.; Costamagna, F.M.; Marcello, I. Exponential growth of new chemicals and evolution of information relevant to risk control. Ann. dell’Istituto Super. di Sanita 2008, 44, 13–15. [Google Scholar]
- Trevan, J.W. The Error of Determination of Toxicity. Proc. R. Soc. Lond. 1927, 101, 483–514. [Google Scholar] [CrossRef]
- Gute, B.D.; Basak, S.C. Predicting acute toxicity (LC50) of benzene derivatives using theoretical molecular descriptors: A hierarchical QSAR approach. SAR QSAR Environ. Res. 1997, 7, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.C.; Grunwald, G.D.; Gute, B.D.; Balasubramanian, K.; Opitz, D. Use of statistical and neural net approaches in predicting toxicity of chemicals. J. Chem. Inf. Comput. Sci. 2000, 40, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.M.; Lilavois, C.R.; Barron, M.G. Prediction of pesticide acute toxicity using two-dimensional chemical descriptors and target species classification. SAR QSAR Environ. Res. 2017, 28, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Madore, M.; Glover, K.P.; Feasel, M.G.; Wallqvist, A. Assessing Deep and Shallow Learning Methods for Quantitative Prediction of Acute Chemical Toxicity. Toxicol. Sci. 2018, 164, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pei, J.; Lai, L. Deep Learning Based Regression and Multiclass Models for Acute Oral Toxicity Prediction with Automatic Chemical Feature Extraction. J. Chem. Inf. Model. 2017, 57, 2672–2685. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Chen, H.; Li, H.; Zhao, Y. In silico prediction of chronic toxicity with chemical category approaches. RSC Adv. 2017, 7, 41330–41338. [Google Scholar] [CrossRef]
- Liu, J.; Xu, C.; Yang, W.; Shu, Y.; Zheng, W.; Zhou, F. Multiple similarly effective solutions exist for biomedical feature selection and classification problems. Sci. Rep. 2017, 7, 12830. [Google Scholar] [CrossRef] [PubMed]
- Van, D.J.; Gaj, S.; Lienhard, M.; Albrecht, M.W.; Kirpiy, A.; Brauers, K.; Claessen, S.; Lizarraga, D.; Lehrach, H.; Herwig, R. RNA-Seq provides new insights in the transcriptome responses induced by the carcinogen benzo[a]pyrene. Br. J. Dermatol. 2012, 130, 568–577. [Google Scholar]
- Liu, R.; Yu, X.; Wallqvist, A. Using Chemical-Induced Gene Expression in Cultured Human Cells to Predict Chemical Toxicity. Chem. Res. Toxicol. 2016, 29, 1883. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.P.; Hou, Z.; Propson, N.E.; Zhang, J.; Engstrom, C.J.; Santos, C.V.; Jiang, P.; Nguyen, B.K.; Bolin, J.M.; Daly, W. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 12516–12521. [Google Scholar] [CrossRef] [PubMed]
- Yamane, J.; Aburatani, S.; Imanishi, S.; Akanuma, H.; Nagano, R.; Kato, T.; Sone, H.; Ohsako, S.; Fujibuchi, W. Prediction of developmental chemical toxicity based on gene networks of human embryonic stem cells. Nucleic Acids Res. 2016, 44, 5515–5528. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, D.L.; Abdulhameed, M.D.; Tawa, G.J.; Baer, C.E.; Permenter, M.G.; Mcdyre, B.C.; Dennis, W.E.; Boyle, M.H.; Hobbs, C.A.; Streicker, M.A. Gene Expression Patterns Associated With Histopathology in Toxic Liver Fibrosis. Toxicol. Sci. 2016, 149, 67–88. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.B.; Lanitis, E.; Dangaj, D.; Buza, E.; Poussin, M.; Stashwick, C.; Scholler, N.; Powell, D.J. Tumor Regression and Delayed Onset Toxicity Following B7-H4 CAR T Cell Therapy. Mol. Therapy J. Am. Soc. Gene Therapy 2016, 24, 1987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.D.; Berntenis, N.; Roth, A.; Ebeling, M. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharmacogenomics J. 2014, 14, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Isik, Z.; Baldow, C.; Cannistraci, C.V.; Schroeder, M. Drug target prioritization by perturbed gene expression and network information. Sci. Rep. 2015, 5, 17417. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, M.; Fortney, K.; Jurisica, I. Network-based characterization of drug-regulated genes, drug targets, and toxicity. Methods 2012, 57, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Abdulhameed, M.D.M.; Wallqvist, A. Molecular Structure-Based Large-Scale Prediction of Chemical-Induced Gene Expression Changes. J. Chem. Inform. Model. 2017, 57, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2011, 39, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug signatures database for gene set analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Flynn, C.; Niepel, M.; Hafner, M.; Muhlich, J.L.; Fernandez, N.F.; Rouillard, A.D.; Tan, C.M.; Chen, E.Y.; Golub, T.R. LINCS Canvas Browser: Interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 2014, 42, W449. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Yu, C.Y.; Li, X.X.; Zhang, P.; Tang, J.; Yang, Q.; Fu, T.; Zhang, X.; Cui, X.; Tu, G. Therapeutic target database update 2018: Enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 2017, 46, D1121–D1127. [Google Scholar]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; King, B.L.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2017. Nucleic Acids Res 2017, 45, D972–D978. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Qiu, C.; Cui, Q. Drug-Path: A database for drug-induced pathways. Database 2015, 2015, bav061. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Chaudhary, K.; Gupta, S.; Singh, H.; Kumar, S.; Gautam, A.; Kapoor, P.; Raghava, G.P.S. CancerDR: Cancer Drug Resistance Database. Sci. Rep. 2013, 3, 1445. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Jia, P.; Dai, Y.; Tao, C.; Zhao, Z.; Zhi, D. Gene2Vec: Distributed Representation of Genes Based on Co-Expression. bioRxiv 2018. [Google Scholar] [CrossRef]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. Comput. Sci. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.; Dean, J. Distributed Representations of Words and Phrases and their Compositionality. Adv. Neural Inf. Process. Syst. 2013, 26, 3111–3119. [Google Scholar]
- Duong, D.; Eskin, E.; Li, J. A novel Word2vec based tool to estimate semantic similarity of genes by using Gene Ontology terms. bioRxiv 2017. [Google Scholar] [CrossRef]
- Danaee, P.; Ghaeini, R.; Hendrix, D.A. A Deep Learning Approach For Cancer Detection and Relevant Gene Identification. Pac. Symp. Biocomput. 2016, 22, 219–229. [Google Scholar]
- Sharifi-Noghabi, H.; Liu, Y.; Erho, N.; Shrestha, R.; Alshalalfa, M.; Davicioni, E.; Collins, C.C.; Ester, M. Deep Genomic Signature for early metastasis prediction in prostate cancer. bioRxiv 2018. [Google Scholar] [CrossRef]
- Aliper, A.; Plis, S.; Artemov, A.; Ulloa, A.; Mamoshina, P.; Zhavoronkov, A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm. 2016, 13, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Gayvert, K.M.; Madhukar, N.S.; Elemento, O. A Data-Driven Approach to Predicting Successes and Failures of Clinical Trials. Cell Chem. Biol. 2016, 23, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.; Chen, J.; Zhong, Z.; Hrycushko, B.A.; Zhou, L.; Jiang, S.B.; Albuquerque, K.; Gu, X. Deep convolutional neural network with transfer learning for rectum toxicity prediction in cervical cancer radiotherapy: A feasibility study. Phys. Med. Biol. 2017, 62, 8246–8263. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, J.; Kim, M.T.; Boison, A.; Sedykh, A.; Moran, K. Big data in chemical toxicity research: The use of high-throughput screening assays to identify potential toxicants. Chem. Res. Toxicol. 2014, 27, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Pasturromay, L.A.; Cedrón, F.; Pazos, A.; Portopazos, A.B. Deep Artificial Neural Networks and Neuromorphic Chips for Big Data Analysis: Pharmaceutical and Bioinformatics Applications. Int. J. Mol. Sci. 2016, 17, 1313. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor Type | Descriptor Name | Description |
|---|---|---|
| Fingerprint-based | ECFP4 | atom type, extended connectivity fingerprint, maximum distance = 4 |
| FCFP4 | functional-class-based, extended connectivity fingerprint, maximum distance = 4 | |
| MACCS | 166 predefined MDL keys (public set) | |
| Connectivity-matrix-based | BCUT | atomic charges, polarizabilities, H-bond donor and acceptor abilities, and H-bonding modes of intermolecular interaction |
| Shape-based | rapid overlay of chemical structures (ROCS), combo Tanimoto (shape and electrostatic score) | shape-based molecular similarity method; molecules are described by smooth Gaussian function and pharmacophore points |
| PMI | normalized principal moment-of-inertia ratios | |
| Pharmacophore-based | GpiDAPH3 | graph-based 3-point pharmacophore, eight atom types computed from three atom properties (in pi system, donor, acceptor) |
| TGD | typed graph distances, atom typing (donor, acceptor, polar, anion, cation, hydrophobe) | |
| TAD | typed atom distances, atom typing (donor, acceptor, polar, anion, cation, hydrophobe) | |
| Bioactivity-based | Bayes affinity fingerprints | bioactivity model based on multicategory Bayes classifier trained on data from ChEMBL v. 14 |
| Physicochemical-property-based | prop2D | physicochemical properties (such as molecular weight, atom counts, partial charges, hydrophobicity etc.) |
| Database | Database Description | Online Websites | Reference |
|---|---|---|---|
| TOXNET | A collection of toxicity databases. | https://toxnet.nlm.nih.gov/ | [97] |
| ToxCast | High-throughput toxicity data on thousands of chemicals. | https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data | [98] |
| Tox21 |
| https://ntp.niehs.nih.gov/results/dbsearch/index.html | [99,100] |
| PubChem |
| https://pubchem.ncbi.nlm.nih.gov/ | [94] |
| DrugBank | Detailed drug data and corresponding drug target information. | https://www.drugbank.ca/ | [101] |
| ToxBank Data Warehouse | Data for systemic toxicity. | http://www.toxbank.net/data-warehouse | [102] |
| ECOTOX | Single chemical environmental toxicity data on aquatic life, terrestrial plants and wildlife. | https://cfpub.epa.gov/ecotox/index.html | [103] |
| SuperToxic | Toxic compound data from literature and web sources. | http://bioinformatics.charite.de/supertoxic/ | [104] |
| Molecular Descriptor | Model | AUC | Reference | |
|---|---|---|---|---|
| Shallow architectures | Dragon descriptors (2489 descriptors) | RF | 0.81 | [119] |
| Pubchem keys | SVM | 0.948 | [83] | |
| MACCS fingerprints | RF | 0.947 | [83] | |
| Deep learning | Molecular fragments learned by CNN | DNN | 0.837 | [88] |
| Unidirectional graph learned by CNN | Graph CNN | 0.867 | [120] | |
| LSTM graph | One-shot learning | 0.84 | [84] |
| Database | Description | Websites | References |
|---|---|---|---|
| GEO database | Gene expression data of drug-treated samples in subsets. | https://www.ncbi.nlm.nih.gov/geo/ | [141,142] |
| Connectivity Map (CMap) |
| https://portals.broadinstitute.org/cmap/ | [140] |
| DSigDB |
| http://tanlab.ucdenver.edu/DSigDB | [143] |
| LINCS Canvas Browser (LCB) |
| http://www.maayanlab.net/LINCS/LCB | [144] |
| Therapeutic target database (TTD) |
| http://bidd.nus.edu.sg/group/ttd/ttd.asp | [145] |
| Comparative Toxicogenomics Database (CTD) |
| http://ctdbase.org/ | [146] |
| Drug-Path | Drug-induced pathways. | http://www.cuilab.cn/drugpath | [147] |
| CancerDR |
| http://crdd.osdd.net/raghava/cancerdr/ | [148] |
| KEGG DRUG |
| https://www.genome.jp/kegg/drug/ | [149] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Wang, G. Machine Learning Based Toxicity Prediction: From Chemical Structural Description to Transcriptome Analysis. Int. J. Mol. Sci. 2018, 19, 2358. https://doi.org/10.3390/ijms19082358
Wu Y, Wang G. Machine Learning Based Toxicity Prediction: From Chemical Structural Description to Transcriptome Analysis. International Journal of Molecular Sciences. 2018; 19(8):2358. https://doi.org/10.3390/ijms19082358
Chicago/Turabian StyleWu, Yunyi, and Guanyu Wang. 2018. "Machine Learning Based Toxicity Prediction: From Chemical Structural Description to Transcriptome Analysis" International Journal of Molecular Sciences 19, no. 8: 2358. https://doi.org/10.3390/ijms19082358
APA StyleWu, Y., & Wang, G. (2018). Machine Learning Based Toxicity Prediction: From Chemical Structural Description to Transcriptome Analysis. International Journal of Molecular Sciences, 19(8), 2358. https://doi.org/10.3390/ijms19082358