Enteric Microbiota–Gut–Brain Axis from the Perspective of Nuclear Receptors



Abstract
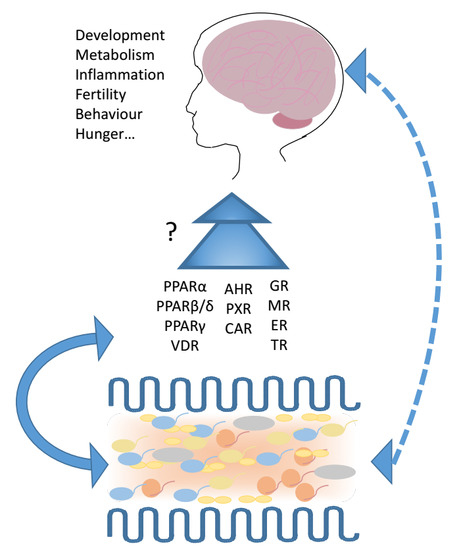
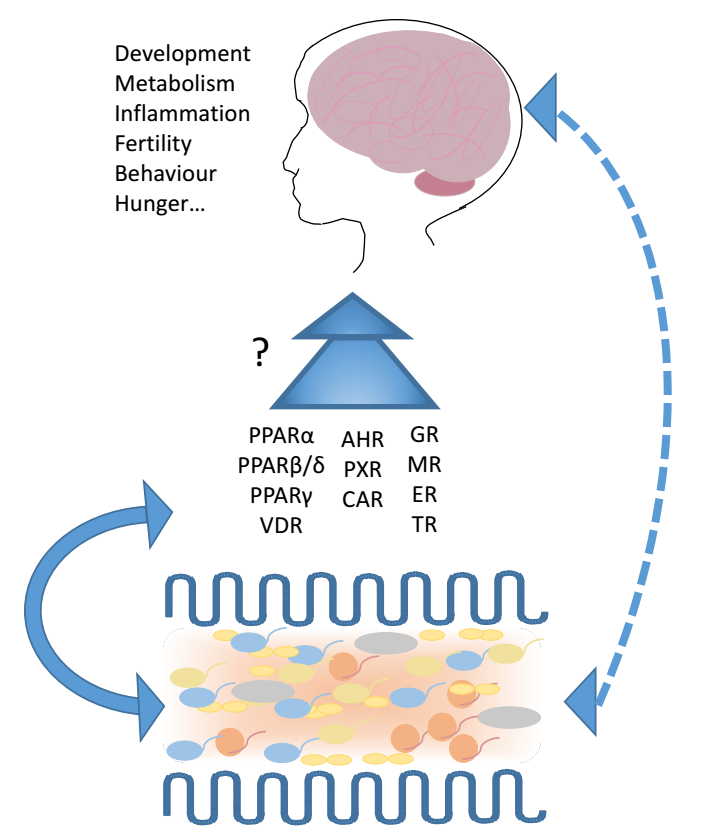{kind=link}
{kind=link}
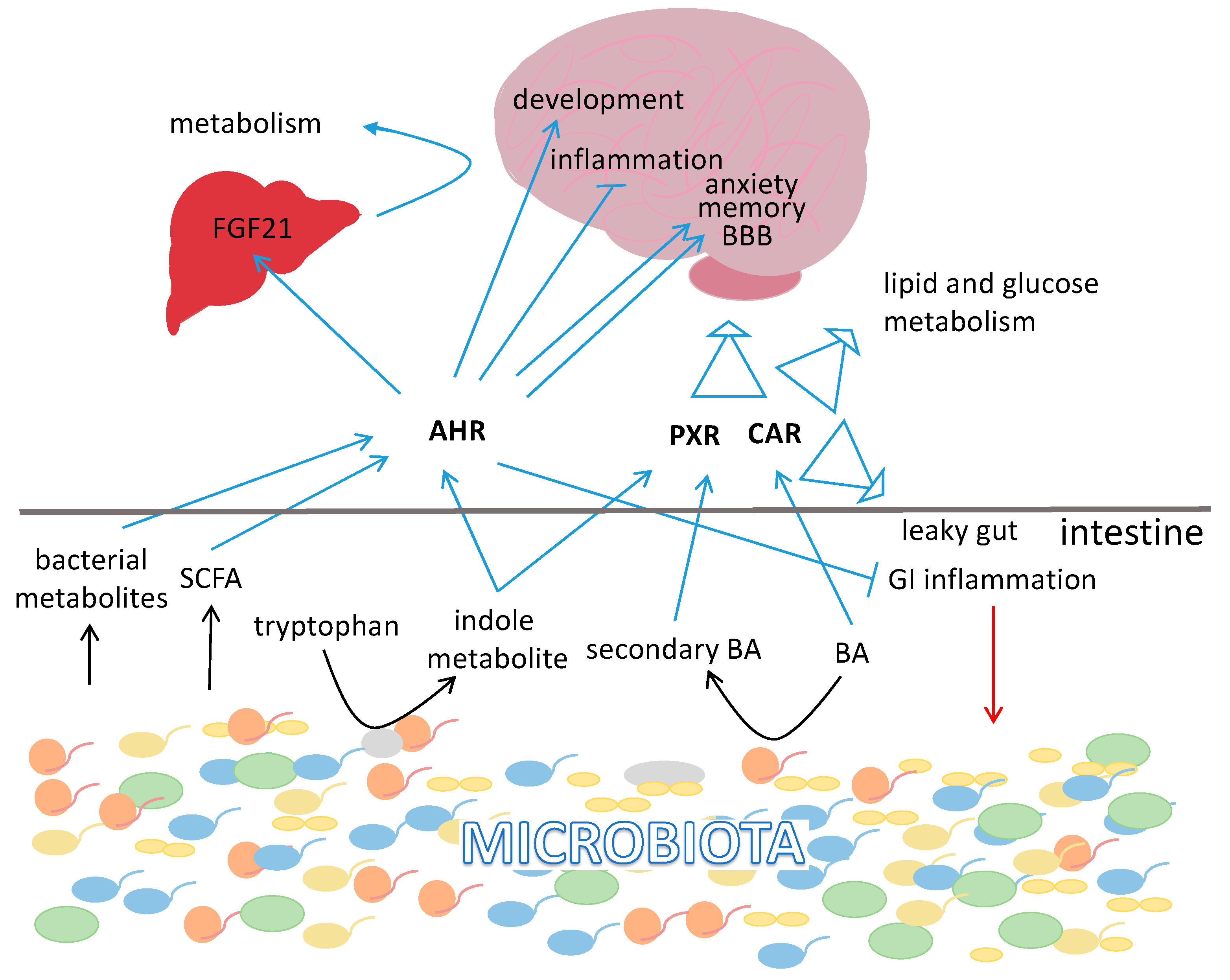{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Gut–Brain Axis
3. Nuclear Receptors
3.1. PPARs
3.1.1. Peroxisome Proliferator Activated Receptor α
3.1.2. Peroxisome Proliferator Activated Receptor β/δ
3.1.3. Peroxisome Proliferator Activated Receptor γ
3.2. Farnesoid X Receptor (FXR)
3.3. Vitamin D Receptor
3.4. Xenobiotic Receptors
3.4.1. Aryl Hydrocarbon Receptor
3.4.2. Pregnane X Receptor
3.4.3. Constitutive Androstane Nuclear Receptor (CAR)
3.5. Steroid Receptors
3.5.1. Glucocorticoid Receptor
3.5.2. Mineralocorticoid Receptor
3.5.3. Estrogen Receptors
3.6. Thyroid Hormone Receptors
4. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Makino, H.; Cetinyurek Yavuz, A.; Ben-Amor, K.; Roelofs, M.; Ishikawa, E.; Kubota, H.; Swinkels, S.; Sakai, T.; Oishi, K.; et al. Early-Life Events, Including Mode of Delivery and Type of Feeding, Siblings and Gender, Shape the Developing Gut Microbiota. PLoS ONE 2016, 11, e0158498. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, E.; Mayer, M.J.; Cotter, P.D.; Narbad, A. Gut microbiota as a source of novel antimicrobials. Gut Microbes 2018, 1–21, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Bjorkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.A.; Nguyen, J.C.; Polglaze, K.E.; Bertrand, P.P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C. Mental health: Thinking from the gut. Nature 2015, 518, S12–S15. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A. The tantalizing links between gut microbes and the brain. Nature 2015, 526, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut microbes and the brain: Paradigm shift in neuroscience. J. Neurosci. 2014, 34, 15490–15496. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Backhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. More than a gut feeling: The microbiota regulates neurodevelopment and behavior. Neuropsychopharmacology 2015, 40, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T.; Lubach, G.R.; Coe, C.L. Prenatal stress alters bacterial colonization of the gut in infant monkeys. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T.; Coe, C.L. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev. Psychobiol. 1999, 35, 146–155. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Marchesi, J.R.; Scully, P.; Codling, C.; Ceolho, A.M.; Quigley, E.M.; Cryan, J.F.; Dinan, T.G. Early life stress alters behavior, immunity, and microbiota in rats: Implications for irritable bowel syndrome and psychiatric illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Kiliaan, A.J.; Saunders, P.R.; Bijlsma, P.B.; Berin, M.C.; Taminiau, J.A.; Groot, J.A.; Perdue, M.H. Stress stimulates transepithelial macromolecular uptake in rat jejunum. Am. J. Physiol. 1998, 275, G1037–G1044. [Google Scholar] [CrossRef] [PubMed]
- Demaude, J.; Salvador-Cartier, C.; Fioramonti, J.; Ferrier, L.; Bueno, L. Phenotypic changes in colonocytes following acute stress or activation of mast cells in mice: Implications for delayed epithelial barrier dysfunction. Gut 2006, 55, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T.; Engler, H.; Sheridan, J.F. Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. J. Neuroimmunol. 2006, 171, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Freestone, P.P.; Williams, P.H.; Haigh, R.D.; Maggs, A.F.; Neal, C.P.; Lyte, M. Growth stimulation of intestinal commensal Escherichia coli by catecholamines: A possible contributory factor in trauma-induced sepsis. Shock 2002, 18, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Freestone, P.P.; Haigh, R.D.; Williams, P.H.; Lyte, M. Involvement of enterobactin in norepinephrine-mediated iron supply from transferrin to enterohaemorrhagic Escherichia coli. FEMS Microbiol. Lett. 2003, 222, 39–43. [Google Scholar] [CrossRef]
- Lyte, M.; Ernst, S. Catecholamine induced growth of gram negative bacteria. Life Sci. 1992, 50, 203–212. [Google Scholar] [CrossRef]
- Lyte, M.; Freestone, P.P.; Neal, C.P.; Olson, B.A.; Haigh, R.D.; Bayston, R.; Williams, P.H. Stimulation of Staphylococcus epidermidis growth and biofilm formation by catecholamine inotropes. Lancet 2003, 361, 130–135. [Google Scholar] [CrossRef]
- Hegde, M.; Wood, T.K.; Jayaraman, A. The neuroendocrine hormone norepinephrine increases Pseudomonas aeruginosa PA14 virulence through the las quorum-sensing pathway. Appl. Microbiol. Biotechnol. 2009, 84, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, V.; Torres, A.G.; Jarvis, B.; Nataro, J.P.; Kaper, J.B. Bacteria-host communication: The language of hormones. Proc. Natl. Acad. Sci. USA 2003, 100, 8951–8956. [Google Scholar] [CrossRef] [PubMed]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Queipo-Ortuno, M.I.; Seoane, L.M.; Murri, M.; Pardo, M.; Gomez-Zumaquero, J.M.; Cardona, F.; Casanueva, F.; Tinahones, F.J. Gut microbiota composition in male rat models under different nutritional status and physical activity and its association with serum leptin and ghrelin levels. PLoS ONE 2013, 8, e65465. [Google Scholar] [CrossRef] [PubMed]
- Parnell, J.A.; Reimer, R.A. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am. J. Clin. Nutr. 2009, 89, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, Y.; Koren, O.; Spor, A.; LeDuc, C.; Gutman, R.; Stombaugh, J.; Knight, R.; Ley, R.E.; Leibel, R.L. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 2012, 20, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Naruszewicz, M.; Johansson, M.L.; Zapolska-Downar, D.; Bukowska, H. Effect of Lactobacillus plantarum 299v on cardiovascular disease risk factors in smokers. Am. J. Clin. Nutr. 2002, 76, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Su, J.; Koprowski, S.; Hsu, A.; Tweddell, J.S.; Rafiee, P.; Gross, G.J.; Salzman, N.H.; Baker, J.E. Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J. 2012, 26, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Pickens, W.; Vinik, A.I.; Shiloach, J. Bacillus subtilis contains multiple forms of somatostatin-like material. Biochem. Biophys. Res. Commun. 1985, 127, 713–719. [Google Scholar] [CrossRef]
- Yadav, H.; Lee, J.H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lunden, G.O.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Backhed, F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013, 14, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Fetissov, S.O.; Hamze Sinno, M.; Coeffier, M.; Bole-Feysot, C.; Ducrotte, P.; Hokfelt, T.; Dechelotte, P. Autoantibodies against appetite-regulating peptide hormones and neuropeptides: Putative modulation by gut microflora. Nutrition 2008, 24, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Grenham, S.; Clarke, G.; Cryan, J.F.; Dinan, T.G. Brain-gut-microbe communication in health and disease. Front. Physiol. 2011, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 2011, 141, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, M.; Lalonde, R.; Violle, N.; Javelot, H.; Desor, D.; Nejdi, A.; Bisson, J.F.; Rougeot, C.; Pichelin, M.; Cazaubiel, M.; et al. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br. J. Nutr. 2011, 105, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Hiramoto, T.; Nishino, R.; Aiba, Y.; Kimura, T.; Yoshihara, K.; Koga, Y.; Sudo, N. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1288–G1295. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M. Microbial endocrinology in the microbiome-gut-brain axis: How bacterial production and utilization of neurochemicals influence behavior. PLoS Pathog. 2013, 9, e1003726. [Google Scholar] [CrossRef] [PubMed]
- Lyte, M. Microbial endocrinology and the microbiota-gut-brain axis. Adv. Exp. Med. Biol. 2014, 817, 3–24. [Google Scholar] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Bienenstock, J.; Dinan, T.G. The probiotic Bifidobacteria infantis: An assessment of potential antidepressant properties in the rat. J. Psychiatr. Res. 2008, 43, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Fischbach, M.A. Small molecules from the human microbiota. Science 2015, 349, 1254766. [Google Scholar] [CrossRef] [PubMed]
- Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Wahli, W. Peroxisome proliferator-activated receptors: Finding the orphan a home. Mol. Cell. Endocrinol. 1994, 100, 149–153. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Duszka, K.; Ellero-Simatos, S.; Ow, G.G.; Defernez, M.; Paramalingam, E.; Tett, A.; Ying, S.; König, J.; Narbad, A.; Kuznetsov, V.A.; et al. Complementary intestinal mucosa and microbiota responses to caloric restriction. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor α-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: The PPARα-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Crawford, P.A.; Crowley, J.R.; Sambandam, N.; Muegge, B.D.; Costello, E.K.; Hamady, M.; Knight, R.; Gordon, J.I. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc. Natl. Acad. Sci. USA 2009, 106, 11276–11281. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, I.; Ripperger, J.A.; Baeriswyl-Aebischer, S.; Albrecht, U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev. 2010, 24, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Canaple, L.; Rambaud, J.; Dkhissi-Benyahya, O.; Rayet, B.; Tan, N.S.; Michalik, L.; Delaunay, F.; Wahli, W.; Laudet, V. Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor α defines a novel positive feedback loop in the rodent liver circadian clock. Mol. Endocrinol. 2006, 20, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Rando, G.; Degueurce, G.; Leuenberger, N.; Michalik, L.; Wahli, W. New insights into the role of PPARs. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.C.; Apte, U.; Anderson, S.P.; Limaye, P.; Yoon, L.; Latendresse, J.; Dunn, C.; Everitt, J.I.; Voss, K.A.; Swanson, C.; et al. Mimetics of caloric restriction include agonists of lipid-activated nuclear receptors. J. Biol. Chem. 2004, 279, 46204–46212. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, C.; Dubois, G.; Duguay, Y.; Helledie, T.; Vu-Dac, N.; Gervois, P.; Soncin, F.; Mandrup, S.; Fruchart, J.C.; Fruchart-Najib, J.; et al. The orphan nuclear receptor Rev-Erbα is a peroxisome proliferator-activated receptor (PPAR) γ target gene and promotes PPARγ-induced adipocyte differentiation. J. Biol. Chem. 2003, 278, 37672–37680. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Shirai, H.; Ishida, N. CLOCK is involved in the circadian transactivation of peroxisome-proliferator-activated receptor α (PPARα) in mice. Biochem. J. 2005, 386, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.F.; Patel, D.; Wiggins, D.; Knight, B.L. The functional efficiency of lipogenic and cholesterogenic gene expression in normal mice and in mice lacking the peroxisomal proliferator-activated receptor-α (PPAR-α). Adv. Enzym. Regul. 2002, 42, 227–247. [Google Scholar] [CrossRef]
- Patel, D.D.; Knight, B.L.; Wiggins, D.; Humphreys, S.M.; Gibbons, G.F. Disturbances in the normal regulation of SREBP-sensitive genes in PPAR α-deficient mice. J. Lipid Res. 2001, 42, 328–337. [Google Scholar] [PubMed]
- Mukherji, A.; Kobiita, A.; Ye, T.; Chambon, P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 2013, 153, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, I.; Suryawanshi, A.; Hong, Y.; Ranganathan, P.; Shanmugam, A.; Ahmad, S.; Swafford, D.; Manicassamy, B.; Ramesh, G.; Koni, P.A.; et al. Homeostatic PPARα Signaling Limits Inflammatory Responses to Commensal Microbiota in the Intestine. J. Immunol. 2016, 196, 4739–4749. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Cuzzocrea, S. Absence of functional peroxisome proliferator-activated receptor-α enhanced ileum permeability during experimental colitis. Shock 2007, 28, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, L.; Mazzon, E.; Bruscoli, S.; Esposito, E.; Crisafulli, C.; Di Paola, R.; Caminiti, R.; Riccardi, C.; Cuzzocrea, S. Peroxisome proliferator-activated receptor-α modulates the anti-inflammatory effect of glucocorticoids in a model of inflammatory bowel disease in mice. Shock 2009, 31, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Mazzon, E.; Paterniti, I.; Dal Toso, R.; Pressi, G.; Caminiti, R.; Cuzzocrea, S. PPAR-α Contributes to the Anti-Inflammatory Activity of Verbascoside in a Model of Inflammatory Bowel Disease in Mice. PPAR Res. 2010, 2010, 917312. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Di Paola, R.; Mazzon, E.; Genovese, T.; Muia, C.; Centorrino, T.; Caputi, A.P. Role of endogenous and exogenous ligands for the peroxisome proliferators activated receptors α (PPAR-α) in the development of inflammatory bowel disease in mice. Lab. Investig. 2004, 84, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodriguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Dipatrizio, N.V.; Guijarro, A.; Schwartz, G.J.; Li, X.; Gaetani, S.; Astarita, G.; Piomelli, D. Sympathetic activity controls fat-induced oleoylethanolamide signaling in small intestine. J. Neurosci. 2011, 31, 5730–5736. [Google Scholar] [CrossRef] [PubMed]
- Magotti, P.; Bauer, I.; Igarashi, M.; Babagoli, M.; Marotta, R.; Piomelli, D.; Garau, G. Structure of human N-acylphosphatidylethanolamine-hydrolyzing phospholipase D: Regulation of fatty acid ethanolamide biosynthesis by bile acids. Structure 2015, 23, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor α gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, S.; Oveisi, F.; Piomelli, D. Modulation of meal pattern in the rat by the anorexic lipid mediator oleoylethanolamide. Neuropsychopharmacology 2003, 28, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Oveisi, F.; Gaetani, S.; Lin, E.; Piomelli, D. Oleoylethanolamide, an endogenous PPAR-α agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 2005, 48, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Caillon, A.; Duszka, K.; Wahli, W.; Rohner-Jeanrenaud, F.; Altirriba, J. The OEA effect on food intake is independent from the presence of PPARα in the intestine and the nodose ganglion, while the impact of OEA on energy expenditure requires the presence of PPARα in mice. Metabolism 2018. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Gaetani, S.; Fu, J.; Oveisi, F.; Burton, K.; Piomelli, D. Regulation of food intake by oleoylethanolamide. Cell. Mol. Life Sci. 2005, 62, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. A fatty gut feeling. Trends Endocrinol. Metab. 2013, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Proulx, K.; Cota, D.; Castaneda, T.R.; Tschop, M.H.; D’Alessio, D.A.; Tso, P.; Woods, S.C.; Seeley, R.J. Mechanisms of oleoylethanolamide-induced changes in feeding behavior and motor activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R729–R737. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez de Fonseca, F.; Navarro, M.; Gomez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodriguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S.; et al. An anorexic lipid mediator regulated by feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, S.; Fu, J.; Cassano, T.; Dipasquale, P.; Romano, A.; Righetti, L.; Cianci, S.; Laconca, L.; Giannini, E.; Scaccianoce, S.; et al. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. J. Neurosci. 2010, 30, 8096–8101. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Michalik, L.; Di-Poi, N.; Ng, C.Y.; Mermod, N.; Roberts, A.B.; Desvergne, B.; Wahli, W. Essential role of Smad3 in the inhibition of inflammation-induced PPARβ/δ expression. EMBO J. 2004, 23, 4211–4221. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Heggeler, B.B.; Grisel, P.; Boucard, N.; Corthesy-Theulaz, I.; Wahli, W.; Desvergne, B. PPARβ/δ regulates paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology 2006, 131, 538–553. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Hollingshead, H.E.; Gonzalez, F.J. Role of peroxisome-proliferator-activated receptor β/δ (PPARβ/δ) in gastrointestinal tract function and disease. Clin. Sci. 2008, 115, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Burdick, A.D.; Kim, D.J.; Peraza, M.A.; Gonzalez, F.J.; Peters, J.M. The role of peroxisome proliferator-activated receptor-β/δ in epithelial cell growth and differentiation. Cell Signal. 2006, 18, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, H.E.; Morimura, K.; Adachi, M.; Kennett, M.J.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peters, J.M. PPARβ/δ protects against experimental colitis through a ligand-independent mechanism. Dig. Dis. Sci. 2007, 52, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W. A gut feeling of the PXR, PPAR and NF-κB connection. J. Intern. Med. 2008, 263, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, M.; Zwarts, I.; Zutphen, T.V.; Dijk, T.H.; Bloks, V.W.; Harkema, L.; Bruin, A.; Downes, M.; Evans, R.M.; Verkade, H.J.; et al. Intestinal PPARδ protects against diet-induced obesity, insulin resistance and dyslipidemia. Sci. Rep. 2017, 7, 846. [Google Scholar] [CrossRef] [PubMed]
- Daoudi, M.; Hennuyer, N.; Borland, M.G.; Touche, V.; Duhem, C.; Gross, B.; Caiazzo, R.; Kerr-Conte, J.; Pattou, F.; Peters, J.M.; et al. PPARβ/δ activation induces enteroendocrine L cell GLP-1 production. Gastroenterology 2011, 140, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Anghel, S.I.; Wahli, W. Fat poetry: A kingdom for PPAR γ. Cell Res. 2007, 17, 486–511. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J. Nuclear receptors. I. PPAR γ in the gastrointestinal tract: Gain or pain? Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G581–G585. [Google Scholar] [CrossRef] [PubMed]
- Dubuquoy, L.; Rousseaux, C.; Thuru, X.; Peyrin-Biroulet, L.; Romano, O.; Chavatte, P.; Chamaillard, M.; Desreumaux, P. PPARγ as a new therapeutic target in inflammatory bowel diseases. Gut 2006, 55, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPARγ and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Harmon, G.S.; Dumlao, D.S.; Ng, D.T.; Barrett, K.E.; Dennis, E.A.; Dong, H.; Glass, C.K. Pharmacological correction of a defect in PPAR-γ signaling ameliorates disease severity in Cftr-deficient mice. Nat. Med. 2010, 16, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Mansen, A.; Guardiola-Diaz, H.; Rafter, J.; Branting, C.; Gustafsson, J.A. Expression of the peroxisome proliferator-activated receptor (PPAR) in the mouse colonic mucosa. Biochem. Biophys. Res. Commun. 1996, 222, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Marion-Letellier, R.; Dechelotte, P.; Iacucci, M.; Ghosh, S. Dietary modulation of peroxisome proliferator-activated receptor γ. Gut 2009, 58, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Reynders, V.; Loitsch, S.; Steinhilber, D.; Stein, J.; Schroder, O. Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NF κ B signalling. Mol. Immunol. 2007, 44, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Wachtershauser, A.; Loitsch, S.M.; Stein, J. PPAR-γ is selectively upregulated in Caco-2 cells by butyrate. Biochem. Biophys. Res. Commun. 2000, 272, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Nepelska, M.; de Wouters, T.; Jacouton, E.; Beguet-Crespel, F.; Lapaque, N.; Dore, J.; Arulampalam, V.; Blottiere, H.M. Commensal gut bacteria modulate phosphorylation-dependent PPARγ transcriptional activity in human intestinal epithelial cells. Sci. Rep. 2017, 7, 43199. [Google Scholar] [CrossRef] [PubMed]
- Voltan, S.; Martines, D.; Elli, M.; Brun, P.; Longo, S.; Porzionato, A.; Macchi, V.; D’Inca, R.; Scarpa, M.; Palu, G.; et al. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-γ in the intestinal mucosa. Gastroenterology 2008, 135, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Are, A.; Aronsson, L.; Wang, S.; Greicius, G.; Lee, Y.K.; Gustafsson, J.A.; Pettersson, S.; Arulampalam, V. Enterococcus faecalis from newborn babies regulate endogenous PPARγ activity and IL-10 levels in colonic epithelial cells. Proc. Natl. Acad. Sci. USA 2008, 105, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Couvigny, B.; de Wouters, T.; Kaci, G.; Jacouton, E.; Delorme, C.; Dore, J.; Renault, P.; Blottiere, H.M.; Guedon, E.; Lapaque, N. Commensal Streptococcus salivarius Modulates PPARγ Transcriptional Activity in Human Intestinal Epithelial Cells. PLoS ONE 2015, 10, e0125371. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Tognini, P.; Liu, Y.; Eckel-Mahan, K.L.; Baldi, P.; Sassone-Corsi, P. Gut microbiota directs PPARγ-driven reprogramming of the liver circadian clock by nutritional challenge. EMBO Rep. 2016, 17, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Cerbone, A.; Toaldo, C.; Laurora, S.; Briatore, F.; Pizzimenti, S.; Dianzani, M.U.; Ferretti, C.; Barrera, G. 4-Hydroxynonenal and PPARγ ligands affect proliferation, differentiation, and apoptosis in colon cancer cells. Free Radic. Biol. Med. 2007, 42, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Martinasso, G.; Oraldi, M.; Trombetta, A.; Maggiora, M.; Bertetto, O.; Canuto, R.A.; Muzio, G. Involvement of PPARs in Cell Proliferation and Apoptosis in Human Colon Cancer Specimens and in Normal and Cancer Cell Lines. PPAR Res. 2007, 2007, 93416. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, S.; Margeli, A.; Vielh, P.; Kouraklis, G. Peroxisome proliferator-activated receptor-γ ligands as cell-cycle modulators. Cancer Treat. Rev. 2004, 30, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.P.; Zhang, X.; Xie, W.F. Differentiation therapy for solid tumors. J. Dig. Dis. 2014, 15, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Lee, J.F.; Wang, S.H.; Chan, U.P.; Ip, P.C.; Lau, W.Y. Apoptosis induced by activation of peroxisome-proliferator activated receptor-γ is associated with Bcl-2 and NF-κB in human colon cancer. Life Sci. 2002, 70, 2631–2646. [Google Scholar] [CrossRef]
- Chen, G.G.; Xu, H.; Lee, J.F.; Subramaniam, M.; Leung, K.L.; Wang, S.H.; Chan, U.P.; Spelsberg, T.C. 15-hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor γ-dependent pathway. Int. J. Cancer 2003, 107, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Han, J.S.; Seo, C.Y.; Park, T.H.; Kwon, H.C.; Jeong, J.S.; Kim, I.H.; Yun, J.; Bae, Y.S.; Kwak, J.Y.; et al. Pioglitazone, a synthetic ligand for PPARγ, induces apoptosis in RB-deficient human colorectal cancer cells. Apoptosis 2006, 11, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome proliferator-activated receptor γ activation can regulate β-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Lichtenstein, G.R.; Stein, R.B.; Deren, J.J.; Judge, T.A.; Fogt, F.; Furth, E.E.; Demissie, E.J.; Hurd, L.B.; Su, C.G.; et al. An open-label trial of the PPAR-γ ligand rosiglitazone for active ulcerative colitis. Am. J. Gastroenterol. 2001, 96, 3323–3328. [Google Scholar] [PubMed]
- Liang, H.L.; Ouyang, Q. A clinical trial of combined use of rosiglitazone and 5-aminosalicylate for ulcerative colitis. World J. Gastroenterol. 2008, 14, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Bassaganya-Riera, J.; Hontecillas, R. CLA and n-3 PUFA differentially modulate clinical activity and colonic PPAR-responsive gene expression in a pig model of experimental IBD. Clin. Nutr. 2006, 25, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas, R.; Wannemeulher, M.J.; Zimmerman, D.R.; Hutto, D.L.; Wilson, J.H.; Ahn, D.U.; Bassaganya-Riera, J. Nutritional regulation of porcine bacterial-induced colitis by conjugated linoleic acid. J. Nutr. 2002, 132, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Hidalgo, M.; Martin, A.R.; Villegas, I.; de la Lastra, C.A. Rosiglitazone, a PPARγ ligand, modulates signal transduction pathways during the development of acute TNBS-induced colitis in rats. Eur. J. Pharmacol. 2007, 562, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Kozar, R.A.; Zou, L.; Weatherall, J.M.; Attuwaybi, B.; Moore-Olufemi, S.D.; Weisbrodt, N.W.; Moore, F.A. Peroxisome proliferator-activated receptor γ mediates protection against cyclooxygenase-2-induced gut dysfunction in a rodent model of mesenteric ischemia/reperfusion. Shock 2005, 24, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Saubermann, L.J.; Nakajima, A.; Wada, K.; Zhao, S.; Terauchi, Y.; Kadowaki, T.; Aburatani, H.; Matsuhashi, N.; Nagai, R.; Blumberg, R.S. Peroxisome proliferator-activated receptor γ agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm. Bowel Dis. 2002, 8, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Su, C.G.; Wen, X.; Bailey, S.T.; Jiang, W.; Rangwala, S.M.; Keilbaugh, S.A.; Flanigan, A.; Murthy, S.; Lazar, M.A.; Wu, G.D. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. J. Clin. Investig. 1999, 104, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Ling, T.W.; Korecka, A.; Li, Y.; D’Arienzo, R.; Bunte, R.M.; Berger, T.; Arulampalam, V.; Chambon, P.; Mak, T.W.; et al. Absence of intestinal PPARγ aggravates acute infectious colitis in mice through a lipocalin-2-dependent pathway. PLoS Pathog. 2014, 10, e1003887. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Wahli, W. PPARs Mediate Lipid Signaling in Inflammation and Cancer. PPAR Res. 2008, 2008, 134059. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Morimura, K.; Gonzalez, F.J. Expression of peroxisome proliferator-activated receptor-γ in macrophage suppresses experimentally induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G657–G666. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Hong, J.; Fraser, A.; Merriman, T.; Krissansen, G. PPAR-γ and Crohn’s disease in New Zealand. Gastroenterology 2006, 130, 2249–2250. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Nakajima, A.; Blumberg, R.S. PPARγ and inflammatory bowel disease: A new therapeutic target for ulcerative colitis and Crohn’s disease. Trends Mol. Med. 2001, 7, 329–331. [Google Scholar] [CrossRef]
- Yasui, Y.; Hosokawa, M.; Sahara, T.; Suzuki, R.; Ohgiya, S.; Kohno, H.; Tanaka, T.; Miyashita, K. Bitter gourd seed fatty acid rich in 9c,11t,13t-conjugated linolenic acid induces apoptosis and up-regulates the GADD45, p53 and PPARγ in human colon cancer Caco-2 cells. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Akinyeke, T.O.; Stewart, L.V. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARγ-independent mechanism. Cancer Biol. Ther. 2011, 11, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Frucht, H. Activation of the PPAR pathway induces apoptosis and COX-2 inhibition in HT-29 human colon cancer cells. Carcinogenesis 2001, 22, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Lin, D.T.; Hart, J.C.; Dannenberg, A.J. Peroxisome proliferator-activated receptor γ ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J. Biol. Chem. 2001, 276, 12440–12448. [Google Scholar] [CrossRef] [PubMed]
- Vandoros, G.P.; Konstantinopoulos, P.A.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papachristou, G.I.; Karamouzis, M.V.; Gkermpesi, M.; Varakis, I.; Papavassiliou, A.G. PPAR-γ is expressed and NF-kB pathway is activated and correlates positively with COX-2 expression in stromal myofibroblasts surrounding colon adenocarcinomas. J. Cancer Res. Clin. Oncol. 2006, 132, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Girnun, G.D.; Smith, W.M.; Drori, S.; Sarraf, P.; Mueller, E.; Eng, C.; Nambiar, P.; Rosenberg, D.W.; Bronson, R.T.; Edelmann, W.; et al. APC-dependent suppression of colon carcinogenesis by PPARγ. Proc. Natl. Acad. Sci. USA 2002, 99, 13771–13776. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Nakajima, A.; Fujisawa, N.; Takahashi, H.; Ikeda, I.; Tomimoto, A.; Yonemitsu, K.; Nakajima, N.; Kudo, C.; Wada, K.; et al. Peroxisome proliferator-activated receptor γ (PPARγ) suppresses colonic epithelial cell turnover and colon carcinogenesis through inhibition of the β-catenin/T cell factor (TCF) pathway. J. Pharmacol. Sci. 2008, 106, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Beisner, J.; Wang, G.; Nuding, S.; Oommen, S.T.; Kelly, D.; Parmentier-Decrucq, E.; Dessein, R.; Merour, E.; Chavatte, P.; et al. Peroxisome proliferator-activated receptor γ activation is required for maintenance of innate antimicrobial immunity in the colon. Proc. Natl. Acad. Sci. USA 2010, 107, 8772–8777. [Google Scholar] [CrossRef] [PubMed]
- Duszka, K.; Oresic, M.; Le May, C.; Konig, J.; Wahli, W. PPARγ Modulates Long Chain Fatty Acid Processing in the Intestinal Epithelium. Int. J. Mol. Sci. 2017, 18, 2559. [Google Scholar] [CrossRef] [PubMed]
- Duszka, K.; Picard, A.; Ellero-Simatos, S.; Chen, J.; Defernez, M.; Paramalingam, E.; Pigram, A.; Vanoaica, L.; Canlet, C.; Parini, P.; et al. Intestinal PPARγ signalling is required for sympathetic nervous system activation in response to caloric restriction. Sci. Rep. 2016, 6, 36937. [Google Scholar] [CrossRef] [PubMed]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Midtvedt, T. Microbial bile acid transformation. Am. J. Clin. Nutr. 1974, 27, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Bouscarel, B. Bile acids and signal transduction: Role in glucose homeostasis. Cell Signal. 2008, 20, 2180–2197. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.; Fu, T.; Choi, S.E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investog. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Kong, B.; Stieger, B.; Tschopp, O.; Schultze, S.M.; Rau, M.; Weber, A.; Mullhaupt, B.; Guo, G.L.; Geier, A. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 2015, 35, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Kosters, A.; Mells, J.E.; Zhang, W.; Setchell, K.D.; Amanso, A.M.; Wynn, G.M.; Xu, T.; Keller, B.T.; Yin, H.; et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci. Transl. Med. 2016, 8, 357ra122. [Google Scholar] [CrossRef] [PubMed]
- Raju, U.; Levitz, M.; Javitt, N.B. Bile acids in human breast cyst fluid: The identification of lithocholic acid. J. Clin. Endocrinol. Metab. 1990, 70, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Javitt, N.B.; Budai, K.; Miller, D.G.; Cahan, A.C.; Raju, U.; Levitz, M. Breast-gut connection: Origin of chenodeoxycholic acid in breast cyst fluid. Lancet 1994, 343, 633–635. [Google Scholar] [CrossRef]
- Swales, K.E.; Korbonits, M.; Carpenter, R.; Walsh, D.T.; Warner, T.D.; Bishop-Bailey, D. The farnesoid X receptor is expressed in breast cancer and regulates apoptosis and aromatase expression. Cancer Res. 2006, 66, 10120–10126. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Lundasen, T.; Galman, C.; Angelin, B.; Rudling, M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 2006, 260, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of FGF19 require Klotho β. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Moschetta, A.; Bookout, A.L.; Peng, L.; Umetani, M.; Holmstrom, S.R.; Suino-Powell, K.; Xu, H.E.; Richardson, J.A.; Gerard, R.D.; et al. Identification of a hormonal basis for gallbladder filling. Nat. Med. 2006, 12, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.J.; Osborne, T.F. FGF15/FGFR4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J. Biol. Chem. 2009, 284, 11110–11120. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, C.; Luo, Y.; Jin, C.; Wang, F.; McKeehan, W.L. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes 2007, 56, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Marcelin, G.; Blouet, C.; Jeong, J.H.; Jo, Y.H.; Schwartz, G.J.; Chua, S., Jr. A gut-brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Mol. Metab. 2018, 8, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Investig. 2013, 123, 4799–4808. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cao, L.; Jiang, C.; Xie, Y.; Cheng, X.; Krausz, K.W.; Qi, Y.; Sun, L.; Shah, Y.M.; Gonzalez, F.J.; et al. PPARα-UGT axis activation represses intestinal FXR-FGF15 feedback signalling and exacerbates experimental colitis. Nat. Commun. 2014, 5, 4573. [Google Scholar] [CrossRef] [PubMed]
- Okamura, A.; Koyanagi, S.; Dilxiat, A.; Kusunose, N.; Chen, J.J.; Matsunaga, N.; Shibata, S.; Ohdo, S. Bile acid-regulated peroxisome proliferator-activated receptor-α (PPARα) activity underlies circadian expression of intestinal peptide absorption transporter PepT1/Slc15a1. J. Biol. Chem. 2014, 289, 25296–25305. [Google Scholar] [CrossRef] [PubMed]
- Overbergh, L.; Stoffels, K.; Waer, M.; Verstuyf, A.; Bouillon, R.; Mathieu, C. Immune regulation of 25-hydroxyvitamin d-1α-hydroxylase in human monocytic THP1 cells: Mechanisms of interferon-γ-mediated induction. J. Clin. Endocrinol. Metab. 2006, 91, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Overbergh, L.; Decallonne, B.; Valckx, D.; Verstuyf, A.; Depovere, J.; Laureys, J.; Rutgeerts, O.; Saint-Arnaud, R.; Bouillon, R.; Mathieu, C. Identification and immune regulation of 25-hydroxyvitamin d-1-α-hydroxylase in murine macrophages. Clin. Exp. Immunol. 2000, 120, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Segersten, U.; Holm, P.K.; Bjorklund, P.; Hessman, O.; Nordgren, H.; Binderup, L.; Akerstrom, G.; Hellman, P.; Westin, G. 25-Hydroxyvitamin D3 1α-hydroxylase expression in breast cancer and use of non-1α-hydroxylated vitamin D analogue. Breast Cancer Res. 2005, 7, R980–R986. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.; Laban, C.; Bustin, S.A.; Ogunkolade, W.; Khalaf, S.; Carpenter, R.; Jenkins, P.J. Expression of 25-hydroxyvitamin d-1-α-hydroxylase, and vitamin D receptor mRNA in normal and malignant breast tissue. Anticancer Res. 2009, 29, 155–157. [Google Scholar] [PubMed]
- Tangpricha, V.; Flanagan, J.N.; Whitlatch, L.W.; Tseng, C.C.; Chen, T.C.; Holt, P.R.; Lipkin, M.S.; Holick, M.F. 25-hydroxyvitamin d-1α-hydroxylase in normal and malignant colon tissue. Lancet 2001, 357, 1673–1674. [Google Scholar] [CrossRef]
- Garland, C.F.; Kim, J.J.; Mohr, S.B.; Gorham, E.D.; Grant, W.B.; Giovannucci, E.L.; Baggerly, L.; Hofflich, H.; Ramsdell, J.W.; Zeng, K.; et al. Meta-analysis of all-cause mortality according to serum 25-hydroxyvitamin D. Am. J. Public Health 2014, 104, e43–e50. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, M.C.; Preziosi, P.; Maamer, M.; Arnaud, S.; Galan, P.; Hercberg, S.; Meunier, P.J. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos. Int. 1997, 7, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M. Vitamin D and genomic stability. Mutat. Res. 2001, 475, 69–87. [Google Scholar] [CrossRef]
- Schwalfenberg, G.K. A review of the critical role of vitamin D in the functioning of the immune system and the clinical implications of vitamin D deficiency. Mol. Nutr. Food Res. 2011, 55, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Amrein, K.; Quraishi, S.A.; Litonjua, A.A.; Gibbons, F.K.; Pieber, T.R.; Camargo, C.A., Jr.; Giovannucci, E.; Christopher, K.B. Evidence for a U-shaped relationship between prehospital vitamin D status and mortality: A cohort study. J. Clin. Endocrinol. Metab. 2014, 99, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Wu-Wong, J.R.; Chen, Y.W.; Nakane, M.; Wolf, M. Differential effects of vitamin d receptor agonists on gene expression in neonatal rat cardiomyocytes. Cardiovasc. Drugs Ther. 2011, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Adachi, R.; Honma, Y.; Masuno, H.; Kawana, K.; Shimomura, I.; Yamada, S.; Makishima, M. Selective activation of vitamin D receptor by lithocholic acid acetate, a bile acid derivative. J. Lipid Res. 2005, 46, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Haussler, C.A.; Bartik, L.; Whitfield, G.K.; Hsieh, J.C.; Slater, S.; Jurutka, P.W. Vitamin D receptor: Molecular signaling and actions of nutritional ligands in disease prevention. Nutr. Rev. 2008, 66, S98–S112. [Google Scholar] [CrossRef] [PubMed]
- Moon, J. The role of vitamin D in toxic metal absorption: A review. J. Am. Coll. Nutr. 1994, 13, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.W.; Liu, P.Y.; Josh, P.; Cui, X. Intracellular distribution of the vitamin D receptor in the brain: Comparison with classic target tissues and redistribution with development. Neuroscience 2014, 268, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, J.; DeLuca, H.F. Where is the vitamin D receptor? Arch. Biochem. Biophys. 2012, 523, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Prufer, K.; Veenstra, T.D.; Jirikowski, G.F.; Kumar, R. Distribution of 1,25-dihydroxyvitamin D3 receptor immunoreactivity in the rat brain and spinal cord. J. Chem. Neuroanat. 1999, 16, 135–145. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Sakiyama, R.; Coty, W.A. Restricted transport of vitamin D and A derivatives through the rat blood-brain barrier. J. Neurochem. 1985, 44, 1138–1141. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.K.; Lin, D.; Zhang, M.Y.; Bikle, D.D.; Shackleton, C.H.; Miller, W.L.; Portale, A.A. Cloning of human 25-hydroxyvitamin d-1 α-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol. Endocrinol. 1997, 11, 1961–1970. [Google Scholar] [PubMed]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1 α-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wei, X.; Ge, X.; Chen, Y.; Li, Y.C. Microbiota-Dependent Induction of Colonic Cyp27b1 Is Associated with Colonic Inflammation: Implications of Locally Produced 1,25-Dihydroxyvitamin D3 in Inflammatory Regulation in the Colon. Endocrinology 2017, 158, 4064–4075. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; de Vries, C.J.M. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE 2017, 12, e0176715. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, T.; Shi, Y.; Tian, F.; Hu, H.; Deb, D.K.; Chen, Y.; Bissonnette, M.; Li, Y.C. Gut Epithelial Vitamin D Receptor Regulates Microbiota-Dependent Mucosal Inflammation by Suppressing Intestinal Epithelial Cell Apoptosis. Endocrinology 2018, 159, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Waddell, A.; Lin, Y.D.; Cantorna, M.T. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. 2015, 8, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Nie, Y.; Zhu, A.; Chen, Z.; Wu, P.; Zhang, L.; Luo, M.; Sun, Q.; Cai, L.; Lai, Y.; et al. Vitamin D Signaling through Induction of Paneth Cell Defensins Maintains Gut Microbiota and Improves Metabolic Disorders and Hepatic Steatosis in Animal Models. Front. Physiol. 2016, 7, 498. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.O.; Claud, E.C.; Chen, D.; Chang, E.B.; Carmeliet, G.; et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2015, 64, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thingholm, L.B.; Skieceviciene, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.A.; Ruhlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Tang, D.H.; Modlin, R.L. Cutting edge: Vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J. Immunol. 2007, 179, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yoon, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Wan, J.; Petrof, E.O.; Claud, E.C.; Chen, D.; Sun, J. Vitamin D receptor pathway is required for probiotic protection in colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G341–G349. [Google Scholar] [CrossRef] [PubMed]
- Bora, S.A.; Kennett, M.J.; Smith, P.B.; Patterson, A.D.; Cantorna, M.T. Regulation of vitamin D metabolism following disruption of the microbiota using broad spectrum antibiotics. J. Nutr. Biochem. 2018, 56, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Eyles, D.; Brown, J.; Mackay-Sim, A.; McGrath, J.; Feron, F. Vitamin D3 and brain development. Neuroscience 2003, 118, 641–653. [Google Scholar] [CrossRef]
- Jiang, P.; Zhang, L.H.; Cai, H.L.; Li, H.D.; Liu, Y.P.; Tang, M.M.; Dang, R.L.; Zhu, W.Y.; Xue, Y.; He, X. Neurochemical effects of chronic administration of calcitriol in rats. Nutrients 2014, 6, 6048–6059. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.P.; Fletcher-Turner, A.; Yurek, D.M.; Cass, W.A. Calcitriol protection against dopamine loss induced by intracerebroventricular administration of 6-hydroxydopamine. Neurochem. Res. 2006, 31, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Wu, J.N.; Cherng, T.L.; Hoffer, B.J.; Chen, H.H.; Borlongan, C.V.; Wang, Y. Vitamin D3 attenuates 6-hydroxydopamine-induced neurotoxicity in rats. Brain Res. 2001, 904, 67–75. [Google Scholar] [CrossRef]
- Nataf, S.; Garcion, E.; Darcy, F.; Chabannes, D.; Muller, J.Y.; Brachet, P. 1,25 Dihydroxyvitamin D3 exerts regional effects in the central nervous system during experimental allergic encephalomyelitis. J. Neuropathol. Exp. Neurol. 1996, 55, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Garcion, E.; Sindji, L.; Montero-Menei, C.; Andre, C.; Brachet, P.; Darcy, F. Expression of inducible nitric oxide synthase during rat brain inflammation: Regulation by 1,25-dihydroxyvitamin D3. Glia 1998, 22, 282–294. [Google Scholar] [CrossRef]
- Cannell, J.J. Autism and vitamin D. Med. Hypotheses 2008, 70, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Soles, C.M. Epidemiologic evidence supporting the role of maternal vitamin D deficiency as a risk factor for the development of infantile autism. Dermatoendocrinol 2009, 1, 223–228. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J. Hypothesis: Is low prenatal vitamin D a risk-modifying factor for schizophrenia? Schizophr. Res. 1999, 40, 173–177. [Google Scholar] [CrossRef]
- Kesby, J.P.; Burne, T.H.; McGrath, J.J.; Eyles, D.W. Developmental vitamin D deficiency alters MK 801-induced hyperlocomotion in the adult rat: An animal model of schizophrenia. Biol. Psychiatry 2006, 60, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kesby, J.P.; Cui, X.; O’Loan, J.; McGrath, J.J.; Burne, T.H.; Eyles, D.W. Developmental vitamin D deficiency alters dopamine-mediated behaviors and dopamine transporter function in adult female rats. Psychopharmacology 2010, 208, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Zhang, S.M.; O’Reilly, E.; Hernan, M.A.; Olek, M.J.; Willett, W.C.; Ascherio, A. Vitamin D intake and incidence of multiple sclerosis. Neurology 2004, 62, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Evatt, M.L.; Delong, M.R.; Khazai, N.; Rosen, A.; Triche, S.; Tangpricha, V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch. Neurol. 2008, 65, 1348–1352. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.E.; Karchner, S.I.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl. Acad. Sci. USA 1997, 94, 13743–13748. [Google Scholar] [CrossRef] [PubMed]
- Diani-Moore, S.; Ram, P.; Li, X.; Mondal, P.; Youn, D.Y.; Sauve, A.A.; Rifkind, A.B. Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 2010, 285, 38801–38810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt-Kamper, C.; Biljes, D.; Merches, K.; Steiner, I.; Daldrup, T.; Bol-Schoenmakers, M.; Pieters, R.H.H.; Esser, C. Indole-3-carbinol, a plant nutrient and AhR-Ligand precursor, supports oral tolerance against OVA and improves peanut allergy symptoms in mice. PLoS ONE 2017, 12, e0180321. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Lee, S.O.; Sridharan, G.; Lee, K.; Davidson, L.A.; Jayaraman, A.; Chapkin, R.S.; Alaniz, R.; Safe, S. Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol. 2014, 85, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Moura-Alves, P.; Fae, K.; Houthuys, E.; Dorhoi, A.; Kreuchwig, A.; Furkert, J.; Barison, N.; Diehl, A.; Munder, A.; Constant, P.; et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature 2014, 512, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Cheng, Y.; Park, H.; Davidson, L.A.; Callaway, E.S.; Chapkin, R.S.; Jayaraman, A.; Asante, A.; Allred, C.; Weaver, E.A.; et al. Short Chain Fatty Acids Enhance Aryl Hydrocarbon (Ah) Responsiveness in Mouse Colonocytes and Caco-2 Human Colon Cancer Cells. Sci. Rep. 2017, 7, 10163. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Christensen, J.; O’Garra, A.; Stockinger, B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J. Exp. Med. 2009, 206, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS ONE 2013, 8, e79819. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Chng, S.H.; Kundu, P.; Dominguez-Brauer, C.; Teo, W.L.; Kawajiri, K.; Fujii-Kuriyama, Y.; Mak, T.W.; Pettersson, S. Ablating the aryl hydrocarbon receptor (AhR) in CD11c+ cells perturbs intestinal epithelium development and intestinal immunity. Sci. Rep. 2016, 6, 23820. [Google Scholar] [CrossRef] [PubMed]
- Korecka, A.; Dona, A.; Lahiri, S.; Tett, A.J.; Al-Asmakh, M.; Braniste, V.; D’Arienzo, R.; Abbaspour, A.; Reichardt, N.; Fujii-Kuriyama, Y.; et al. Bidirectional communication between the Aryl hydrocarbon Receptor (AhR) and the microbiome tunes host metabolism. NPJ Biofilms Microbiomes 2016, 2, 16014. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Le May, C.; Pineau, T.; Bigot, K.; Kohl, C.; Girard, J.; Pegorier, J.P. Reduced hepatic fatty acid oxidation in fasting PPARα null mice is due to impaired mitochondrial hydroxymethylglutaryl-CoA synthase gene expression. FEBS Lett. 2000, 475, 163–166. [Google Scholar] [CrossRef]
- Shaban, Z.; Soliman, M.; El-Shazly, S.; El-Bohi, K.; Abdelazeez, A.; Kehelo, K.; Kim, H.S.; Muzandu, K.; Ishizuka, M.; Kazusaka, A.; et al. AhR and PPARα: Antagonistic effects on CYP2B and CYP3A, and additive inhibitory effects on CYP2C11. Xenobiotica 2005, 35, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Yan, J.; Liu, K.; Garbacz, W.G.; Wang, P.; Xu, M.; Ma, X.; Xie, W. Activation of aryl hydrocarbon receptor dissociates fatty liver from insulin resistance by inducing fibroblast growth factor 21. Hepatology 2015, 61, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Remillard, R.B.; Bunce, N.J. Linking dioxins to diabetes: Epidemiology and biologic plausibility. Environ. Health Perspect. 2002, 110, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Villard, P.H.; Caverni, S.; Baanannou, A.; Khalil, A.; Martin, P.G.; Penel, C.; Pineau, T.; Seree, E.; Barra, Y. PPARα transcriptionally induces AhR expression in Caco-2, but represses AhR pro-inflammatory effects. Biochem. Biophys. Res. Commun. 2007, 364, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Fallone, F.; Villard, P.H.; Decome, L.; Seree, E.; Meo, M.; Chacon, C.; Durand, A.; Barra, Y.; Lacarelle, B. PPARα activation potentiates AhR-induced CYP1A1 expression. Toxicology 2005, 216, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Tomita, T. Thiazolidinediones are potent inducers of fibroblast growth factor 21 expression in the liver. Biol. Pharm. Bull. 2011, 34, 1120–1121. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.; Xu, A. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Owen, B.M.; Ding, X.; Morgan, D.A.; Coate, K.C.; Bookout, A.L.; Rahmouni, K.; Kliewer, S.A.; Mangelsdorf, D.J. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 2014, 20, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; de Groot, M.H.; Owen, B.M.; Lee, S.; Gautron, L.; Lawrence, H.L.; Ding, X.; Elmquist, J.K.; Takahashi, J.S.; Mangelsdorf, D.J.; et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 2013, 19, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Regnier, M.; Fouche, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Owen, B.M.; Song, P.; Hernandez, G.; Zhang, Y.; Zhou, Y.; Scott, W.T.; Paratala, B.; Turner, T.; Smith, A.; et al. FGF21 Regulates Sweet and Alcohol Preference. Cell Metab. 2016, 23, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zechner, C.; Hernandez, G.; Canovas, J.; Xie, Y.; Sondhi, V.; Wagner, M.; Stadlbauer, V.; Horvath, A.; Leber, B.; et al. The Hormone FGF21 Stimulates Water Drinking in Response to Ketogenic Diet and Alcohol. Cell Metab. 2018, 27, 1338–1347.e4. [Google Scholar] [CrossRef] [PubMed]
- Filbrandt, C.R.; Wu, Z.; Zlokovic, B.; Opanashuk, L.; Gasiewicz, T.A. Presence and functional activity of the aryl hydrocarbon receptor in isolated murine cerebral vascular endothelial cells and astrocytes. Neurotoxicology 2004, 25, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Hartz, A.M.; Potin, S.; Coumoul, X.; Yousif, S.; Scherrmann, J.M.; Bauer, B.; Decleves, X. Aryl hydrocarbon receptor-dependent upregulation of Cyp1b1 by TCDD and diesel exhaust particles in rat brain microvessels. Fluids Barriers CNS 2011, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Powell-Coffman, J.A. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev. Biol. 2004, 270, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Powell-Coffman, J.A.; Jin, Y. The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Development 2004, 131, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Howard, C.V.; Strahle, U.; Cossins, A. Neurodevelopmental defects in zebrafish (Danio rerio) at environmentally relevant dioxin (TCDD) concentrations. Toxicol. Sci. 2003, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, J.M.; Stockton, P.S.; Sieber, S.; Foley, J.; Portier, C.J. AhR-mediated gene expression in the developing mouse telencephalon. Reprod. Toxicol. 2009, 28, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.A.; Gasiewicz, T.A.; Opanashuk, L.A. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: Implications for development and dioxin neurotoxicity. Toxicol. Sci. 2005, 83, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Ding, Y.; Tohyama, C. AhR signaling activation disrupts migration and dendritic growth of olfactory interneurons in the developing mouse. Sci. Rep. 2016, 6, 26386. [Google Scholar] [CrossRef] [PubMed]
- Latchney, S.E.; Hein, A.M.; O’Banion, M.K.; DiCicco-Bloom, E.; Opanashuk, L.A. Deletion or activation of the aryl hydrocarbon receptor alters adult hippocampal neurogenesis and contextual fear memory. J. Neurochem. 2013, 125, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhai, Z.; Powell-Coffman, J.A. The Caenorhabditis elegans AHR-1 transcription complex controls expression of soluble guanylate cyclase genes in the URX neurons and regulates aggregation behavior. Dev. Biol. 2006, 298, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Pocivavsek, A.; Wu, H.Q.; Elmer, G.I.; Bruno, J.P.; Schwarcz, R. Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur. J. Neurosci. 2012, 35, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hawkins, B.T.; Miller, D.S. Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 2011, 25, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hawkins, B.T.; Miller, D.S. Activating PKC-β1 at the blood-brain barrier reverses induction of P-glycoprotein activity by dioxin and restores drug delivery to the CNS. J. Cereb. Blood Flow Metab. 2011, 31, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, Q.; Ma, Y.; Li, L.; Yu, K.; Zhang, Z.; Chen, G.; Li, X.; Xiao, W.; Xu, P.; et al. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int. J. Biol. Sci. 2018, 14, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Andrysik, Z.; Prochazkova, J.; Kabatkova, M.; Umannova, L.; Simeckova, P.; Kohoutek, J.; Kozubik, A.; Machala, M.; Vondracek, J. Aryl hydrocarbon receptor-mediated disruption of contact inhibition is associated with connexin43 downregulation and inhibition of gap junctional intercellular communication. Arch. Toxicol. 2013, 87, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Ezan, P.; Andre, P.; Cisternino, S.; Saubamea, B.; Boulay, A.C.; Doutremer, S.; Thomas, M.A.; Quenech’du, N.; Giaume, C.; Cohen-Salmon, M. Deletion of astroglial connexins weakens the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Benson, J.M.; Shepherd, D.M. Dietary ligands of the aryl hydrocarbon receptor induce anti-inflammatory and immunoregulatory effects on murine dendritic cells. Toxicol. Sci. 2011, 124, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y.; et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Nam, K.N.; Son, M.S.; Kang, H.; Hong, J.W.; Kim, J.W.; Lee, E.H. Indirubin-3′-oxime inhibits inflammatory activation of rat brain microglia. Neurosci. Lett. 2011, 487, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, D. Activation of pregnane X receptor by pregnenolone 16 α-carbonitrile prevents high-fat diet-induced obesity in AKR/J mice. PLoS ONE 2012, 7, e38734. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Negishi, M.; Kodama, S. The roles of nuclear receptors CAR and PXR in hepatic energy metabolism. Drug Metab. Pharmacokinet. 2008, 23, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Migliorati, M.; Barbanti, M.; Cipriani, S.; Palladino, G.; Distrutti, E.; Renga, B.; Fiorucci, S. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem. Pharmacol. 2010, 80, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Yasuda, K.; Lamba, J.K.; Assem, M.; Davila, J.; Strom, S.; Schuetz, E.G. PXR (NR1I2): Splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol. Appl. Pharmacol. 2004, 199, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Fukuen, S.; Fukuda, T.; Matsuda, H.; Sumida, A.; Yamamoto, I.; Inaba, T.; Azuma, J. Identification of the novel splicing variants for the hPXR in human livers. Biochem. Biophys. Res. Commun. 2002, 298, 433–438. [Google Scholar] [CrossRef]
- Gray, M.A.; Pollock, C.B.; Schook, L.B.; Squires, E.J. Characterization of porcine pregnane X receptor, farnesoid X receptor and their splice variants. Exp. Biol. Med. 2010, 235, 718–736. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Maglich, J.M.; Stoltz, C.M.; Goodwin, B.; Hawkins-Brown, D.; Moore, J.T.; Kliewer, S.A. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol. Pharmacol. 2002, 62, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Chrencik, J.E.; Orans, J.; Moore, L.B.; Xue, Y.; Peng, L.; Collins, J.L.; Wisely, G.B.; Lambert, M.H.; Kliewer, S.A.; Redinbo, M.R. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol. Endocrinol. 2005, 19, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Javitt, N.B. Cholestasis in rats induced by taurolithocholate. Nature 1966, 210, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. Prevention by catatoxic steroids of lithocholic acid-induced biliary concrements in the rat. Proc. Soc. Exp. Biol. Med. 1972, 141, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Dring, M.M.; Goulding, C.A.; Trimble, V.I.; Keegan, D.; Ryan, A.W.; Brophy, K.M.; Smyth, C.M.; Keeling, P.W.; O’Donoghue, D.; O’Sullivan, M.; et al. The pregnane X receptor locus is associated with susceptibility to inflammatory bowel disease. Gastroenterology 2006, 130, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Zhao, A.; Erickson, S.L.; Mukherjee, S.; Lau, A.J.; Alston, L.; Chang, T.K.; Mani, S.; Hirota, S.A. Pregnane X Receptor Activation Attenuates Inflammation-Associated Intestinal Epithelial Barrier Dysfunction by Inhibiting Cytokine-Induced Myosin Light-Chain Kinase Expression and c-Jun N-Terminal Kinase 1/2 Activation. J. Pharmacol. Exp. Ther. 2016, 359, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Terc, J.; Hansen, A.; Alston, L.; Hirota, S.A. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur. J. Pharm. Sci. 2014, 55, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Renga, B.; Palladino, G.; Claudio, D.; Ricci, P.; Distrutti, E.; Barbanti, M.; Baldelli, F.; Fiorucci, S. Inhibition of NF-κB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur. J. Pharmacol. 2011, 668, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Dou, W.; Zhang, J.; Zhang, E.; Sun, A.; Ding, L.; Chou, G.; Wang, Z.; Mani, S. Chrysin ameliorates chemically induced colitis in the mouse through modulation of a PXR/NF-κB signaling pathway. J. Pharmacol. Exp. Ther. 2013, 345, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, B.; Lakhal, L.; Payrastre, L.; Ghosh, C.; Pascussi, J.M.; Gangarossa, G.; Marchi, N. Pregnane X Receptor Deletion Modifies Recognition Memory and Electroencephalographic Activity. Neuroscience 2018, 370, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Yang, X.; Hartz, A.M.; Olson, E.R.; Zhao, R.; Kalvass, J.C.; Pollack, G.M.; Miller, D.S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006, 70, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Pascussi, J.M.; Busson-Le Coniat, M.; Maurel, P.; Vilarem, M.J. Transcriptional analysis of the orphan nuclear receptor constitutive androstane receptor (NR1I3) gene promoter: Identification of a distal glucocorticoid response element. Mol. Endocrinol. 2003, 17, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.D.; Hollingshead, B.D.; Omiecinski, C.J.; Perdew, G.H. Aryl-hydrocarbon receptor activation regulates constitutive androstane receptor levels in murine and human liver. Hepatology 2007, 46, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lichti, K.; Kim, I.; Gonzalez, F.J.; Staudinger, J.L. Regulation of constitutive androstane receptor and its target genes by fasting, cAMP, hepatocyte nuclear factor α, and the coactivator peroxisome proliferator-activated receptor γ coactivator-1α. J. Biol. Chem. 2006, 281, 26540–26551. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, S.; Sobhany, M.; Moore, R.; Perera, L.; Pedersen, L.; Sueyoshi, T.; Negishi, M. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal. 2013, 6, ra31. [Google Scholar] [CrossRef] [PubMed]
- Lundin, A.; Bok, C.M.; Aronsson, L.; Bjorkholm, B.; Gustafsson, J.A.; Pott, S.; Arulampalam, V.; Hibberd, M.; Rafter, J.; Pettersson, S. Gut flora, Toll-like receptors and nuclear receptors: A tripartite communication that tunes innate immunity in large intestine. Cell. Microbiol. 2008, 10, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Assenat, E.; Gerbal-Chaloin, S.; Larrey, D.; Saric, J.; Fabre, J.M.; Maurel, P.; Vilarem, M.J.; Pascussi, J.M. Interleukin 1β inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology 2004, 40, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Kakizaki, S.; Yoshinari, K.; Negishi, M. Estrogen activation of the nuclear orphan receptor CAR (constitutive active receptor) in induction of the mouse Cyp2b10 gene. Mol. Endocrinol. 2000, 14, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Robbins, D.; Chen, T. Modulation of xenobiotic receptors by steroids. Molecules 2013, 18, 7389–7406. [Google Scholar] [CrossRef] [PubMed]
- Bjorkholm, B.; Bok, C.M.; Lundin, A.; Rafter, J.; Hibberd, M.L.; Pettersson, S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS ONE 2009, 4, e6958. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, T.; Sueyoshi, T.; Zelko, I.; Moore, R.; Washburn, K.; Negishi, M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell. Biol. 1999, 19, 6318–6322. [Google Scholar] [CrossRef] [PubMed]
- Honkakoski, P.; Zelko, I.; Sueyoshi, T.; Negishi, M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell. Biol. 1998, 18, 5652–5658. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, S.S.; Ramsden, R.; Stoner, M.A.; Verlinde, C.; Hassett, C.; Omiecinski, C.J. Alternatively spliced isoforms of the human constitutive androstane receptor. Nucleic Acids Res. 2003, 31, 3194–3207. [Google Scholar] [CrossRef] [PubMed]
- Koyano, S.; Kurose, K.; Saito, Y.; Ozawa, S.; Hasegawa, R.; Komamura, K.; Ueno, K.; Kamakura, S.; Kitakaze, M.; Nakajima, T.; et al. Functional characterization of four naturally occurring variants of human pregnane X receptor (PXR): One variant causes dramatic loss of both DNA binding activity and the transactivation of the CYP3A4 promoter/enhancer region. Drug Metab. Dispos. 2004, 32, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Lamba, V.; Yasuda, K.; Lin, Y.S.; Assem, M.; Thompson, E.; Strom, S.; Schuetz, E. Expression of constitutive androstane receptor splice variants in human tissues and their functional consequences. J. Pharmacol. Exp. Ther. 2004, 311, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Yasuda, K.; Assem, M.; Cline, C.; Barber, J.; Li, C.W.; Kholodovych, V.; Ai, N.; Chen, J.D.; Welsh, W.J.; et al. The major human pregnane X receptor (PXR) splice variant, PXR.2, exhibits significantly diminished ligand-activated transcriptional regulation. Drug Metab. Dispos. 2009, 37, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.D.; Redinbo, M.R. Xenobiotic-sensing nuclear receptors involved in drug metabolism: A structural perspective. Drug Metab. Rev. 2013, 45, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Lin, M.; Hollister, K.; Fan, M.; Termini, J.; Sherman, M.A.; Forman, B.M. A structural model of the constitutive androstane receptor defines novel interactions that mediate ligand-independent activity. Mol. Cell. Biol. 2002, 22, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Jyrkkarinne, J.; Windshugel, B.; Ronkko, T.; Tervo, A.J.; Kublbeck, J.; Lahtela-Kakkonen, M.; Sippl, W.; Poso, A.; Honkakoski, P. Insights into ligand-elicited activation of human constitutive androstane receptor based on novel agonists and three-dimensional quantitative structure-activity relationship. J. Med. Chem. 2008, 51, 7181–7192. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Kobayashi, K.; Moore, R.; Kawamoto, T.; Negishi, M. Identification of the nuclear receptor CAR:HSP90 complex in mouse liver and recruitment of protein phosphatase 2A in response to phenobarbital. FEBS Lett. 2003, 548, 17–20. [Google Scholar] [CrossRef]
- Chen, S.; Wang, K.; Wan, Y.J. Retinoids activate RXR/CAR-mediated pathway and induce CYP3A. Biochem. Pharmacol. 2010, 79, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Koike, C.; Moore, R.; Negishi, M. Localization of the nuclear receptor CAR at the cell membrane of mouse liver. FEBS Lett. 2005, 579, 6733–6736. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.B.; Parks, D.J.; Jones, S.A.; Bledsoe, R.K.; Consler, T.G.; Stimmel, J.B.; Goodwin, B.; Liddle, C.; Blanchard, S.G.; Willson, T.M.; et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J. Biol. Chem. 2000, 275, 15122–15127. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; D’Costa, D.J.; Robertson, G.R.; Liddle, C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol. 2002, 62, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Wortham, M.; Czerwinski, M.; He, L.; Parkinson, A.; Wan, Y.J. Expression of constitutive androstane receptor, hepatic nuclear factor 4 α, and P450 oxidoreductase genes determines interindividual variability in basal expression and activity of a broad scope of xenobiotic metabolism genes in the human liver. Drug Metab. Dispos. 2007, 35, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Faucette, S.; Sueyoshi, T.; Moore, R.; Ferguson, S.; Negishi, M.; LeCluyse, E.L. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J. Biol. Chem. 2003, 278, 14146–14152. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Hamadeh, H.K.; Webb, H.K.; Yamamoto, Y.; Sueyoshi, T.; Afshari, C.A.; Lehmann, J.M.; Negishi, M. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol. Pharmacol. 2002, 61, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; LeCluyse, E.L.; Negishi, M.; Goldstein, J.A. Regulation of human CYP2C9 by the constitutive androstane receptor: Discovery of a new distal binding site. Mol. Pharmacol. 2002, 62, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Gerbal-Chaloin, S.; Daujat, M.; Pascussi, J.M.; Pichard-Garcia, L.; Vilarem, M.J.; Maurel, P. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, J.; Kojima, H.; Ueda, A.; Kakizaki, S.; Yoshinari, K.; Gong, Q.H.; Owens, I.S.; Negishi, M.; Sueyoshi, T. The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology 2001, 33, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; He, J.; Zhai, Y.; Wada, T.; Xie, W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 2009, 284, 25984–25992. [Google Scholar] [CrossRef] [PubMed]
- Yarushkin, A.A.; Kachaylo, E.M.; Pustylnyak, V.O. The constitutive androstane receptor activator 4-[(4R,6R)-4,6-diphenyl-1,3-dioxan-2-yl]-N,N-dimethylaniline inhibits the gluconeogenic genes PEPCK and G6Pase through the suppression of HNF4α and FOXO1 transcriptional activity. Br. J. Pharmacol. 2013, 168, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Kachaylo, E.M.; Yarushkin, A.A.; Pustylnyak, V.O. Constitutive androstane receptor activation by 2,4,6-triphenyldioxane-1,3 suppresses the expression of the gluconeogenic genes. Eur. J. Pharmacol. 2012, 679, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Looser, R.; Kaufmann, M.; Blattler, S.M.; Rencurel, F.; Huang, W.; Moore, D.D.; Meyer, U.A. Regulatory cross-talk between drug metabolism and lipid homeostasis: Constitutive androstane receptor and pregnane X receptor increase Insig-1 expression. Mol. Pharmacol. 2008, 73, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Moore, R.; Negishi, M.; Sueyoshi, T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J. Biol. Chem. 2007, 282, 9768–9776. [Google Scholar] [CrossRef] [PubMed]
- Maglich, J.M.; Watson, J.; McMillen, P.J.; Goodwin, B.; Willson, T.M.; Moore, J.T. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J. Biol. Chem. 2004, 279, 19832–19838. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.M.; Flannigan, K.L.; Erickson, S.L.; Vicentini, F.A.; Zamponi, A.; Hirota, C.L.; Alston, L.; Altier, C.; Ghosh, S.; Rioux, K.P.; et al. Constitutive androstane receptor regulates the intestinal mucosal response to injury. Br. J. Pharmacol. 2017, 174, 1857–1871. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, B.; Gangarossa, G.; Mselli-Lakhal, L.; Rousset, M.C.; de Bock, F.; Lassere, F.; Ghosh, C.; Pascussi, J.M.; Janigro, D.; Marchi, N. Lack of CAR impacts neuronal function and cerebrovascular integrity in vivo. Exp. Neurol. 2016, 283, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. RXRα, PXR and CAR xenobiotic receptors mediate the apoptotic and neurotoxic actions of nonylphenol in mouse hippocampal cells. J. Steroid Biochem. Mol. Biol. 2016, 156, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.H.; Xia, J.; Sun, X.C.; Li, X.N.; Zhang, C.; Zhao, H.S.; Zhu, S.Y.; Li, J.L. A novel nuclear xenobiotic receptors (AhR/PXR/CAR)-mediated mechanism of DEHP-induced cerebellar toxicity in quails (Coturnix japonica) via disrupting CYP enzyme system homeostasis. Environ. Pollut. 2017, 226, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Son, G.H.; Kim, K. Circadian rhythm of adrenal glucocorticoid: Its regulation and clinical implications. Biochim. Biophys. Acta 2011, 1812, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Van Cauter, E.; Blackman, J.D.; Roland, D.; Spire, J.P.; Refetoff, S.; Polonsky, K.S. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J. Clin. Investig. 1991, 88, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Czeisler, C.A.; Klerman, E.B. Circadian and sleep-dependent regulation of hormone release in humans. Recent Prog. Horm. Res. 1999, 54, 97–130. [Google Scholar] [PubMed]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Thiboutot, D.; Jabara, S.; McAllister, J.M.; Sivarajah, A.; Gilliland, K.; Cong, Z.; Clawson, G. Human skin is a steroidogenic tissue: Steroidogenic enzymes and cofactors are expressed in epidermis, normal sebocytes, and an immortalized sebocyte cell line (SEB-1). J. Investig. Dermatol. 2003, 120, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Lechner, O.; Wiegers, G.J.; Oliveira-Dos-Santos, A.J.; Dietrich, H.; Recheis, H.; Waterman, M.; Boyd, R.; Wick, G. Glucocorticoid production in the murine thymus. Eur. J. Immunol. 2000, 30, 337–346. [Google Scholar] [CrossRef]
- Pazirandeh, A.; Xue, Y.; Rafter, I.; Sjovall, J.; Jondal, M.; Okret, S. Paracrine glucocorticoid activity produced by mouse thymic epithelial cells. FASEB J. 1999, 13, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Miyamori, I.; Yoneda, T.; Iki, K.; Hatakeyama, H.; Blair, I.A.; Hsieh, F.Y.; Takeda, R. Synthesis of corticosterone in the vascular wall. Endocrinology 1994, 135, 2283–2286. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, N.; Bianchi, P.; Gennari-Moser, C.; Kassahn, D.; Schoonjans, K.; Corazza, N.; Brunner, T. Local glucocorticoid production in the mouse lung is induced by immune cell stimulation. Allergy 2012, 67, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, C.E.; Zhou, M.Y.; Cozza, E.N.; Morita, H.; Eddleman, F.C.; Gomez-Sanchez, E.P. Corticosteroid synthesis in the central nervous system. Endocr. Res. 1996, 22, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Cima, I.; Noti, M.; Fuhrer, A.; Jakob, S.; Dubuquoy, L.; Schoonjans, K.; Brunner, T. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J. Exp. Med. 2006, 203, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Cima, I.; Corazza, N.; Dick, B.; Fuhrer, A.; Herren, S.; Jakob, S.; Ayuni, E.; Mueller, C.; Brunner, T. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J. Exp. Med. 2004, 200, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.J.; Ridlon, J.M. Glucocorticoids and gut bacteria: “The GALF Hypothesis” in the metagenomic era. Steroids 2017, 125, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; McQueen, A.; Chen, T.C.; Wang, J.C. Regulation of Glucose Homeostasis by Glucocorticoids. Adv. Exp. Med. Biol. 2015, 872, 99–126. [Google Scholar] [PubMed]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Vegiopoulos, A.; Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol. Cell. Endocrinol. 2007, 275, 43–61. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; De Kloet, E.R.; Rostene, W. Adrenal steroid receptors and actions in the nervous system. Physiol. Rev. 1986, 66, 1121–1188. [Google Scholar] [CrossRef] [PubMed]
- Spencer, R.L.; Young, E.A.; Choo, P.H.; McEwen, B.S. Adrenal steroid type I and type II receptor binding: Estimates of in vivo receptor number, occupancy, and activation with varying level of steroid. Brain Res. 1990, 514, 37–48. [Google Scholar] [CrossRef]
- Morimoto, M.; Morita, N.; Ozawa, H.; Yokoyama, K.; Kawata, M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: An immunohistochemical and in situ hybridization study. Neurosci. Res. 1996, 26, 235–269. [Google Scholar] [CrossRef]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Encio, I.J.; Detera-Wadleigh, S.D. The genomic structure of the human glucocorticoid receptor. J. Biol. Chem. 1991, 266, 7182–7188. [Google Scholar] [PubMed]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Beger, C.; Gerdes, K.; Lauten, M.; Tissing, W.J.; Fernandez-Munoz, I.; Schrappe, M.; Welte, K. Expression and structural analysis of glucocorticoid receptor isoform γ in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. Br. J. Haematol. 2003, 122, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Calhoun, W.J. Differential regulation of the transcriptional activity of the glucocorticoid receptor through site-specific phosphorylation. Biologics 2008, 2, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Duma, D.; Jewell, C.M.; Cidlowski, J.A. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J. Steroid Biochem. Mol. Biol. 2006, 102, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar] [CrossRef] [PubMed]
- Beato, M. Gene regulation by steroid hormones. Cell 1989, 56, 335–344. [Google Scholar] [CrossRef]
- Freedman, L.P. Anatomy of the steroid receptor zinc finger region. Endocr. Rev. 1992, 13, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Cidlowski, J.A. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann. N. Y. Acad. Sci. 2009, 1179, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Nasuhara, Y.; Stevens, D.A.; Barnes, P.J. Ligand-induced differentiation of glucocorticoid receptor (GR) trans-repression and transactivation: Preferential targetting of NF-κB and lack of I-κB involvement. Br. J. Pharmacol. 1999, 127, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R.I.; Gualberto, A.; Jewell, C.M.; Cidlowski, J.A.; Baldwin, A.S., Jr. Characterization of mechanisms involved in transrepression of NF-κ B by activated glucocorticoid receptors. Mol. Cell. Biol. 1995, 15, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Wahli, W. A trilogy of glucocorticoid receptor actions. Proc. Natl. Acad. Sci. USA 2016, 113, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, E.F.; Lamberts, S.W. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog. Horm. Res. 2004, 59, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lee, M.S.; Baik, S.M.; Cho, E.B.; Son, G.H.; Seong, J.Y.; Lee, K.H.; Kim, K. Nova-1 mediates glucocorticoid-induced inhibition of pre-mRNA splicing of gonadotropin-releasing hormone transcripts. J. Biol. Chem. 2009, 284, 12792–12800. [Google Scholar] [CrossRef] [PubMed]
- Park, O.H.; Do, E.; Kim, Y.K. A new function of glucocorticoid receptor: Regulation of mRNA stability. BMB Rep. 2015, 48, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Shah, R.R.; Cidlowski, J.A. Glucocorticoids modulate microRNA expression and processing during lymphocyte apoptosis. J. Biol. Chem. 2010, 285, 36698–36708. [Google Scholar] [CrossRef] [PubMed]
- Savory, J.G.; Prefontaine, G.G.; Lamprecht, C.; Liao, M.; Walther, R.F.; Lefebvre, Y.A.; Hache, R.J. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol. Cell. Biol. 2001, 21, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, J.; Sauter, N.K.; Pearce, D. Steroid receptor heterodimerization demonstrated in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 12480–12484. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D.; Choudhury, Q.; Flower, R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000, 130, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Mulla, A.; Morris, J.F.; Christian, H.C.; Flower, R.J.; Buckingham, J.C. Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor, protein kinase C, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase. Endocrinology 2003, 144, 1164–1174. [Google Scholar] [PubMed]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joels, M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol. Cell. Endocrinol. 2012, 350, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights from Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Chrousos, G.P.; Kino, T. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol. Metab. 2010, 21, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, B.; Nakahata, Y.; Sahar, S.; Kaluzova, M.; Gauthier, D.; Pham, K.; Patel, N.; Hirayama, J.; Sassone-Corsi, P. Chromatin remodeling and circadian control: Master regulator CLOCK is an enzyme. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Chrousos, G.P.; Kino, T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: Potential physiological implications. FASEB J. 2009, 23, 1572–1583. [Google Scholar] [CrossRef] [PubMed]
- Balsalobre, A.; Brown, S.A.; Marcacci, L.; Tronche, F.; Kellendonk, C.; Reichardt, H.M.; Schutz, G.; Schibler, U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 2000, 289, 2344–2347. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr. Dev. 2013, 24, 41–56. [Google Scholar] [PubMed]
- Wiley, J.W.; Higgins, G.A.; Athey, B.D. Stress and glucocorticoid receptor transcriptional programming in time and space: Implications for the brain-gut axis. Neurogastroenterol. Motil. 2016, 28, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M.; Pan, X. Clock genes, intestinal transport and plasma lipid homeostasis. Trends Endocrinol. Metab. 2009, 20, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Hoogerwerf, W.A.; Hellmich, H.L.; Cornelissen, G.; Halberg, F.; Shahinian, V.B.; Bostwick, J.; Savidge, T.C.; Cassone, V.M. Clock gene expression in the murine gastrointestinal tract: Endogenous rhythmicity and effects of a feeding regimen. Gastroenterology 2007, 133, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Sladek, M.; Rybova, M.; Jindrakova, Z.; Zemanova, Z.; Polidarova, L.; Mrnka, L.; O’Neill, J.; Pacha, J.; Sumova, A. Insight into the circadian clock within rat colonic epithelial cells. Gastroenterology 2007, 133, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Hussain, M.M. Clock is important for food and circadian regulation of macronutrient absorption in mice. J. Lipid Res. 2009, 50, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zeevi, D.; Levy, M.; Zilberman-Schapira, G.; Suez, J.; Tengeler, A.C.; Abramson, L.; Katz, M.N.; Korem, T.; Zmora, N.; et al. Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 2014, 159, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; Gibbons, S.M.; Martinez, K.; Hutchison, A.L.; Huang, E.Y.; Cham, C.M.; Pierre, J.F.; Heneghan, A.F.; Nadimpalli, A.; Hubert, N.; et al. Effects of diurnal variation of gut microbes and high-fat feeding on host circadian clock function and metabolism. Cell Host Microbe 2015, 17, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Korem, T.; Dohnalova, L.; Shapiro, H.; Jaitin, D.A.; David, E.; Winter, D.R.; Gury-BenAri, M.; Tatirovsky, E.; et al. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell 2016, 167, 1495–1510.e12. [Google Scholar] [CrossRef] [PubMed]
- Imai, E.; Stromstedt, P.E.; Quinn, P.G.; Carlstedt-Duke, J.; Gustafsson, J.A.; Granner, D.K. Characterization of a complex glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. Mol. Cell. Biol. 1990, 10, 4712–4719. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Seitz, S.; Baschant, U.; Schilling, A.F.; Illing, A.; Stride, B.; Kirilov, M.; Mandic, V.; Takacz, A.; Schmidt-Ullrich, R.; et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010, 11, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Yang, S.; Zhang, T.; Sun, H.; Wang, Y.; Xue, H.; Zhou, D. Hypoxia enhances glucocorticoid-induced apoptosis and cell cycle arrest via the PI3K/Akt signaling pathway in osteoblastic cells. J. Bone Miner. Metab. 2015, 33, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Zhao, H.; Kitaura, H.; Bhattacharyya, S.; Brewer, J.A.; Muglia, L.J.; Ross, F.P.; Teitelbaum, S.L. Glucocorticoids suppress bone formation via the osteoclast. J. Clin. Investig. 2006, 116, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta 2014, 1842, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Chinenov, Y.; Gupte, R.; Rogatsky, I. Nuclear receptors in inflammation control: Repression by GR and beyond. Mol. Cell. Endocrinol. 2013, 380, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Busillo, J.M.; Cidlowski, J.A. The five Rs of glucocorticoid action during inflammation: Ready, reinforce, repress, resolve, and restore. Trends Endocrinol. Metab. 2013, 24, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Faubion, W.A., Jr.; Loftus, E.V., Jr.; Harmsen, W.S.; Zinsmeister, A.R.; Sandborn, W.J. The natural history of corticosteroid therapy for inflammatory bowel disease: A population-based study. Gastroenterology 2001, 121, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.P.; Muglia, L.J. Effects of genetically altered brain glucocorticoid receptor action on behavior and adrenal axis regulation in mice. Front. Neuroendocrinol. 2006, 27, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, A.; Tuckermann, J.P. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: Lessons from conditional knockout mice. Mol. Cell. Endocrinol. 2007, 275, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Gallant, C.; Kenny, P. Oral glucocorticoids and their complications. A review. J. Am. Acad. Dermatol. 1986, 14, 161–177. [Google Scholar] [CrossRef]
- Heimdal, K.; Hirschberg, H.; Slettebo, H.; Watne, K.; Nome, O. High incidence of serious side effects of high-dose dexamethasone treatment in patients with epidural spinal cord compression. J. Neurooncol. 1992, 12, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Cryan, J.; Shanahan, F.; Keeling, P.W.; Quigley, E.M. IBS: An epigenetic perspective. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Elsenbruch, S. Abdominal pain in Irritable Bowel Syndrome: A review of putative psychological, neural and neuro-immune mechanisms. Brain Behav. Immun. 2011, 25, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A. The neurobiology of stress and gastrointestinal disease. Gut 2000, 47, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J.; Pillemer, S.R.; Kalogeras, K.T.; Cash, J.M.; Michelson, D.; Kling, M.A.; Sternberg, E.M.; Gold, P.W.; Chrousos, G.P.; Wilder, R.L. Hypothalamic-pituitary-adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994, 37, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.O.; Vlachoyiannopoulos, P.G.; Skopouli, F.N.; Tzioufas, A.G.; Moutsopoulos, H.M. Hypofunction of the stress axis in Sjogren’s syndrome. J. Rheumatol. 1998, 25, 1508–1514. [Google Scholar] [PubMed]
- Buske-Kirschbaum, A.; Jobst, S.; Wustmans, A.; Kirschbaum, C.; Rauh, W.; Hellhammer, D. Attenuated free cortisol response to psychosocial stress in children with atopic dermatitis. Psychosom. Med. 1997, 59, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Nanthakumar, N.N.; Meng, D.; Newburg, D.S. Glucocorticoids and microbiota regulate ontogeny of intestinal fucosyltransferase 2 requisite for gut homeostasis. Glycobiology 2013, 23, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.D.; Foller, M.; Rexhepaj, R.; Pathare, G.; Minnich, K.; Tuckermann, J.P.; Lang, F.; Reichardt, H.M. Glucocorticoids enhance intestinal glucose uptake via the dimerized glucocorticoid receptor in enterocytes. Endocrinology 2012, 153, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Thiesen, A.; Wild, G.E.; Tappenden, K.A.; Drozdowski, L.; Keelan, M.; Thomson, B.K.; McBurney, M.I.; Clandinin, M.T.; Thomson, A.B. The locally acting glucocorticosteroid budesonide enhances intestinal sugar uptake following intestinal resection in rats. Gut 2003, 52, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Boivin, M.A.; Ye, D.; Kennedy, J.C.; Al-Sadi, R.; Shepela, C.; Ma, T.Y. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G590–G598. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, T.; Williams, G.; Petit, E.; Borowsky, M.; Walker, W.A. Hydrocortisone induces changes in gene expression and differentiation in immature human enterocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G425–G432. [Google Scholar] [CrossRef] [PubMed]
- Filaretova, L.P.; Filaretov, A.A.; Makara, G.B. Corticosterone increase inhibits stress-induced gastric erosions in rats. Am. J. Physiol. 1998, 274, G1024–G1030. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Mitra, R.; Shankaranarayana Rao, B.S.; Chattarji, S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. 2002, 22, 6810–6818. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.G.; Davies, H.A.; Sandi, C.; Kraev, I.V.; Rogachevsky, V.V.; Peddie, C.J.; Rodriguez, J.J.; Cordero, M.I.; Donohue, H.S.; Gabbott, P.L.; et al. Stress suppresses and learning induces plasticity in CA3 of rat hippocampus: A three-dimensional ultrastructural study of thorny excrescences and their postsynaptic densities. Neuroscience 2005, 131, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Gould, E.; McEwen, B.S. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992, 588, 341–345. [Google Scholar] [CrossRef]
- Samarasinghe, R.A.; Di Maio, R.; Volonte, D.; Galbiati, F.; Lewis, M.; Romero, G.; DeFranco, D.B. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, 16657–16662. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Arango-Lievano, M.; Lambert, W.M.; Bath, K.G.; Garabedian, M.J.; Chao, M.V.; Jeanneteau, F. Neurotrophic-priming of glucocorticoid receptor signaling is essential for neuronal plasticity to stress and antidepressant treatment. Proc. Natl. Acad. Sci. USA 2015, 112, 15737–15742. [Google Scholar] [CrossRef] [PubMed]
- Blandino, P., Jr.; Barnum, C.J.; Deak, T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1β responses to stress. J. Neuroimmunol. 2006, 173, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Emmetsberger, J.; Tsirka, S.E. Microglial inhibitory factor (MIF/TKP) mitigates secondary damage following spinal cord injury. Neurobiol. Dis. 2012, 47, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.K.; DeVries, A.C.; Kigerl, K.A.; Dahlman, J.M.; Popovich, P.G. Stress exacerbates neuropathic pain via glucocorticoid and NMDA receptor activation. Brain Behav. Immun. 2009, 23, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lim, G.; Zeng, Q.; Sung, B.; Yang, L.; Mao, J. Central glucocorticoid receptors modulate the expression and function of spinal NMDA receptors after peripheral nerve injury. J. Neurosci. 2005, 25, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Sandi, C.; Merino, J.J.; Cordero, M.I.; Touyarot, K.; Venero, C. Effects of chronic stress on contextual fear conditioning and the hippocampal expression of the neural cell adhesion molecule, its polysialylation, and L1. Neuroscience 2001, 102, 329–339. [Google Scholar] [CrossRef]
- Dubovsky, A.N.; Arvikar, S.; Stern, T.A.; Axelrod, L. The neuropsychiatric complications of glucocorticoid use: Steroid psychosis revisited. Psychosomatics 2012, 53, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Diorio, J.; Tannenbaum, B.; Caldji, C.; Francis, D.; Freedman, A.; Sharma, S.; Pearson, D.; Plotsky, P.M.; Meaney, M.J. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science 1997, 277, 1659–1662. [Google Scholar] [CrossRef] [PubMed]
- Caldji, C.; Tannenbaum, B.; Sharma, S.; Francis, D.; Plotsky, P.M.; Meaney, M.J. Maternal care during infancy regulates the development of neural systems mediating the expression of fearfulness in the rat. Proc. Natl. Acad. Sci. USA 1998, 95, 5335–5340. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Scott, L.V. Anatomy of melancholia: Focus on hypothalamic-pituitary-adrenal axis overactivity and the role of vasopressin. J. Anat. 2005, 207, 259–264. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, C.S.; Vermetten, E.; Geuze, E.; Kavelaars, A.; Heijnen, C.J.; Westenberg, H.G. Assessment of HPA-axis function in posttraumatic stress disorder: Pharmacological and non-pharmacological challenge tests, a review. J. Psychiatr. Res. 2006, 40, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Schechter, D.S.; Moser, D.A.; Paoloni-Giacobino, A.; Stenz, L.; Gex-Fabry, M.; Aue, T.; Adouan, W.; Cordero, M.I.; Suardi, F.; Manini, A.; et al. Methylation of NR3C1 is related to maternal PTSD, parenting stress and maternal medial prefrontal cortical activity in response to child separation among mothers with histories of violence exposure. Front. Psychol. 2015, 6, 690. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.J.; Knable, M.B.; O’Grady, J.; Orthmann, J.; Weickert, C.S. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol. Psychiatry 2002, 7, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T. Influence of stressor-induced nervous system activation on the intestinal microbiota and the importance for immunomodulation. Adv. Exp. Med. Biol. 2014, 817, 255–276. [Google Scholar] [PubMed]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Bharwani, A.; Mian, M.F.; Foster, J.A.; Surette, M.G.; Bienenstock, J.; Forsythe, P. Structural & functional consequences of chronic psychosocial stress on the microbiome & host. Psychoneuroendocrinology 2016, 63, 217–227. [Google Scholar] [PubMed]
- De Palma, G.; Blennerhassett, P.; Lu, J.; Deng, Y.; Park, A.J.; Green, W.; Denou, E.; Silva, M.A.; Santacruz, A.; Sanz, Y.; et al. Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat. Commun. 2015, 6, 7735. [Google Scholar] [CrossRef] [PubMed]
- De Palma, G.; Collins, S.M.; Bercik, P.; Verdu, E.F. The microbiota-gut-brain axis in gastrointestinal disorders: Stressed bugs, stressed brain or both? J. Physiol. 2014, 592, 2989–2997. [Google Scholar] [CrossRef] [PubMed]
- Moloney, R.D.; Desbonnet, L.; Clarke, G.; Dinan, T.G.; Cryan, J.F. The microbiome: Stress, health and disease. Mamm. Genome 2014, 25, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, L.C.; Zhang, C.; Wu, Y.; Hu, W.; Zhang, Q.; Wang, J.; Li, S.; Zhao, L. Gender-based differences in host behavior and gut microbiota composition in response to high fat diet and stress in a mouse model. Sci. Rep. 2017, 7, 10776. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.Y.; Inoue, T.; Leone, V.A.; Dalal, S.; Touw, K.; Wang, Y.; Musch, M.W.; Theriault, B.; Higuchi, K.; Donovan, S.; et al. Using corticosteroids to reshape the gut microbiome: Implications for inflammatory bowel diseases. Inflamm. Bowel Dis. 2015, 21, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Unsal, H.; Balkaya, M.; Unsal, C.; Biyik, H.; Basbulbul, G.; Poyrazoglu, E. The short-term effects of different doses of dexamethasone on the numbers of some bacteria in the ileum. Dig. Dis. Sci. 2008, 53, 1842–1845. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, P.; Kunze, W.A.; Bienenstock, J. On communication between gut microbes and the brain. Curr. Opin. Gastroenterol. 2012, 28, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Ait-Belgnaoui, A.; Durand, H.; Cartier, C.; Chaumaz, G.; Eutamene, H.; Ferrier, L.; Houdeau, E.; Fioramonti, J.; Bueno, L.; Theodorou, V. Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 2012, 37, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Burokas, A.; Arboleya, S.; Moloney, R.D.; Peterson, V.L.; Murphy, K.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Targeting the Microbiota-Gut-Brain Axis: Prebiotics Have Anxiolytic and Antidepressant-like Effects and Reverse the Impact of Chronic Stress in Mice. Biol. Psychiatry 2017, 82, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Gareau, M.G.; Jury, J.; MacQueen, G.; Sherman, P.M.; Perdue, M.H. Probiotic treatment of rat pups normalises corticosterone release and ameliorates colonic dysfunction induced by maternal separation. Gut 2007, 56, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, U.A.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.M.; Gomez-Sanchez, C.E. The ubiquitous mineralocorticoid receptor: Clinical implications. Curr. Hypertens. Rep. 2012, 14, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; Patel, P.D.; Akil, H.; Watson, S.J. Localization and regulation of glucocorticoid and mineralocorticoid receptor messenger RNAs in the hippocampal formation of the rat. Mol. Endocrinol. 1989, 3, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Vallee, S.M.; Grillo, C.A.; Gonzalez, S.; Cosen-Binker, L.; de Kloet, E.R.; McEwen, B.S.; De Nicola, A.F. Further studies in deoxycorticosterone acetate treated rats: Brain content of mineralocorticoid and glucocorticoid receptors and effect of steroid antagonists on salt intake. Neuroendocrinology 1995, 61, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.B. Sex difference in glucocorticoid binding in rat pituitary is estrogen dependent. Life Sci. 1990, 46, 1399–1406. [Google Scholar] [CrossRef]
- Castren, M.; Patchev, V.K.; Almeida, O.F.; Holsboer, F.; Trapp, T.; Castren, E. Regulation of rat mineralocorticoid receptor expression in neurons by progesterone. Endocrinology 1995, 136, 3800–3806. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Le Menuet, D.; Lombes, M. Characterization of the human mineralocorticoid receptor gene 5′-regulatory region: Evidence for differential hormonal regulation of two alternative promoters via nonclassical mechanisms. Mol. Endocrinol. 1996, 10, 1549–1560. [Google Scholar] [PubMed]
- Wang, J.; Li, X.; Ke, Y.; Lu, Y.; Wang, F.; Fan, N.; Sun, H.; Zhang, H.; Liu, R.; Yang, J.; et al. GPR48 increases mineralocorticoid receptor gene expression. J. Am. Soc. Nephrol. 2012, 23, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.P.; Patel, P.D.; Thompson, R.C.; Akil, H.; Watson, S.J. 5′-Heterogeneity of the mineralocorticoid receptor messenger ribonucleic acid: Differential expression and regulation of splice variants within the rat hippocampus. Endocrinology 1993, 133, 2344–2350. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Keightley, M.C.; Kotelevtsev, Y.; Conway, G.S.; Soubrier, F.; Fuller, P.J. Human mineralocorticoid receptor genomic structure and identification of expressed isoforms. J. Biol. Chem. 1995, 270, 21016–21020. [Google Scholar] [CrossRef] [PubMed]
- Hesen, W.; Karst, H.; Meijer, O.; Cole, T.J.; Schmid, W.; de Kloet, E.R.; Schutz, G.; Joels, M. Hippocampal cell responses in mice with a targeted glucocorticoid receptor gene disruption. J. Neurosci. 1996, 16, 6766–6774. [Google Scholar] [CrossRef] [PubMed]
- Karst, H.; Wadman, W.J.; Joels, M. Corticosteroid receptor-dependent modulation of calcium currents in rat hippocampal CA1 neurons. Brain Res. 1994, 649, 234–242. [Google Scholar] [CrossRef]
- Kerr, D.S.; Campbell, L.W.; Thibault, O.; Landfield, P.W. Hippocampal glucocorticoid receptor activation enhances voltage-dependent Ca2+ conductances: Relevance to brain aging. Proc. Natl. Acad. Sci. USA 1992, 89, 8527–8531. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Feve, B.; Claes, A.; Viengchareun, S.; Lombes, M.; Zennaro, M.C. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007, 21, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Marzolla, V.; Armani, A.; Zennaro, M.C.; Cinti, F.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol. Cell. Endocrinol. 2012, 350, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Central regulation of blood pressure by the mineralocorticoid receptor. Mol. Cell. Endocrinol. 2012, 350, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.P.; Holmes, M.C.; de Kloet, E.R.; Chapman, K.E.; Seckl, J.R. Mineralocorticoid and glucocorticoid receptor balance in control of HPA axis and behaviour. Psychoneuroendocrinology 2013, 38, 648–658. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joels, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.M.; Storring, J.M.; Kushwaha, N.; Albert, P.R. Heterodimerization of mineralocorticoid and glucocorticoid receptors at a novel negative response element of the 5-HT1A receptor gene. J. Biol. Chem. 2001, 276, 14299–14307. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, D.; Gresko, N.; Carrel, M.; Loffing-Cueni, D.; Habermehl, D.; Gomez-Sanchez, C.; Rossier, B.C.; Loffing, J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: Differential effect of corticosteroids along the distal tubule. Am. J. Physiol. Renal Physiol. 2010, 299, F1473–F1485. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Brennan, F.E.; Burgess, J.S. Acute differential regulation by corticosteroids of epithelial sodium channel subunit and Nedd4 mRNA levels in the distal colon. Pflugers Arch. 2000, 441, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Lastra-Gonzalez, G.; Manrique-Acevedo, C.; Sowers, J.R. The role of aldosterone in cardiovascular disease in people with diabetes and hypertension: An update. Curr. Diabetes Rep. 2008, 8, 203–207. [Google Scholar] [CrossRef]
- Zennaro, M.C.; Caprio, M.; Feve, B. Mineralocorticoid receptors in the metabolic syndrome. Trends Endocrinol. Metab. 2009, 20, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Vogt, B.; Burnier, M. Aldosterone and cardiovascular risk. Curr. Hypertens. Rep. 2009, 11, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Garg, R.; Adler, G.K. Mineralocorticoid receptor antagonists and the metabolic syndrome. Curr. Hypertens. Rep. 2010, 12, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Cheskis, B.J. Regulation of cell signalling cascades by steroid hormones. J. Cell Biochem. 2004, 93, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Gekle, M. New aspects of rapid aldosterone signaling. Mol. Cell. Endocrinol. 2009, 308, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Gekle, M. Non-classical actions of the mineralocorticoid receptor: Misuse of EGF receptors? Mol. Cell. Endocrinol. 2007, 277, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W. Non-genomic actions of aldosterone: Role in hypertension. Curr. Opin Nephrol. Hypertens. 2001, 10, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, A.; Ding, G.; Chen, R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am. J. Physiol. Renal Physiol. 2009, 296, F1323–F1333. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.W.; Schuster, C.; Gassner, B.; Freudinger, R.; Mildenberger, S.; Troppmair, J.; Gekle, M. Human epidermal growth factor receptor-1 expression renders Chinese hamster ovary cells sensitive to alternative aldosterone signaling. J. Biol. Chem. 2002, 277, 45892–45897. [Google Scholar] [CrossRef] [PubMed]
- Michea, L.; Delpiano, A.M.; Hitschfeld, C.; Lobos, L.; Lavandero, S.; Marusic, E.T. Eplerenone blocks nongenomic effects of aldosterone on the Na+/H+ exchanger, intracellular Ca2+ levels, and vasoconstriction in mesenteric resistance vessels. Endocrinology 2005, 146, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Krozowski, Z.S.; Funder, J.W. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc. Natl. Acad. Sci. USA 1983, 80, 6056–6060. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, H.D.; Van Koppen, C.; Van Ittersum, M.; De Kloet, E.R. Specificity of the adrenal steroid receptor system in rat hippocampus. Endocrinology 1982, 110, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Reul, J.M. Feedback action and tonic influence of corticosteroids on brain function: A concept arising from the heterogeneity of brain receptor systems. Psychoneuroendocrinology 1987, 12, 83–105. [Google Scholar] [CrossRef]
- Karst, H.; Berger, S.; Turiault, M.; Tronche, F.; Schutz, G.; Joels, M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA 2005, 102, 19204–19207. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. 11β-hydroxysteroid dehydrogenases: Changing glucocorticoid action. Curr. Opin. Pharmacol. 2004, 4, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Dou, W.; Lou, J.; Leng, Y.; Shen, J. Discovery of novel inhibitors of 11β-hydroxysteroid dehydrogenase type 1 by docking and pharmacophore modeling. Bioorg. Med. Chem. Lett. 2008, 18, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Kang, Y.M.; Yu, Y.; Wei, S.G.; Schmidt, T.J.; Johnson, A.K.; Felder, R.B. 11β-hydroxysteroid dehydrogenase type 2 activity in hypothalamic paraventricular nucleus modulates sympathetic excitation. Hypertension 2006, 48, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gomez-Sanchez, C.E.; Penman, A.; May, P.J.; Gomez-Sanchez, E. Expression of mineralocorticoid and glucocorticoid receptors in preautonomic neurons of the rat paraventricular nucleus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R328–R340. [Google Scholar] [CrossRef] [PubMed]
- Geerling, J.C.; Loewy, A.D. Aldosterone-sensitive neurons in the nucleus of the solitary tract: Bidirectional connections with the central nucleus of the amygdala. J. Comp. Neurol. 2006, 497, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Chantong, B.; Kratschmar, D.V.; Nashev, L.G.; Balazs, Z.; Odermatt, A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-κB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J. Neuroinflamm. 2012, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, D.M.; Lopez, J.F.; Morano, M.I.; Kwak, S.P.; Watson, S.J.; Akil, H. Alpha, β, and γ mineralocorticoid receptor messenger ribonucleic acid splice variants: Differential expression and rapid regulation in the developing hippocampus. Endocrinology 1998, 139, 3165–3177. [Google Scholar] [CrossRef] [PubMed]
- Munier, M.; Meduri, G.; Viengchareun, S.; Leclerc, P.; Le Menuet, D.; Lombes, M. Regulation of mineralocorticoid receptor expression during neuronal differentiation of murine embryonic stem cells. Endocrinology 2010, 151, 2244–2254. [Google Scholar] [CrossRef] [PubMed]
- Gass, P.; Kretz, O.; Wolfer, D.P.; Berger, S.; Tronche, F.; Reichardt, H.M.; Kellendonk, C.; Lipp, H.P.; Schmid, W.; Schutz, G. Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep. 2000, 1, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Brinks, V.; van der Mark, M.H.; de Kloet, E.R.; Oitzl, M.S. Differential MR/GR activation in mice results in emotional states beneficial or impairing for cognition. Neural Plast. 2007, 2007, 90163. [Google Scholar] [CrossRef] [PubMed]
- Sterlemann, V.; Ganea, K.; Liebl, C.; Harbich, D.; Alam, S.; Holsboer, F.; Muller, M.B.; Schmidt, M.V. Long-term behavioral and neuroendocrine alterations following chronic social stress in mice: Implications for stress-related disorders. Horm. Behav. 2008, 53, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.R.; Kamphuis, W.; Wang, S.; Wang, Q.; Lucassen, P.J.; Zhou, J.N.; Swaab, D.F. Aberrant stress hormone receptor balance in the human prefrontal cortex and hypothalamic paraventricular nucleus of depressed patients. Psychoneuroendocrinology 2013, 38, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Chen, H.T.; Chang, H.Y.; Yang, C.Y.; Hsiao, M.C.; Cheng, M.L.; Chen, J.C. Mineralocorticoid receptor antagonist spironolactone prevents chronic corticosterone induced depression-like behavior. Psychoneuroendocrinology 2013, 38, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Okuhara, D.Y.; Beck, S.G. Corticosteroids alter 5-hydroxytryptamine1A receptor-effector pathway in hippocampal subfield CA3 pyramidal cells. J. Pharmacol. Exp. Ther. 1998, 284, 1227–1233. [Google Scholar] [PubMed]
- Beck, S.G.; Choi, K.C.; List, T.J.; Okuhara, D.Y.; Birnsteil, S. Corticosterone alters 5-HT1A receptor-mediated hyperpolarization in area CA1 hippocampal pyramidal neurons. Neuropsychopharmacology 1996, 14, 27–33. [Google Scholar] [CrossRef]
- Anacker, C.; Zunszain, P.A.; Carvalho, L.A.; Pariante, C.M. The glucocorticoid receptor: Pivot of depression and of antidepressant treatment? Psychoneuroendocrinology 2011, 36, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Maletic, V.; Robinson, M.; Oakes, T.; Iyengar, S.; Ball, S.G.; Russell, J. Neurobiology of depression: An integrated view of key findings. Int. J. Clin. Pract. 2007, 61, 2030–2040. [Google Scholar] [CrossRef] [PubMed]
- Yagi, S.; Akaike, M.; Aihara, K.; Iwase, T.; Yoshida, S.; Sumitomo-Ueda, Y.; Ikeda, Y.; Ishikawa, K.; Matsumoto, T.; Sata, M. High plasma aldosterone concentration is a novel risk factor of cognitive impairment in patients with hypertension. Hypertens. Res. 2011, 34, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.L.; Hibberd, C.; Noble, J.; Seckl, J.R. The effect of chronic fluoxetine treatment on brain corticosteroid receptor mRNA expression and spatial memory in young and aged rats. Brain Res. Mol. Brain Res. 2002, 106, 117–123. [Google Scholar] [CrossRef]
- Tytherleigh, M.Y.; Vedhara, K.; Lightman, S.L. Mineralocorticoid and glucocorticoid receptors and their differential effects on memory performance in people with Addison’s disease. Psychoneuroendocrinology 2004, 29, 712–723. [Google Scholar] [CrossRef]
- Yau, J.L.; Olsson, T.; Morris, R.G.; Meaney, M.J.; Seckl, J.R. Glucocorticoids, hippocampal corticosteroid receptor gene expression and antidepressant treatment: Relationship with spatial learning in young and aged rats. Neuroscience 1995, 66, 571–581. [Google Scholar] [CrossRef]
- Yau, J.L.; Noble, J.; Hibberd, C.; Rowe, W.B.; Meaney, M.J.; Morris, R.G.; Seckl, J.R. Chronic treatment with the antidepressant amitriptyline prevents impairments in water maze learning in aging rats. J. Neurosci. 2002, 22, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Cornelisse, S.; Joels, M.; Smeets, T. A randomized trial on mineralocorticoid receptor blockade in men: Effects on stress responses, selective attention, and memory. Neuropsychopharmacology 2011, 36, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor α and β subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef] [PubMed]
- Riant, E.; Waget, A.; Cogo, H.; Arnal, J.F.; Burcelin, R.; Gourdy, P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 2009, 150, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.C. The emergence of the metabolic syndrome with menopause. J. Clin. Endocrinol. Metab. 2003, 88, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Mezei, G.; Nie, Y.; Rao, Y.; Choi, C.S.; Bechmann, I.; Leranth, C.; Toran-Allerand, D.; Priest, C.A.; Roberts, J.L.; et al. Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat. Med. 2007, 13, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wren, B.G. The benefits of oestrogen following menopause: Why hormone replacement therapy should be offered to postmenopausal women. Med. J. Aust. 2009, 190, 321–325. [Google Scholar] [PubMed]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, B.J.; Feigelson, H.S.; Flores, R.; Gail, M.H.; Xu, X.; Ravel, J.; Goedert, J.J. Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J. Clin. Endocrinol. Metab. 2014, 99, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Indias, I.; Sanchez-Alcoholado, L.; Sanchez-Garrido, M.A.; Martin-Nunez, G.M.; Perez-Jimenez, F.; Tena-Sempere, M.; Tinahones, F.J.; Queipo-Ortuno, M.I. Neonatal Androgen Exposure Causes Persistent Gut Microbiota Dysbiosis Related to Metabolic Disease in Adult Female Rats. Endocrinology 2016, 157, 4888–4898. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: A cross-sectional study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Dominianni, C.; Sinha, R.; Goedert, J.J.; Pei, Z.; Yang, L.; Hayes, R.B.; Ahn, J. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Burrows, M.; Khan, A.A.; Graham, L.; Volchkov, P.; Becker, L.; Antonopoulos, D.; Umesaki, Y.; Chervonsky, A.V. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013, 39, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Kisiela, M.; Skarka, A.; Ebert, B.; Maser, E. Hydroxysteroid dehydrogenases (HSDs) in bacteria: A bioinformatic perspective. J. Steroid Biochem. Mol. Biol. 2012, 129, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Raftogianis, R.; Creveling, C.; Weinshilboum, R.; Weisz, J. Estrogen metabolism by conjugation. J. Natl. Cancer Inst. Monogr. 2000, 2000, 113–124. [Google Scholar] [CrossRef]
- McBain, A.J.; Macfarlane, G.T. Ecological and physiological studies on large intestinal bacteria in relation to production of hydrolytic and reductive enzymes involved in formation of genotoxic metabolites. J. Med. Microbiol. 1998, 47, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Reddy, B.S.; Weisburger, J.H.; Wynder, E.L. Fecal bacterial β-glucuronidase: Control by diet. Science 1974, 183, 416–417. [Google Scholar] [CrossRef] [PubMed]
- Cenci, G.; Caldini, G.; Mastrandrea, V.; Votoni, M.R. Carbohydrate enriched diets and bacterial glycosidases in rat faeces. Microbios 1993, 76, 143–151. [Google Scholar] [PubMed]
- Eriyamremu, G.E.; Osagie, V.E.; Alufa, O.I.; Osaghae, M.O.; Oyibu, F.A. Early biochemical events in mice exposed to cycas and fed a Nigerian-like diet. Ann. Nutr. Metab. 1995, 39, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Goldin, B.R.; Gorbach, S.L. Alterations of the intestinal microflora by diet, oral antibiotics, and Lactobacillus: Decreased production of free amines from aromatic nitro compounds, azo dyes, and glucuronides. J. Natl. Cancer Inst. 1984, 73, 689–695. [Google Scholar] [PubMed]
- Martin, F.; Peltonen, J.; Laatikainen, T.; Pulkkinen, M.; Adlercreutz, H. Excretion of progesterone metabolites and estriol in faeces from pregnant women during ampicillin administration. J. Steroid Biochem. 1975, 6, 1339–1346. [Google Scholar] [CrossRef]
- Adlercreutz, H.; Martin, F.; Tikkanen, M.J.; Pulkkinen, M. Effect of ampicillin administration on the excretion of twelve oestrogens in pregnancy urine. Acta Endocrinol. 1975, 80, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Hamalainen, E.; Korpela, J.T.; Adlercreutz, H. Effect of oxytetracycline administration on intestinal metabolism of oestrogens and on plasma sex hormones in healthy men. Gut 1987, 28, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Interactions and competition within the microbial community of the human colon: Links between diet and health. Environ. Microbiol. 2007, 9, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Dabek, M.; McCrae, S.I.; Stevens, V.J.; Duncan, S.H.; Louis, P. Distribution of β-glucosidase and β-glucuronidase activity and of β-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol. Ecol. 2008, 66, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Kubota, Y.; Miyaoka, M.; Saitoh, T.; Mizuno, F.; Benno, Y. Comparison of four microbial enzymes in Clostridia and Bacteroides isolated from human feces. Microbiol. Immunol. 2002, 46, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Van Eldere, J.R.; De Pauw, G.; Eyssen, H.J. Steroid sulfatase activity in a Peptococcus niger strain from the human intestinal microflora. Appl. Environ. Microbiol. 1987, 53, 1655–1660. [Google Scholar] [PubMed]
- Frankenfeld, C.L.; Atkinson, C.; Wahala, K.; Lampe, J.W. Obesity prevalence in relation to gut microbial environments capable of producing equol or O-desmethylangolensin from the isoflavone daidzein. Eur. J. Clin. Nutr. 2014, 68, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, C.H.; Armstrong, A.; Clavijo, A.P.; Martin, B.R.; Barnes, S.; Weaver, C.M. Fecal bacterial community changes associated with isoflavone metabolites in postmenopausal women after soy bar consumption. PLoS ONE 2014, 9, e108924. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Tochiya, M.; Sasaki, Y.; Muranaka, K.; Yamakage, H.; Himeno, A.; Shimatsu, A.; Inaguma, A.; Ueno, T.; Uchiyama, S.; et al. Effects of natural S-equol supplements on overweight or obesity and metabolic syndrome in the Japanese, based on sex and equol status. Clin. Endocrinol. 2013, 78, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Velicer, C.M.; Heckbert, S.R.; Lampe, J.W.; Potter, J.D.; Robertson, C.A.; Taplin, S.H. Antibiotic use in relation to the risk of breast cancer. JAMA 2004, 291, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.X. Recurrent antibiotic exposure may promote cancer formation--Another step in understanding the role of the human microbiota? Eur. J. Cancer 2015, 51, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Muranaka, Y.; Fujimura, R.; Ishida, H.; Tazume, S.; Shimamura, T. Normalization of reproductive function in germfree mice following bacterial contamination. Exp. Anim. 1998, 47, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.A. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Sauerwein, H.; Pfaffl, M.; Hagen-Mann, K.; Malucelli, A.; Meyer, H.H. Expression of estrogen and androgen receptor in the bovine gastrointestinal tract. Dtsch. Tierarztl. Wochenschr. 1995, 102, 164–168. [Google Scholar] [PubMed]
- Pfaffl, M.W.; Lange, I.G.; Meyer, H.H. The gastrointestinal tract as target of steroid hormone action: Quantification of steroid receptor mRNA expression (AR, ERα, ERβ and PR) in 10 bovine gastrointestinal tract compartments by kinetic RT-PCR. J. Steroid Biochem. Mol. Biol. 2003, 84, 159–166. [Google Scholar] [CrossRef]
- Singh, S.; Sheppard, M.C.; Langman, M.J. Sex differences in the incidence of colorectal cancer: An exploration of oestrogen and progesterone receptors. Gut 1993, 34, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.F.; Jazaeri, A.A.; Shupnik, M.A.; Jazaeri, O.; Rice, L.W. Selective loss of estrogen receptor β in malignant human colon. Cancer Res. 2000, 60, 245–248. [Google Scholar] [PubMed]
- Hess, R.A.; Gist, D.H.; Bunick, D.; Lubahn, D.B.; Farrell, A.; Bahr, J.; Cooke, P.S.; Greene, G.L. Estrogen receptor (α and β) expression in the excurrent ducts of the adult male rat reproductive tract. J. Androl. 1997, 18, 602–611. [Google Scholar] [PubMed]
- Hileman, S.M.; Handa, R.J.; Jackson, G.L. Distribution of estrogen receptor-β messenger ribonucleic acid in the male sheep hypothalamus. Biol. Reprod. 1999, 60, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.W.; Hoskin, E.; Yudkovitz, J.; Pear, L.; Wilkinson, H.A.; Hayashi, S.; Pfaff, D.W.; Ogawa, S.; Rohrer, S.P.; Schaeffer, J.M.; et al. Immunolocalization of estrogen receptor β in the mouse brain: Comparison with estrogen receptor α. Endocrinology 2003, 144, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, N.; Nappi, R.E.; Drolet, G.; Labrie, C.; Rivest, S. Expression and neuropeptidergic characterization of estrogen receptors (ERα and ERβ) throughout the rat brain: Anatomical evidence of distinct roles of each subtype. J. Neurobiol. 1998, 36, 357–378. [Google Scholar] [CrossRef]
- Shughrue, P.; Scrimo, P.; Lane, M.; Askew, R.; Merchenthaler, I. The distribution of estrogen receptor-β mRNA in forebrain regions of the estrogen receptor-α knockout mouse. Endocrinology 1997, 138, 5649–5652. [Google Scholar] [CrossRef] [PubMed]
- Shughrue, P.J.; Komm, B.; Merchenthaler, I. The distribution of estrogen receptor-β mRNA in the rat hypothalamus. Steroids 1996, 61, 678–681. [Google Scholar] [CrossRef]
- Wang, J.M.; Liu, L.; Brinton, R.D. Estradiol-17β-induced human neural progenitor cell proliferation is mediated by an estrogen receptor β-phosphorylated extracellularly regulated kinase pathway. Endocrinology 2008, 149, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, C.; Ding, Z.L.; Ling, M.; Wang, T.; Wang, W.; Huang, B. 17β-Oestradiol promotes differentiation of human embryonic stem cells into dopamine neurons via cross-talk between insulin-like growth factors-1 and oestrogen receptor β. J. Cell. Mol. Med. 2017, 21, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Eng, V.; Taylor, J.; Lubahn, D.B.; Korach, K.S.; Pfaff, D.W. Roles of estrogen receptor-α gene expression in reproduction-related behaviors in female mice. Endocrinology 1998, 139, 5070–5081. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Korach, K.S. Oestrogen receptor knockout mice: Roles for oestrogen receptors α and β in reproductive tissues. Reproduction 2003, 125, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Herbison, A.E.; Pape, J.R. New evidence for estrogen receptors in gonadotropin-releasing hormone neurons. Front. Neuroendocrinol. 2001, 22, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Dorling, A.A.; Todman, M.G.; Korach, K.S.; Herbison, A.E. Critical role for estrogen receptor α in negative feedback regulation of gonadotropin-releasing hormone mRNA expression in the female mouse. Neuroendocrinology 2003, 78, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Heine, P.A.; Taylor, J.A.; Iwamoto, G.A.; Lubahn, D.B.; Cooke, P.S. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 12729–12734. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Koda, M.; Ando, F.; Niino, N.; Ohta, S.; Shimokata, H. Association of polymorphisms in the estrogen receptor α gene with body fat distribution. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.L.; Ibarra, M.J. Effects of ovariectomy on duodenal calcium transport in the rat: Altered ability to adapt to low-calcium diet. Proc. Soc. Exp. Biol. Med. 1987, 185, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Hope, W.G.; Bruns, M.E.; Thomas, M.L. Regulation of duodenal insulin-like growth factor I and active calcium transport by ovariectomy in female rats. Proc Soc. Exp. Biol. Med. 1992, 200, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Nedungadi, T.P.; Zhu, L.; Sobhani, N.; Irani, B.G.; Davis, K.E.; Zhang, X.; Zou, F.; Gent, L.M.; Hahner, L.D.; et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011, 14, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Krezel, W.; Dupont, S.; Krust, A.; Chambon, P.; Chapman, P.F. Increased anxiety and synaptic plasticity in estrogen receptor β -deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 12278–12282. [Google Scholar] [CrossRef] [PubMed]
- Imwalle, D.B.; Gustafsson, J.A.; Rissman, E.F. Lack of functional estrogen receptor β influences anxiety behavior and serotonin content in female mice. Physiol. Behav. 2005, 84, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Donner, N.; Handa, R.J. Estrogen receptor β regulates the expression of tryptophan-hydroxylase 2 mRNA within serotonergic neurons of the rat dorsal raphe nuclei. Neuroscience 2009, 163, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.A.; Fleischer, R.; Schaeffer, J.M.; Rohrer, S.P.; Hickey, G.J. 17 Beta-estradiol-induced antidepressant-like effect in the forced swim test is absent in estrogen receptor-β knockout (BERKO) mice. Psychopharmacology 2005, 179, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Walf, A.A.; Rhodes, M.E.; Frye, C.A. Antidepressant effects of ERβ-selective estrogen receptor modulators in the forced swim test. Pharmacol. Biochem. Behav. 2004, 78, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Walf, A.A.; Frye, C.A. ERβ-selective estrogen receptor modulators produce antianxiety behavior when administered systemically to ovariectomized rats. Neuropsychopharmacology 2005, 30, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.D.; Rovis, T.; Chung, W.C.; Handa, R.J. Novel actions of estrogen receptor-β on anxiety-related behaviors. Endocrinology 2005, 146, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Burgess, L.H.; Handa, R.J. Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor-mediated functions in female rats. Endocrinology 1992, 131, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.A.; Xu, C.; Dorsa, D.M. Differential transcriptional regulation of rat vasopressin gene expression by estrogen receptor α and β. Endocrinology 2000, 141, 4056–4064. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.J.; Suzuki, S.; Miller, L.K.; Handa, R.; Uht, R.M. Estrogen receptor (ER)β isoforms rather than ERα regulate corticotropin-releasing hormone promoter activity through an alternate pathway. J. Neurosci. 2004, 24, 10628–10635. [Google Scholar] [CrossRef] [PubMed]
- Siiteri, P.K.; Murai, J.T.; Hammond, G.L.; Nisker, J.A.; Raymoure, W.J.; Kuhn, R.W. The serum transport of steroid hormones. Recent Prog. Horm. Res. 1982, 38, 457–510. [Google Scholar] [PubMed]
- Barrett Mueller, K.; Lu, Q.; Mohammad, N.N.; Luu, V.; McCurley, A.; Williams, G.H.; Adler, G.K.; Karas, R.H.; Jaffe, I.Z. Estrogen receptor inhibits mineralocorticoid receptor transcriptional regulatory function. Endocrinology 2014, 155, 4461–4472. [Google Scholar] [CrossRef] [PubMed]
- Quinkler, M.; Meyer, B.; Bumke-Vogt, C.; Grossmann, C.; Gruber, U.; Oelkers, W.; Diederich, S.; Bahr, V. Agonistic and antagonistic properties of progesterone metabolites at the human mineralocorticoid receptor. Eur. J. Endocrinol. 2002, 146, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Watzka, M.; Beyenburg, S.; Blumcke, I.; Elger, C.E.; Bidlingmaier, F.; Stoffel-Wagner, B. Expression of mineralocorticoid and glucocorticoid receptor mRNA in the human hippocampus. Neurosci. Lett. 2000, 290, 121–124. [Google Scholar] [CrossRef]
- Kudielka, B.M.; Kirschbaum, C. Sex differences in HPA axis responses to stress: A review. Biol. Psychol. 2005, 69, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Llorente, R.; Miguel-Blanco, C.; Aisa, B.; Lachize, S.; Borcel, E.; Meijer, O.C.; Ramirez, M.J.; De Kloet, E.R.; Viveros, M.P. Long term sex-dependent psychoneuroendocrine effects of maternal deprivation and juvenile unpredictable stress in rats. J. Neuroendocrinol. 2011, 23, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Karandrea, D.; Kittas, C.; Kitraki, E. Contribution of sex and cellular context in the regulation of brain corticosteroid receptors following restraInt. stress. Neuroendocrinology 2000, 71, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kitraki, E.; Kremmyda, O.; Youlatos, D.; Alexis, M.N.; Kittas, C. Gender-dependent alterations in corticosteroid receptor status and spatial performance following 21 days of restraint stress. Neuroscience 2004, 125, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Cyranowski, J.M.; Frank, E.; Young, E.; Shear, M.K. Adolescent onset of the gender difference in lifetime rates of major depression: A theoretical model. Arch. Gen. Psychiatry 2000, 57, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Piccinelli, M.; Wilkinson, G. Gender differences in depression: Critical review. Br. J. Psychiatry 2000, 177, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Handa, R.J. Regulation of estrogen receptor-β expression in the female rat hypothalamus: Differential effects of dexamethasone and estradiol. Endocrinology 2004, 145, 3658–3670. [Google Scholar] [CrossRef] [PubMed]
- Isgor, C.; Cecchi, M.; Kabbaj, M.; Akil, H.; Watson, S.J. Estrogen receptor β in the paraventricular nucleus of hypothalamus regulates the neuroendocrine response to stress and is regulated by corticosterone. Neuroscience 2003, 121, 837–845. [Google Scholar] [CrossRef]
- Leret, M.L.; Molina-Holgado, F.; Gonzalez, M.I. The effect of perinatal exposure to estrogens on the sexually dimorphic response to novelty. Physiol. Behav. 1994, 55, 371–373. [Google Scholar] [CrossRef]
- Palermo-Neto, J.; Dorce, V.A. Influences of estrogen and/or progesterone on some dopamine related behavior in rats. Gen. Pharmacol. 1990, 21, 83–87. [Google Scholar] [CrossRef]
- Smanik, P.A.; Liu, Q.; Furminger, T.L.; Ryu, K.; Xing, S.; Mazzaferri, E.L.; Jhiang, S.M. Cloning of the human sodium lodide symporter. Biochem. Biophys. Res. Commun. 1996, 226, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Levy, O.; Carrasco, N. Cloning and characterization of the thyroid iodide transporter. Nature 1996, 379, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M.; Ando, S.; Feng, X.; Liu, Y.; Maruvada, P.; Xia, X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol. Cell. Endocrinol. 2006, 246, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Brent, G.A. Mechanisms of thyroid hormone action. J. Clin. Investig. 2012, 122, 3035–3043. [Google Scholar] [CrossRef] [PubMed]
- Braverman, L.E.; Ingbar, S.H.; Sterling, K. Conversion of thyroxine (T4) to triiodothyronine (T3) in athyreotic human subjects. J. Clin. Investig. 1970, 49, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Kohrle, J. The selenoenzyme family of deiodinase isozymes controls local thyroid hormone availability. Rev. Endocr. Metab. Disord. 2000, 1, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Iervasi, G.; Ferrazzi, P.; Francesconi, D.; Chopra, I.J. A study of iodothyronine 5′-monodeiodinase activities in normal and pathological tissues in man and their comparison with activities in rat tissues. Life Sci. 2000, 68, 191–202. [Google Scholar] [CrossRef]
- Galton, V.A.; McCarthy, P.T.; St Germain, D.L. The ontogeny of iodothyronine deiodinase systems in liver and intestine of the rat. Endocrinology 1991, 128, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Jolin, T. The role of somatostatin and/or dopamine in basal and TRH-stimulated TSH release in food-restricted rats. Acta Endocrinol. 1991, 125, 186–191. [Google Scholar] [CrossRef] [PubMed]
- DeRuyter, H.; Burman, K.D.; Wartofsky, L.; Smallridge, R.C. Thyrotropin secretion in starved rats is enhanced by somatostatin antiserum. Horm. Metab. Res. 1984, 16, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.J.; Young, W.S., 3rd; Weinberger, C. Differential expression of α and β thyroid hormone receptor genes in rat brain and pituitary. Proc. Natl. Acad. Sci. USA 1989, 86, 7250–7254. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J. Thyroid hormone receptors in brain development and function. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Horlein, A.J.; Naar, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Guigon, C.J.; Zhao, L.; Lu, C.; Willingham, M.C.; Cheng, S.Y. Regulation of β-catenin by a novel nongenomic action of thyroid hormone β receptor. Mol. Cell. Biol. 2008, 28, 4598–4608. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Whybrow, P.C. Thyroid hormone, neural tissue and mood modulation. World J. Biol. Psychiatry 2001, 2, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Farsetti, A.; Desvergne, B.; Hallenbeck, P.; Robbins, J.; Nikodem, V.M. Characterization of myelin basic protein thyroid hormone response element and its function in the context of native and heterologous promoter. J. Biol. Chem. 1992, 267, 15784–15788. [Google Scholar] [PubMed]
- Farsetti, A.; Mitsuhashi, T.; Desvergne, B.; Robbins, J.; Nikodem, V.M. Molecular basis of thyroid hormone regulation of myelin basic protein gene expression in rodent brain. J. Biol. Chem. 1991, 266, 23226–23232. [Google Scholar] [PubMed]
- Koibuchi, N.; Fukuda, H.; Chin, W.W. Promoter-specific regulation of the brain-derived neurotropic factor gene by thyroid hormone in the developing rat cerebellum. Endocrinology 1999, 140, 3955–3961. [Google Scholar] [CrossRef] [PubMed]
- Graff, M.N.; Baas, D.; Puymirat, J.; Sarlieve, L.L.; Delaunoy, J.P. The α and β thyroid receptors are expressed by cultured ependymal cells. Correlation with the effect of l-3,5,3′-triiodothyronine on glutamine synthetase mRNAs. Neurosci. Lett. 1993, 150, 174–178. [Google Scholar] [CrossRef]
- Garcia-Fernandez, L.F.; Rausell, E.; Urade, Y.; Hayaishi, O.; Bernal, J.; Munoz, A. Hypothyroidism alters the expression of prostaglandin D2 synthase/β trace in specific areas of the developing rat brain. Eur. J. Neurosci. 1997, 9, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Yauk, C.L.; Rowan-Carroll, A.; You, S.H.; Zoeller, R.T.; Lambert, I.; Wade, M.G. Identification of thyroid hormone receptor binding sites and target genes using ChIP-on-chip in developing mouse cerebellum. PLoS ONE 2009, 4, e4610. [Google Scholar] [CrossRef] [PubMed]
- Gagne, R.; Green, J.R.; Dong, H.; Wade, M.G.; Yauk, C.L. Identification of thyroid hormone receptor binding sites in developing mouse cerebellum. BMC Genom. 2013, 14, 341. [Google Scholar] [CrossRef] [PubMed]
- Nayak, B.; Hodak, S.P. Hyperthyroidism. Endocrinol. Metab. Clin. N. Am. 2007, 36, 617–656. [Google Scholar] [CrossRef] [PubMed]
- Zamoner, A.; Heimfarth, L.; Pessoa-Pureur, R. Congenital hypothyroidism is associated with intermediate filament misregulation, glutamate transporters down-regulation and MAPK activation in developing rat brain. Neurotoxicology 2008, 29, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Joffe, R.T.; Sokolov, S.T. Thyroid hormones, the brain, and affective disorders. Crit. Rev. Neurobiol. 1994, 8, 45–63. [Google Scholar] [PubMed]
- Rabie, A.; Favre, C.; Clavel, M.C.; Legrand, J. Effects of thyroid dysfunction on the development of the rat cerebellum, with special reference to cell death within the internal granular layer. Brain Res. 1977, 120, 521–531. [Google Scholar] [CrossRef]
- Rabie, A.; Legrand, J. Effects of thyroid hormone and undernourishment on the amount of synaptosomal fraction in the cerebellum of the young rat. Brain Res. 1973, 61, 267–278. [Google Scholar] [CrossRef]
- Zhou, L.; Li, X.; Ahmed, A.; Wu, D.; Liu, L.; Qiu, J.; Yan, Y.; Jin, M.; Xin, Y. Gut microbe analysis between hyperthyroid and healthy individuals. Curr. Microbiol. 2014, 69, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Lauritano, E.C.; Bilotta, A.L.; Gabrielli, M.; Scarpellini, E.; Lupascu, A.; Laginestra, A.; Novi, M.; Sottili, S.; Serricchio, M.; Cammarota, G.; et al. Association between hypothyroidism and small intestinal bacterial overgrowth. J. Clin. Endocrinol. Metab. 2007, 92, 4180–4184. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Green, W.L.; Huang, W.S.; Hays, M.T.; Chopra, I.J. Alternate pathways of thyroid hormone metabolism. Thyroid 2005, 15, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Ventura, M.; Melo, M.; Carrilho, F. Selenium and Thyroid Disease: From Pathophysiology to Treatment. Int. J. Endocrinol. 2017, 2017, 1297658. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; DiStefano, J.J., 3rd; Yamada, H.; Yen, Y.M. Steady state organ distribution and metabolism of thyroxine and 3,5,3′-triiodothyronine in intestines, liver, kidneys, blood, and residual carcass of the rat in vivo. Endocrinology 1993, 133, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.M.; Suen, V.M.; Souza, I.M.; De Oliveira, J.E.; Marchini, J.S. Patients with severe bowel malabsorption do not have changes in iodine status. Nutrition 2005, 21, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Michalaki, M.; Volonakis, S.; Mamali, I.; Kalfarentzos, F.; Vagenakis, A.G.; Markou, K.B. Dietary iodine absorption is not influenced by malabsorptive bariatric surgery. Obes. Surg. 2014, 24, 1921–1925. [Google Scholar] [CrossRef] [PubMed]
- Vought, R.L.; Brown, F.A.; Sibinovic, K.H.; McDaniel, E.G. Effect of changing intestinal bacterial flora on thyroid function in the rat. Horm. Metab. Res. 1972, 4, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Van, M. Absorption of Thyroxine from the Small Intestine of Rats. Endocrinology 1964, 74, 694–700. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, J.J., 3rd; de Luze, A.; Nguyen, T.T. Binding and degradation of 3,5,3′-triiodothyronine and thyroxine by rat intestinal bacteria. Am. J. Physiol. 1993, 264, E966–E972. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, G.; Covelli, I.; Roche, J. The fixation of thyroid hormones by Escherichia coli and its mechanism. Gen. Comp. Endocrinol. 1963, 3, 15–25. [Google Scholar] [CrossRef]
- Mehdi, Y.; Hornick, J.L.; Istasse, L.; Dufrasne, I. Selenium in the environment, metabolism and involvement in body functions. Molecules 2013, 18, 3292–3311. [Google Scholar] [CrossRef] [PubMed]
- Drutel, A.; Archambeaud, F.; Caron, P. Selenium and the thyroid gland: More good news for clinicians. Clin. Endocrinol. 2013, 78, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Lavu, R.V.; Van De Wiele, T.; Pratti, V.L.; Tack, F.; Du Laing, G. Selenium bioaccessibility in stomach, small intestine and colon: Comparison between pure Se compounds, Se-enriched food crops and food supplements. Food Chem. 2016, 197, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Hrdina, J.; Banning, A.; Kipp, A.; Loh, G.; Blaut, M.; Brigelius-Flohe, R. The gastrointestinal microbiota affects the selenium status and selenoprotein expression in mice. J. Nutr. Biochem. 2009, 20, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Kasaikina, M.V.; Kravtsova, M.A.; Lee, B.C.; Seravalli, J.; Peterson, D.A.; Walter, J.; Legge, R.; Benson, A.K.; Hatfield, D.L.; Gladyshev, V.N. Dietary selenium affects host selenoproteome expression by influencing the gut microbiota. FASEB J. 2011, 25, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; DiStefano, J.J., 3rd; Huang, L.M.; Yamada, H.; Cahnmann, H.J. 5′- and 5-deiodinase activities in adult rat cecum and large bowel contents inhibited by intestinal microflora. Am. J. Physiol. 1993, 265, E521–E524. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Combs, G.F., Jr. Effects of selenium on colonic fermentation in the rat. Biol. Trace Elem. Res. 1997, 56, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Gladyshev, V.N. The prokaryotic selenoproteome. EMBO Rep. 2004, 5, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Rother, M.; Resch, A.; Wilting, R.; Bock, A. Selenoprotein synthesis in archaea. Biofactors 2001, 14, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Khangulov, S.V.; Stadtman, T.C. Properties of the selenium- and molybdenum-containing nicotinic acid hydroxylase from Clostridium barkeri. Biochemistry 1996, 35, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Schrader, T.; Rienhofer, A.; Andreesen, J.R. Selenium-containing xanthine dehydrogenase from Eubacterium barkeri. Eur. J. Biochem. 1999, 264, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Visser, T.J. Pathways of thyroid hormone metabolism. Acta Med. Austriaca 1996, 23, 10–16. [Google Scholar] [PubMed]
- Boelen, A.; Kwakkel, J.; Thijssen-Timmer, D.C.; Alkemade, A.; Fliers, E.; Wiersinga, W.M. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J. Endocrinol. 2004, 182, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Boelen, A.; Kwakkel, J.; Platvoet-ter Schiphorst, M.; Mentrup, B.; Baur, A.; Koehrle, J.; Wiersinga, W.M. Interleukin-18, a proinflammatory cytokine, contributes to the pathogenesis of non-thyroidal illness mainly via the central part of the hypothalamus-pituitary-thyroid axis. Eur. J. Endocrinol. 2004, 151, 497–502. [Google Scholar] [CrossRef] [PubMed]
- de Herder, W.W.; Hazenberg, M.P.; Pennock-Schroder, A.M.; Hennemann, G.; Visser, T.J. Rapid and bacteria-dependent in vitro hydrolysis of iodothyronine-conjugates by intestinal contents of humans and rats. Med. Biol. 1986, 64, 31–35. [Google Scholar] [PubMed]
- Hazenberg, M.P.; de Herder, W.W.; Visser, T.J. Hydrolysis of iodothyronine conjugates by intestinal bacteria. FEMS Microbiol. Rev. 1988, 4, 9–16. [Google Scholar] [PubMed]
- Otten, M.H.; Herder, W.W.; Hazenberg, M.P.; Boom, M.; Hennemann, G. Iodothyronine sulfatase activity of two anaerobic bacterial strains from rat intestinal microflora. FEMS Microbiol. Lett. 1983, 18, 75–77. [Google Scholar] [CrossRef]
- Rutgers, M.; Heusdens, F.A.; Bonthuis, F.; de Herder, W.W.; Hazenberg, M.P.; Visser, T.J. Enterohepatic circulation of triiodothyronine (T3) in rats: Importance of the microflora for the liberation and reabsorption of T3 from biliary T3 conjugates. Endocrinology 1989, 125, 2822–2830. [Google Scholar] [CrossRef] [PubMed]
- Beigneux, A.P.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. Sick euthyroid syndrome is associated with decreased TR expression and DNA binding in mouse liver. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E228–E236. [Google Scholar] [CrossRef] [PubMed]
- Castro, I.; Quisenberry, L.; Calvo, R.M.; Obregon, M.J.; Lado-Abeal, J. Septic shock non-thyroidal illness syndrome causes hypothyroidism and conditions for reduced sensitivity to thyroid hormone. J. Mol. Endocrinol. 2013, 50, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Virili, C.; Bassotti, G.; Santaguida, M.G.; Iuorio, R.; Del Duca, S.C.; Mercuri, V.; Picarelli, A.; Gargiulo, P.; Gargano, L.; Centanni, M. Atypical celiac disease as cause of increased need for thyroxine: A systematic study. J. Clin. Endocrinol. Metab. 2012, 97, E419–E422. [Google Scholar] [CrossRef] [PubMed]
- Centanni, M.; Marignani, M.; Gargano, L.; Corleto, V.D.; Casini, A.; Delle Fave, G.; Andreoli, M.; Annibale, B. Atrophic body gastritis in patients with autoimmune thyroid disease: An underdiagnosed association. Arch. Intern. Med. 1999, 159, 1726–1730. [Google Scholar] [CrossRef] [PubMed]
- Cindoruk, M.; Tuncer, C.; Dursun, A.; Yetkin, I.; Karakan, T.; Cakir, N.; Soykan, I. Increased colonic intraepithelial lymphocytes in patients with Hashimoto’s thyroiditis. J. Clin. Gastroenterol. 2002, 34, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Carbonara, O.; Torella, R.; Mezzogiorno, A.; Esposito, V.; Demagistris, L.; Secondulfo, M.; Carratu, R.; Iafusco, D.; Carteni, M. Ultrastructural changes in enterocytes in subjects with Hashimoto’s thyroiditis. Gut 2004, 53, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, K.O.; Olowookorun, M.O. Gastric acid secretion and parietal cell mass: Effects of thyroidectomy and thyroxine. Am. J. Physiol. 1989, 256, G975–G978. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.C.; Johnson, L.R. Role of thyroxine in functional gastric development. Am. J. Physiol. 1986, 251, G111–G116. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009, 7, e003. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR α and ROR γ. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yissachar, N.; Zhou, Y.; Ung, L.; Lai, N.Y.; Mohan, J.F.; Ehrlicher, A.; Weitz, D.A.; Kasper, D.L.; Chiu, I.M.; Mathis, D.; et al. An Intestinal Organ Culture System Uncovers a Role for the Nervous System in Microbe-Immune Crosstalk. Cell 2017, 168, 1135–1148. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duszka, K.; Wahli, W. Enteric Microbiota–Gut–Brain Axis from the Perspective of Nuclear Receptors. Int. J. Mol. Sci. 2018, 19, 2210. https://doi.org/10.3390/ijms19082210
Duszka K, Wahli W. Enteric Microbiota–Gut–Brain Axis from the Perspective of Nuclear Receptors. International Journal of Molecular Sciences. 2018; 19(8):2210. https://doi.org/10.3390/ijms19082210
Chicago/Turabian StyleDuszka, Kalina, and Walter Wahli. 2018. "Enteric Microbiota–Gut–Brain Axis from the Perspective of Nuclear Receptors" International Journal of Molecular Sciences 19, no. 8: 2210. https://doi.org/10.3390/ijms19082210
APA StyleDuszka, K., & Wahli, W. (2018). Enteric Microbiota–Gut–Brain Axis from the Perspective of Nuclear Receptors. International Journal of Molecular Sciences, 19(8), 2210. https://doi.org/10.3390/ijms19082210