The Efficacy of Amifostine against Multiple-Dose Doxorubicin-Induced Toxicity in Rats

 , ,
, , 
Abstract
1. Introduction
2. Results
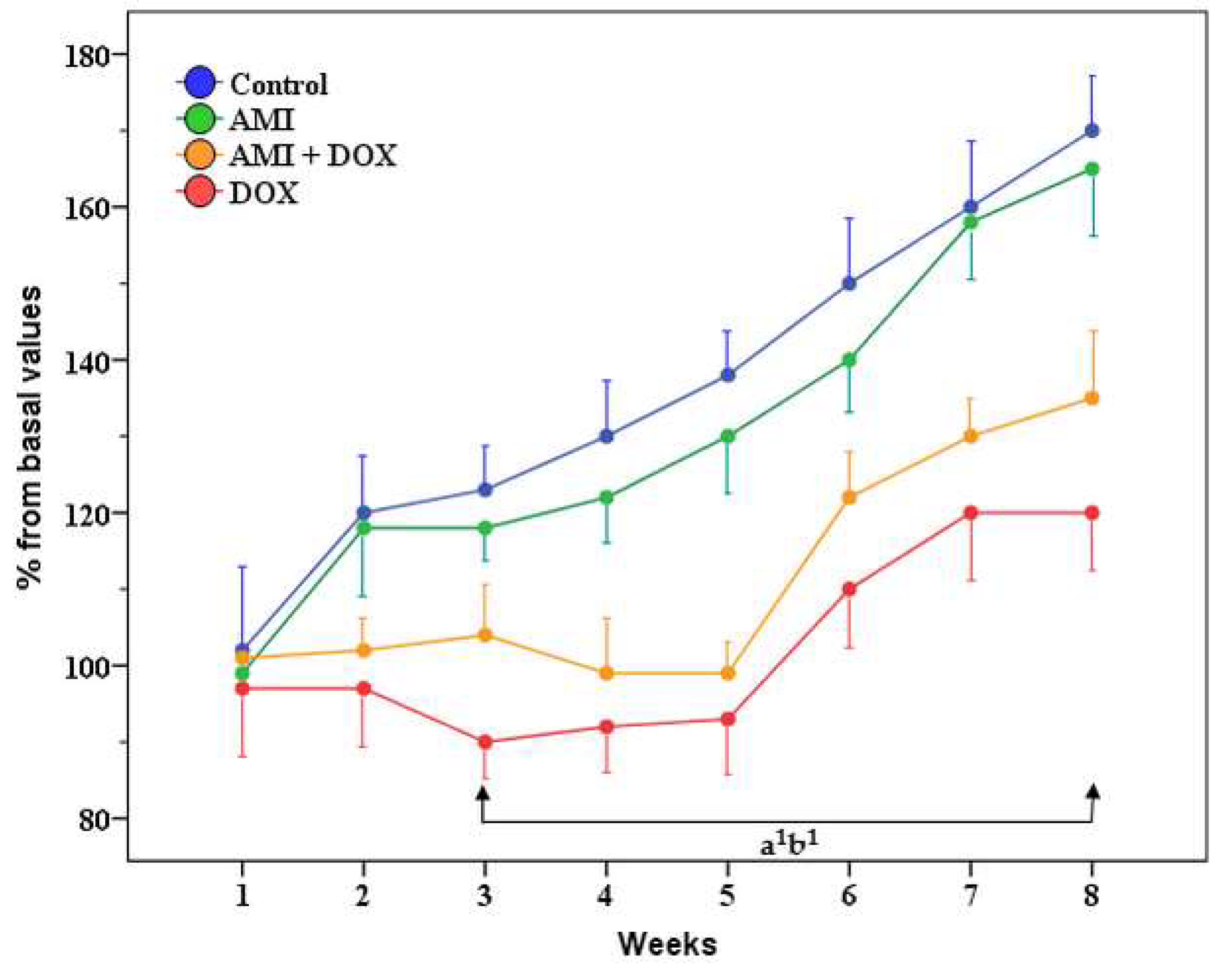
2.1. General Health Condition, Body Weight, Liver and Kidney Weight of the Experimental Animals
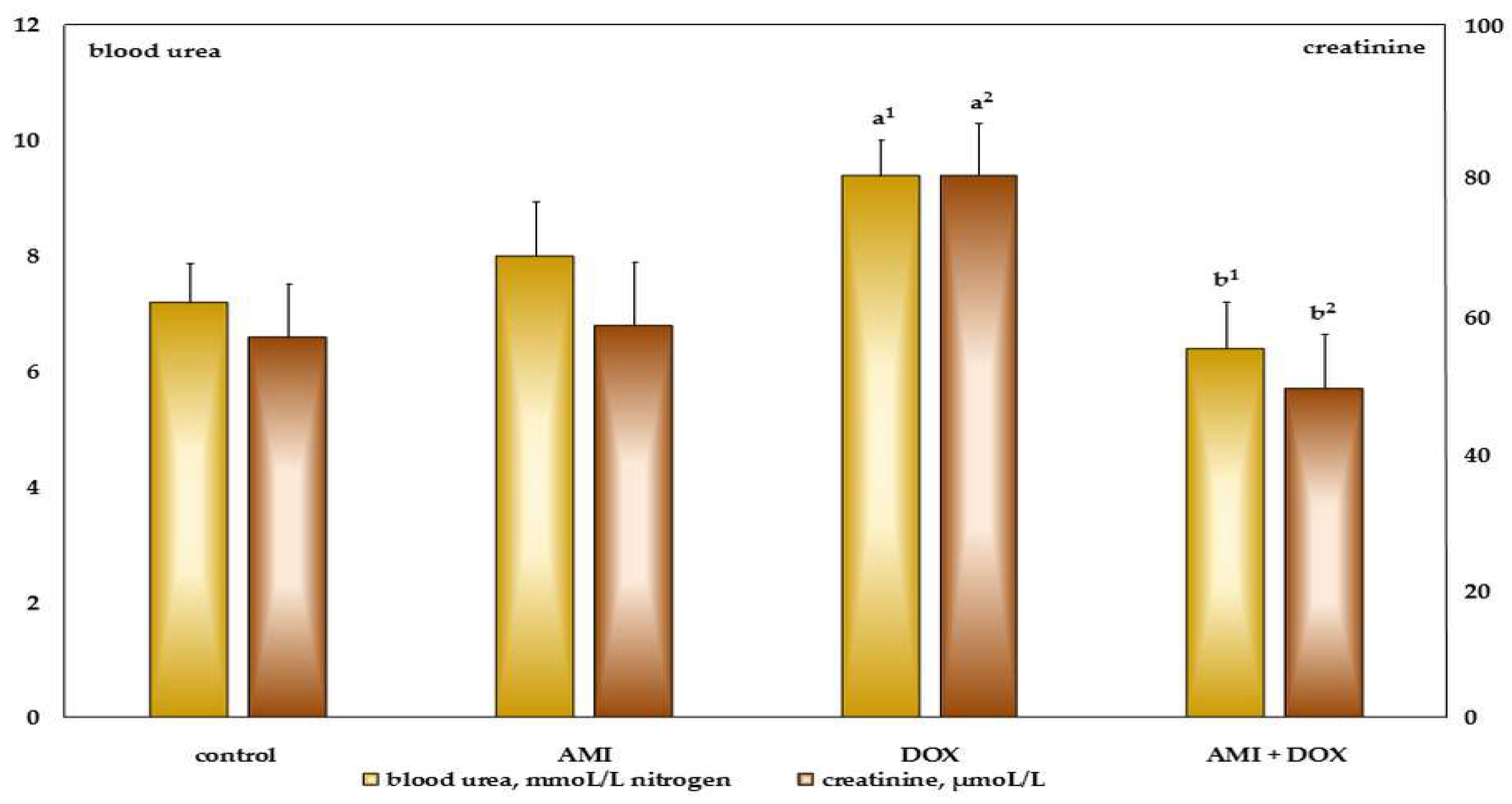
2.2. The Effects of Different Treatment on Biochemical Parameters in the Blood Serum of the Experimental Animals
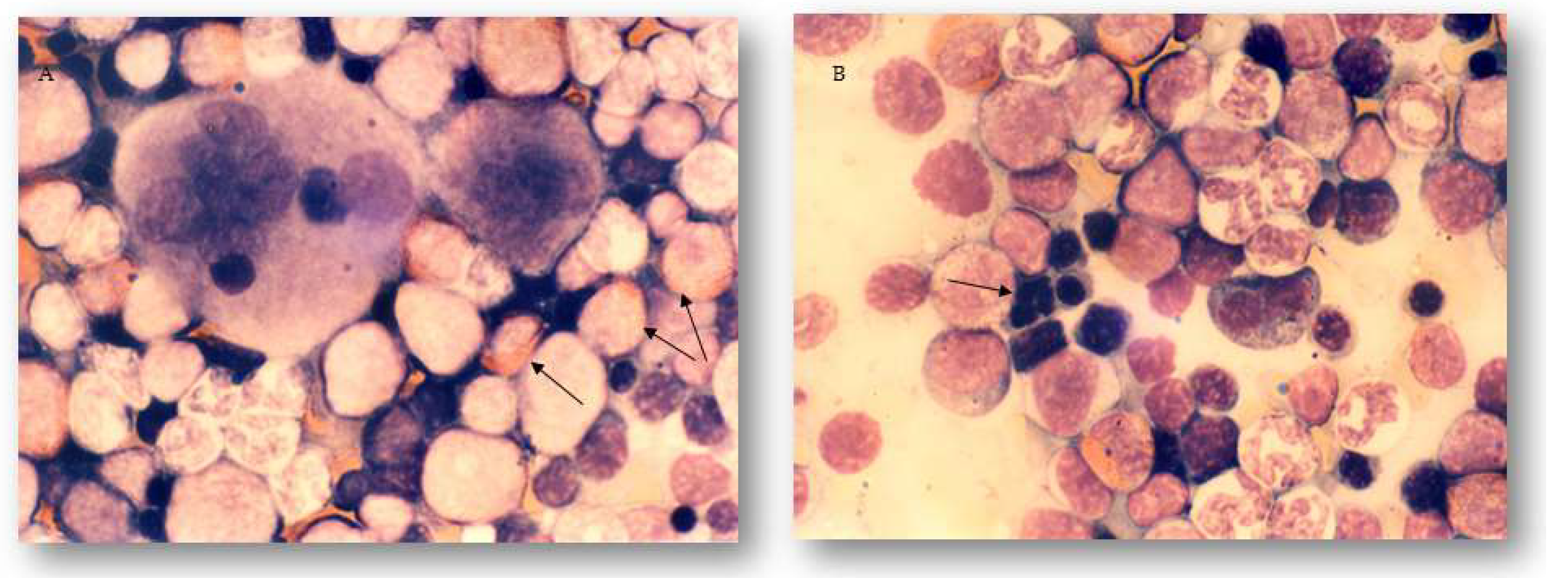
2.3. The Effects of Different Treatments on Granulopoiesis and Erythropoiesis in the Bone Marrow and Peripheral Blood of Experimental Animals
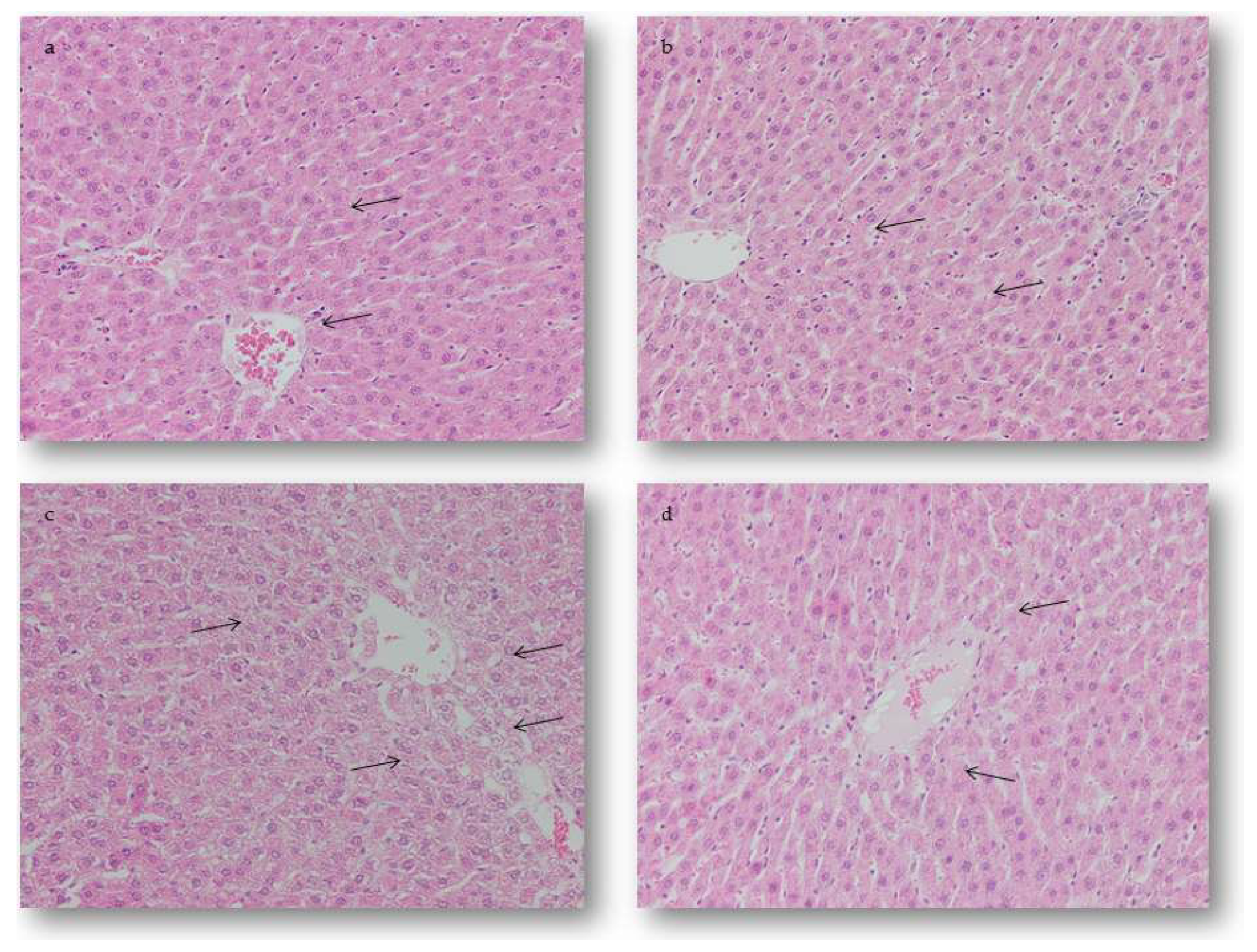
2.4. Pathohistological Examination of Experimental Animal Tissue Alterations
2.4.1. Hepatic Alterations
2.4.2. Renal Alterations
2.5. Semiquanitative Analysis of the Experimental Animal Tissue Alterations
2.5.1. Quantification of Hepatic Alterations
2.5.2. Quantification of Renal Alterations
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Experimental Animals
4.3. Experimental Design
4.4. General Health Condition and Weight Changes
4.5. Biochemical Assays
4.6. Bone Marrow and Peripheral Blood Sample Preparation
4.7. Histopathological Study and Semiquantitative Analysis
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bonadonna, G.; Valagudda, P. Primary Chemotherapy in Operable Breast Cancer. Semin. Oncol. 1998, 23, 464–474. [Google Scholar] [CrossRef]
- Dollery, C. Cyclophosphamide. In Therapeutic Drugs, 2nd ed.; Dollery, C., Ed.; Churchill Livingstone: Edinburgh, UK, 1999; pp. 349–354, ISBN-10: 0443051488, ISBN-13: 978-0443051487. [Google Scholar]
- Hortobagyi, G.N. Anthracyclines in the Treatment of Cancer. An Overview. Drugs 1997, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gewitz, D.A. A Critical Evaluation of the Mechanisms of Action Proposed for the Antitumor Effects of the Anthracycline Antibiotics Adriamycin and Daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Minoti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Development in Antitumor Activity and Cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Mross, K. New Anthracycline Derivates: What for? Eur. J. Cancer Clin. Oncol. 1991, 27, 1542–1544. [Google Scholar] [CrossRef]
- Sayed-Ahmed, M.M.; Al-Shabanah, O.A.; Hafez, M.M.; Aleisa, A.M.; Al-Rejaie, S.S. Inhibition of Gene Expression of Heart Fatty Acid Binding Protein and Organic Cation/Carnitine Transporter in Doxorubicin Cardiomyopathic Rat Model. Eur. J. Pharmacol. 2010, 640, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Martindale. The Complete Drug Reference [CD-ROM], 36th ed.; Pharmaceutical Press: London, UK, 2011; ISBN-13: 978-0853698425, ISBN-10: 0853698422. [Google Scholar]
- Saad, Y.S.; Najjar, A.T.; Al-Rikabi, A.C. The Preventive Role of Deferoxamine against Acute Doxorubicin Induced Cardiac, Renal and Hepatic Toxicity in Rats. Pharmacol. Res. 2000, 43, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Mihailović-Stanojević, N.; Jovović, D.; Miloradović, Z.; Grujić-Milovanović, J.; Marković-Lipovski, J. Reduced Progression of Adriamycin Nephropathy in Spontaneously Hypertensive Rats Treated by Losartan. Nephrol. Dial. Transplant. 2009, 24, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.W.; Kalinovsky, T.; Roomi, N.W.; Rath, M.; Niedzwiecki, A. Prevention of Adriamycin-Induced Hepatic and Renal Toxicity in Male BALB/c Mice by a Nutrient Mixture. Exp. Ther. Med. 2014, 7, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Dragojević-Simić, V.; Dobrić, S.; Jaćević, V.; Bokonjić, D.; Milosavljević, I.; Kovačević, A.; Mikić, D. Efficacy of amifostine in protection against doxorubicin-induced acute cardiotoxic effects in rats. Vojnosanit. Pregl. 2013, 70, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.H.; Zhang, J.; Chadwick, D.P.; Ferrans, V.J. Comparison of the Protective Effects of Amifostine and Dexrazoxane against the Toxicity of Doxorubicin in Spontaneously Hypertensive Rats. Cancer Chemother. Pharmacol. 2000, 45, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Okunewick, J.P.; Buffo, M.J.; Kociban, D.L. Comparative Toxicity of Mitoxantrone and Doxorubicin on Hematopoietic Stem Cells. Exp. Hematol. 1985, 13, 23–30. [Google Scholar] [PubMed]
- Pugazhendhi, A.; Jebakumar, T.N.; Edison, I.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to Different Experimental Organ Systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- El-Sayyad, H.I.; Ismail, M.F.; Shalaby, F.M.; Abou-El-Magd, R.F.; Gaur, R.L.; Fernando, A.; Raj, M.H.G.; Ouhtit, A. Histopathological effects of cisplatin, doxorubicin and 5-flurouracil (5-FU) on the liver of male albino rats. Int. J. Biol. Sci. 2009, 5, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Ayla, S.; Seckin, I.; Tanriverdi, G.; Cengiz, M.; Eser, M.; Soner, B.C.; Oktem, G. Doxorubicin induced nephrotoxicity: Protective effect of nicotinamide. Int. J. Cell Biol. 2011, 2011, 390238. [Google Scholar] [CrossRef] [PubMed]
- Lahoti, T.S.; Patel, D.; Thekkemadom, V.; Beckett, R.; Ray, S.D. Doxorubicin-induced in vivo nephrotoxicity involves oxidative stress-mediated multiple pro- and anti-apoptotic signaling pathways. Curr. Neurovasc. Res. 2012, 9, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Camaggi, C.M.; Comparsi, R.; Srtocchi, E.; Testoni, F.; Angelelli, B.; Pannuti, F. Epirubicin and Doxorubicin Comparative Metabolism and Pharmacokinetics. Cancer Chemother. Pharmacol. 1988, 21, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ganey, P.E.; Kauffman, F.C.; Thurman, R.G. Oxigen Dependent Hepatotoxicity Due to Doxorubicin: Role of Reducing Equivalent Supply in Perfused Rat Liver. Mol. Pharmacol. 1988, 34, 695–701. [Google Scholar] [PubMed]
- Ballet, F.; Vrignaud, P.; Robert, J.; Rey, C.; Poupon, R. Hepatic Extraction, Metabolism and Biliary Excretion of Doxorubicin in the Isolated Perfused Rat Liver. Cancer Chemother. Pharmacol. 1987, 19, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Dodion, P.; Bernstein, A.L.; Fox, B.M.; Bachur, N.R. Loss of Fluorescence by Anthracycline Antibiotics: Effects of Xanthine Oxidase and Identification of the Nonfluorescent Metabolites. Cancer Res. 1987, 47, 1036–1039. [Google Scholar] [PubMed]
- Aryal, B.; Jeong, J.; Rao, V.A. Doxorubicin-Induced Carbonylation of Cardiac Myosin Binding Protein C Promote Cardiotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 2011–2016. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.C.; Hsu, Y.C.; Chen, C.C.; Lin, Y.F.; Wu, C.C. Oxidative Stress and Nucleic Acid Oxidation in Patients with Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2013, 2013, 301982. [Google Scholar] [CrossRef] [PubMed]
- Karanovic, D.; Grujic-Milanovic, J.; Miloradovic, Z.; Ivanov, M.; Jovovic, D.J.; Vajic, U.J.; Zivotic, M.; Markovic-Lipkovski, J.; Mihailovic-Stanojevic, N. Effects of Single and Combined Losartan and Tempol Treatments on Oxidative Stress, Kidney Structure and Function in Spontaneously Hypertensive Rats with Early Course of Proteinuric Nephropathy. PLoS ONE 2016, 11, e0161706. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, D.B.; Jovović, D.; Varagić, J.; Dimitrijević, J.; Dragojlović, Z.; Djukanović, L. Slowing Progression of Chronic Renal Insufficiency with Captopril in Rats with Spontaneous Arterial Hypertension and Adriamycin Nephropathy. Srp. Arh. Celok. Lek. 2002, 130, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Alshabanah, O.A.; Hafez, M.M.; Al-Harbi, M.M.; Hassan, Z.K.; Al Rejaie, S.S.; Asiri, Y.A.; Sayed-Ahmed, M.M. Doxorubicin Toxicity Can be Ameliorated during Antioxidant L-Carnitine Supplementation. Oxid. Med. Cell. Longev. 2014, 2, 428–433. [Google Scholar] [CrossRef]
- Dragojevic-Simic, V.; Jacevic, V.; Dobric, S.; Djordjevic, A.; Bokonjic, D.; Bajcetic, M.; Injac, R. Anti-Inflammatory Activity of Fullerenol C60(OH)24 Nano-Particles in a Model of Acute Inflammation in Rats. Dig. J. Nanomater. Biostruct. 2011, 6, 819–827. [Google Scholar]
- Dorr, R.T.; Holmes, B.C. Dosing considerations with amifostine: A Review of the Literature and Clinical Experience. Semin. Oncol. 1999, 26, 108–119. [Google Scholar] [PubMed]
- Sterba, M.; Popelova, O.; Vavrova, A.; Jirkovsky, E.; Kovaríkova, P.; Gersl, V.; Simunek, T. Oxidative Stress, Redox Signaling, and Metal Chelation in Anthracycline Cardiotoxicity and Pharmacological Cardioprotection. Antioxid. Redox Signal. 2013, 18, 899–929. [Google Scholar] [CrossRef] [PubMed]
- Jacevic, V.; Djordjevic, A.; Srdjenovic, B.; Milic-Tores, V.; Segrt, Z.; Dragojevic-Simic, V.; Kuca, K. Fullerenol Nanoparticles Prevents Doxorubicin-Induced Acute Hepatotoxicity in Rats. Exp. Mol. Pathol. 2017, 102, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Dragojevic-Simic, V.M.; Dobric, S.L.; Bokonjic, D.R.; Vucinic, Z.M.; Sinovec, S.M.; Jacevic, V.M.; Dogovic, N.P. Amifostine Protection against Doxorubicin Cardiotoxicity in Rats. Anti Cancer Drugs 2004, 15, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.M.; Goa, K.L. Amifostine: A review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Potential as a Radioprotector and Cytotoxic Chemoprotector. Drugs 1995, 50, 1001–1031. [Google Scholar] [CrossRef] [PubMed]
- Capizzi, R.L. The Preclinical Basis for Broad-Spectrum Selective Cytoprotection of Normal Tissue from Cytotoxic Therapies by Amifostine. Semin. Oncol. 1999, 26, 3–21. [Google Scholar] [CrossRef]
- Kouvaris, J.; Kouloullas, V.; Vlahos, L. Amifostine: The First Selective-Target and Broad-Spectrum Radioprotector. Oncology 2007, 12, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Kligerman, M.M.; Turrisi, A.T.; Urtasun, R.C.; Norfleet, A.L.; Phillips, T.L.; Barkley, T.; Rubin, P. Final Report On Phase I Trial of WR-2721 before Protracted Fractionated Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 1988, 14, 1119–1122. [Google Scholar] [CrossRef]
- Wasserman, T.H.; Phillips, T.L.; Ross, G.; Kane, L.J. Differential Protection against Cytotoxic Chemotherapeutic Effects on Bone Marrow CFUs by WR-2721. Cancer Clin. Trial 1981, 4, 3–6. [Google Scholar]
- Shpall, E.J.; Stemmer, S.M.; Hami, L.; Franklin, W.A.; Shaw, L.; Bonner, H.S.; Bearman, S.I.; Peters, W.P.; Bast, R.C.; McCulloch, W. Amifostine (WR-2721) Shortens the Engraftment Period of 4-hydroperoxy-Cyclophosphamide-Purged Bone Marrow in Breast Cancer Patients Receiving High-Dose Chemotherapy with Autologous Bone Marrow Support. Blood 1994, 83, 3132–3137. [Google Scholar] [PubMed]
- Rossi, F.; Filipelli, W.; Russo, S.; Filippelli, A.; Berrino, L. Cardiotoxicity of Doxorubicin: Effects of Drugs Inhibiting the Release of Vasoactive Substances. Pharmacol. Toxicol. 1994, 75, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.H.; Zhang, J.; Ferrans, V. Comparison of the Protective Effects of Desferrioxamine and ICRF-187 against Doxorubicin-Induced Toxicity in Spontaneously Hypertensive Rats. Cancer Chemother. Pharmacol. 1994, 35, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.H.; Ferrans, V.; Young, R.S.K.; Hamlin, R.L. Effects of Pretreatment with ICRF-187 on the Total Cumulative Dose of Doxorubicin Tolerated by Beagle Dogs. Cancer Res. 1988, 48, 6918–6925. [Google Scholar] [PubMed]
- Herman, E.H.; El-Hage, A.; Ferrans, V. Protective Effects of ICRF-187 on Doxorubicin-Induced Cardiac and Renal Toxicity in Spontaneously Hypertensive Rats. Toxicol. Appl. Pharmacol. 1988, 92, 42–53. [Google Scholar] [CrossRef]
- Alderton, P.M.; Gross, J.; Green, M.D. Comparative Study of Doxorubicin, Mitoxantrone, and Epirubicin in Combination with ICRF-187 (ADR-529) in Chronic Cardiotoxicity Animal Model. Cancer Res. 1992, 52, 194–201. [Google Scholar] [PubMed]
- Green, D.; Bensley, D.; Schein, P. Preclinical Evaluation of WR-151327: An Orally Active Chemotherapy Protector. Cancer Res. 1994, 54, 738–741. [Google Scholar] [PubMed]
- Meriweather, V.D.; Bachur, N.R. Inhibition of DNA and RNA Metabolism by Daunomycin and Adriamycin in L1210 Mouse Leukemia. Cancer Res. 1972, 32, 1137–1142. [Google Scholar]
- Castaldi, G.; Zavagli, G. Migration of the Macrophages to the Thymus after Cyclophosphamide. Br. J. Exp. Pathol. 1972, 53, 28–30. [Google Scholar] [PubMed]
- List, A.F.; Heaton, R.; Glinsmann-Gibson, B.; Capizzi, R.L. Amifostine Stimulates Formation of Multipotent and Erythroid Bone Marrow Progenitors. Leukemia 1998, 12, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Douau, L.; Hu, C.; Giarratana, M.C. Amifostine Improves the Antileukemic Therapeutic Index of Mafosfamide: Implications for Bone Marrow Purging. Blood 1995, 86, 2849–2855. [Google Scholar]
- Romano, M.F.; Lamberti, A.; Bisogni, R.; Garbi, C.; Pagnano, A.M.; Auletta, P.; Tassone, P.; Turco, M.C.; Venuta, S. Amifostine Inhibits Hematopoietic Progenitor Cell Apoptosis by Activating NF-κB/Rel Transcription Factors. Blood 1999, 94, 4060–4066. [Google Scholar] [PubMed]
- List, A.F.; Heaton, R.; Glinsmann-Gibson, B. Amifostine is a Potent Stimulant of Hematopoietic Progenitors. Proc. Am. Assoc. Cancer Res. 1995, 36, 291. [Google Scholar]
- Ridgway, D.D.; Borzy, S.M.; Bagby, C.M. Granulocyte Macrophage Colony-Stimulating Activity Production by Cultured Human Thymic Nonlymphoid Cells Is Regulated by Endogenous Interleukin 1. Blood 1988, 72, 1230–1236. [Google Scholar] [PubMed]
- Schwartz, N.G.; Patchen, L.M.; Neta, R.; MacVittie, T.J. Radioprotection of Mice with Interleukin 1: Relationship to the Number of Spleen Colony-Forming Units. Radiat. Res. 1989, 119, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Neta, R.; Szein, B.M.; Oppenheim, J.J.; Gillis, S.; Douches, D.S. The In Vivo Effects of Interleukin 1. I. Bone Marrow Cells Are Induced to Cycle after Administration of Interleukin 1. J. Immunol. 1987, 139, 1861–1866. [Google Scholar] [PubMed]
- Schmalbach, T.K.; Borch, R.F. Mechanism of Diethyldithiocarbamate Modulation of Murine Bone Marrow Toxicity. Cancer Res. 1990, 50, 6218–6221. [Google Scholar] [PubMed]
- Eppstein, D.A.; Kurahara, C.G.; Bruno, A.B.; Terrell, G. Prevention of Doxorubicin-Induced Hematotoxicity in Mice by Interleukin 1. Cancer Res. 1989, 49, 3955–3960. [Google Scholar] [PubMed]
- Bertani, T.; Poggi, A.; Pozzoni, R.; Delani, F.; Sacchi, G.; Thoua, Y.; Mecca, G.; Remuzzi, G.; Donati, M.B. Adriamycin-Induced Nephrotic Syndrome in Rats. Sequence of Pathologic Events. Lab. Investig. 1982, 46, 16–23. [Google Scholar] [PubMed]
- Okuda, S.; Oh, Y.; Tsuruda, H.; Onoyama, K.; Fujumi, S.; Fujishima, M. Adriamycin-Induced Nephropathy as a Model of Chronic Progressive Glomerular Disease. Kidney Int. 1986, 29, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Rašković, A.; Stilinović, N.; Kolarović, J.; Vasović, V.; Vukmirović, S.; Mikov, M. The Protective Effects of Silymarin against Doxorubicin-Induced Cardiotoxicity and Hepatotoxicity in Rats. Molecules 2011, 16, 8601–8613. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ye, J.; Qin, Z.; Ding, X. Protective Effects of Madecassoside against Doxorubicin Induced Nephrotoxicity in Vivo and in Vitro. Sci. Rep. 2015, 5, 18314. [Google Scholar] [CrossRef] [PubMed]
- King, P.; Perry, M. Hepatotoxicity of Chemotherapy. Oncologist 2001, 6, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Tkacs, N.C. Current Concepts of Neurohormonal Activation in Heart Failure: Mediators and Mechanisms. AACN Adv. Crit. Care 2008, 19, 364–385. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.E.; Dunlap, M.E. Renal Hemodynamics in Heart Failure: Implications for Treatment. Curr. Opin. Nephrol. Hypertens. 2008, 17, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M. Secondary Endothelial Dysfunction: Hypertension and Heart Failure. J. Mol. Cell. Cardiol. 1999, 31, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Chaggar, P.S.; Malkin, C.J.; Shaw, S.M.; Williams, S.G.; Channer, K.S. Neuroendocrine Effects on the Heart and Targets for Therapeutic Manipulation in Heart Failure. Cardiovasc. Ther. 2009, 27, 187–193. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.; Mansour, A.M.; El-Sawy, W.S. Protective Effect of Proanthocyanidins against Doxorubicin-Induced Nephrotoxicity in Rats. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, D.; Seke, M.; Labudovic-Borovic, M.; Jovic, D.; Borisev, I.; Srdjenovic, B.; Rakocevic, Z.; Pavlovic, V.; Djordjevic, A. Hepatoprotective Effect of Fullerenol/Doxorubicin Nanocomposite in Acute Treatment of Healthy Rats. Exp. Mol. Pathol. 2018, 104, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Marzatico, F.; Porta, C.; Moroni, M.; Bertorelli, L.; Borasio, E.; Finotti, N.; Pansarasa, O.; Castagna, L. In Vitro Antioxidant Properties of Amifostine (WR-2721, Ethyol). Cancer Chemother. Pharmacol. 2000, 45, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Bjelogrlic, S.K.; Lukic, S.T.; Djuricic, S.M. Activity of Dexrazoxane and Amifostine against Late Cardiotoxicity Induced by the Combination of Doxorubicin and Cyclophosphamide In Vivo. Basic Clin. Pharmacol. Toxicol. 2013, 113, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Shokrzadeh, M.; Ghassemi-Barghi, N. Antioxidant and Genoprotective Effects of Amifostine against Irinotecan Toxicity in Human Hepatoma Cells. Int. J. Cancer Res. Ther. 2018, 3, 1–5. [Google Scholar]
- Jacevic, V.; Kuca, K.; Milovanovic, Z.; Bocarov-Stancic, A.; Rancic, I.; Bokonjic, D.; Dragojevic-Simic, V.; Segrt, Z. Gastroprotective Effects of Amifostine in Rats Treated by T-2 Toxin. Toxicol. Rev. 2018, 37, 123–127. [Google Scholar] [CrossRef]
- Jensen, R.A. Doxorubicin Cardiotoxicity: Contractile Changes after Long Term Treatment in the Rat. J. Pharmacol. Exp. Ther. 1986, 236, 197–203. [Google Scholar] [PubMed]
- Jacevic, V.; Jovic, D.; Kuca, K.; Dragojevic-Simic, V.; Dobric, S.; Trajkovic, S.; Borisev, I.; Segrt, Z.; Milovanovic, Z.; Bokonjic, D.; et al. Effects of Fullerenol Nanoparticles and Amifostine on Radiation-Induced Tissue Damages: Histopathological Analysis. J. Appl. Biomed. 2016, 14, 285–297. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Liver (%) | Kidney (%) |
|---|---|---|
| Control | 2.79 ± 0.36 | 0.26 ± 0.02 |
| AMI | 2.86 ± 0.30 | 0.26 ± 0.01 |
| DOX | 3.36 ± 0.22 a1 | 0.37 ± 0.08 a1 |
| AMI + DOX | 3.29 ± 0.36 | 0.31 ± 0.01 |
| Parameter | Control | AMI | DOX | AMI + DOX |
|---|---|---|---|---|
| Myeloblasts | 0.55 ± 0.41 | 0.54 ± 0.60 | 0.70 ± 0.20 | 0.53 ± 0.82 |
| Promyelocytes | 3.05 ± 0.41 | 3.21 ± 1.14 | 2.90 ± 1.23 | 3.56 ± 0.97 |
| Myelocytes | 5.27 ± 0.47 | 5.61 ± 1.55 | 6.22 ± 1.61 | 5.58 ± 1.82 |
| Metamyelocytes and band cells | 14.64 ± 6.35 | 12.65 ± 5.42 | 6.90 ± 3.13 a1 | 10.68 ± 2.05 b1 |
| Neutrophils | 41.32 ± 3.81 | 44.07 ± 6.59 | 37.15 ± 4.19 | 45.63 ± 3.42 b2 |
| Eosinophils | 8.35 ± 4.59 | 10.60 ± 3.91 | 19.45 ± 4.86 a1 | 12.36 ± 3.68 b1 |
| Lymphocytes | 23.05 ± 9.74 | 19.77 ± 8.68 | 20.76 ± 8.98 | 17.95 ± 4.32 |
| Monocytes | 3.32 ± 1.42 | 2.31 ± 0.88 | 4.05 ± 1.47 | 2.65 ± 1.65 |
| Plasma cells | 1.07 ± 1.35 | 1.58 ± 0.44 | 1.55 ± 1.51 | 0.91 ± 0.84 |
| Mast cells | 0.50 ± 0.52 | 0.00 ± 0.00 | 0.07 ± 0.15 a1 | 0.00 ± 0.00 a1 |
| White/red cell line | 2.15 ± 0. 56 | 2.65 ± 0.42 | 2.04 ± 0.86 | 2.65 ± 0.93 a2 |
| Parameter | Control | AMI | DOX | AMI + DOX |
|---|---|---|---|---|
| Neutrophils | 19.40 ± 5.02 | 20.00 ± 4.60 | 14.00 ± 6.16 | 24.33 ± 4.76 b1 |
| Eosinophils | 1.80 ± 0.83 | 3.66 ± 4.58 | 3.00 ± 1.82 | 1.66 ± 1.21 |
| Lymphocytes | 71.40 ± 8.70 | 70.00 ± 3.03 | 67.25 ± 10.68 | 60.00 ± 6.41 a1 |
| Monocytes | 7.40 ± 4.03 | 8.00 ± 3.03 | 15.75 ± 4.19 a1 | 14.00 ± 4.14 a1 |
| Treatment (mg/kg) | Hepatic Damage Score (HDS) (10 Livers/Group × 4 Slices/Liver) | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | ||
| 1. Control | 30 | 10 | 0 | 0 | 0 | 0 | 0.25 ± 0.46 |
| 2. AMI | 0 | 25 | 15 | 0 | 0 | 0 | 1.37 ± 0.75 a2 |
| 3. DOX | 0 | 0 | 0 | 0 | 15 | 25 | 4.62 ± 0.51 a3 b3 |
| 4. AMI + DOX | 0 | 0 | 0 | 15 | 25 | 0 | 3.62 ± 0.51 a3 b3 c1 |
| Treatment (mg/kg) | Renal Damages Score (RDS) (10 Kidneys/Group × 4 Slices/Kidney) | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | ||
| 1. Control | 30 | 10 | 0 | 0 | 0 | 0 | 0.25 ± 0.46 |
| 2. AMI | 0 | 20 | 20 | 0 | 0 | 0 | 1.50 ± 0.53 a3 |
| 3. DOX | 0 | 0 | 0 | 0 | 10 | 30 | 4.75 ± 0.46 a3 b3 |
| 4. AMI + DOX | 0 | 0 | 0 | 30 | 10 | 0 | 3.25 ± 0.46 a3 b3 c2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaćević, V.; Dragojević-Simić, V.; Tatomirović, Ž.; Dobrić, S.; Bokonjić, D.; Kovačević, A.; Nepovimova, E.; Vališ, M.; Kuča, K. The Efficacy of Amifostine against Multiple-Dose Doxorubicin-Induced Toxicity in Rats. Int. J. Mol. Sci. 2018, 19, 2370. https://doi.org/10.3390/ijms19082370
Jaćević V, Dragojević-Simić V, Tatomirović Ž, Dobrić S, Bokonjić D, Kovačević A, Nepovimova E, Vališ M, Kuča K. The Efficacy of Amifostine against Multiple-Dose Doxorubicin-Induced Toxicity in Rats. International Journal of Molecular Sciences. 2018; 19(8):2370. https://doi.org/10.3390/ijms19082370
Chicago/Turabian StyleJaćević, Vesna, Viktorija Dragojević-Simić, Željka Tatomirović, Silva Dobrić, Dubravko Bokonjić, Aleksandra Kovačević, Eugenie Nepovimova, Martin Vališ, and Kamil Kuča. 2018. "The Efficacy of Amifostine against Multiple-Dose Doxorubicin-Induced Toxicity in Rats" International Journal of Molecular Sciences 19, no. 8: 2370. https://doi.org/10.3390/ijms19082370
APA StyleJaćević, V., Dragojević-Simić, V., Tatomirović, Ž., Dobrić, S., Bokonjić, D., Kovačević, A., Nepovimova, E., Vališ, M., & Kuča, K. (2018). The Efficacy of Amifostine against Multiple-Dose Doxorubicin-Induced Toxicity in Rats. International Journal of Molecular Sciences, 19(8), 2370. https://doi.org/10.3390/ijms19082370