PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation


 , ,
, ,
Abstract
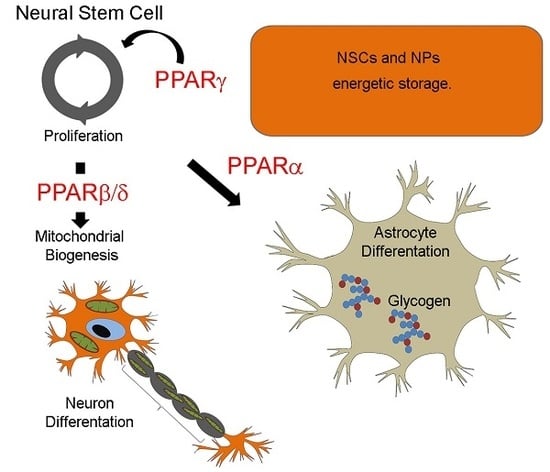
1. Introduction
2. Metabolic States in Neural Stem Cells Lineage
3. Roles of PPARs in the Energetic Metabolic Switch Occurring during Neurogenesis and Neuronal Maturation
4. Roles of PPARβ/δ in Neurogenesis and Neuronal Maturation
5. Roles of PPARγ in Neurogenesis and Neuronal Maturation
6. Energy Metabolism Imbalance in Neurodegenerative Disorders
7. Roles of PPARs in Neurodegenerative Disorders
8. Conclusions
Funding
Conflicts of Interest
References
- Bond, A.M.; Ming, G.L.; Song, H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Cavallucci, V.; Pani, G. Nutrients, neurogenesis and brain ageing: From disease mechanisms to therapeutic opportunities. Biochem. Pharmacol. 2017, 141, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.T.; Schafer, S.T.; Gage, F.H. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell 2016, 167, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Prigione, A. Mitochondrial metabolism in early neural fate and its relevance for neuronal disease modeling. Curr. Opin. Cell Biol. 2017, 49, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Skerrett, R.; Malm, T.; Landreth, G. Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis. 2014, 72, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Farioli-vecchioli, S.; Cerù, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.W.; Tanen, M.; Figueroa, D.J.; Biswas, C.; Zycband, E.; Moller, D.E.; Austin, C.P.; Berger, J.P. Localization of PPARδ; in murine central nervous system: Expression in oligodendrocytes and neurons. Brain Res. 2003, 975, 10–21. [Google Scholar] [CrossRef]
- Cullingford, T.E.; Bhakoo, K.; Peuchen, S.; Dolphin, C.T.; Patel, R.; Clark, J.B. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor α, β, and γ and the retinoid X receptor α, β, and γ in rat central nervous system. J. Neurochem. 1998, 70, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Benedetti, E.; Cristiano, L.; Sebastiani, P.; D’Amico, M.A.; D’Angelo, B.; Di Loreto, S. Expression of peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RXRs) in rat cortical neurons. Neuroscience 2005, 130, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Levi, L.; Siegel, R.; Noy, N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ). J. Biol. Chem. 2012, 287, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, E.; Di Loreto, S.; D’Angelo, B.; Cristiano, L.; d’Angelo, M.; Antonosante, A.; Fidoamore, A.; Golini, R.; Cinque, B.; Cifone, M.G.; et al. The PPARβ/δ Agonist GW0742 Induces Early Neuronal Maturation of Cortical Post-Mitotic Neurons: Role of PPARβ/δ in Neuronal Maturation. J. Cell. Physiol. 2015, 23, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Saluja, I.; Granneman, J.G.; Skoff, R.P. PPAR delta agonists stimulate oligodendrocyte differentiation in tissue culture. Glia 2001, 33, 191–204. [Google Scholar] [CrossRef]
- Cimini, A.; Bernardo, A.; Cifone, G.; Di Muzio, L.; Di Loreto, S. TNFα downregulates PPARδ expression in oligodendrocyte progenitor cells: Implications for demyelinating diseases. Glia 2003, 41, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, L.; Cimini, A.; Moreno, S.; Ragnelli, A.M.; Cerù, M.P. Peroxisome proliferator-activated receptors (PPARs) and related transcription factors in differentiating astrocyte cultures. Neuroscience 2005, 131, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Nakajima, A.; Katayama, K.; Kudo, C.; Shibuya, A.; Kubota, N.; Terauchi, Y.; Tachibana, M.; Miyoshi, H.; Kamisaki, Y.; et al. Peroxisome proliferator-activated receptor γ-mediated regulation of neural stem cell proliferation and differentiation. J. Biol. Chem. 2006, 81, 12673–12681. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of nuclear receptors with the Wnt/β-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 2008, 26, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Song, S.; Choi, C. Peroxisome proliferator activated receptor γ agonists suppress TNFα-induced ICAM-1 expression by endothelial cells in a manner potentially dependent on inhibition of reactive oxygen species. Immunol. Lett. 2008, 117, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Cristiano, L.; Benedetti, E.; D’Angelo, B.; Cerù, M.P. PPAR expression in adult mouse neural stem cells (NSC). Modulation of PPARs during astroglial differentiation. PPAR Res. 2007, 2007, 48242. [Google Scholar] [CrossRef] [PubMed]
- Temple, S. The development of neural stem cells. Nature 2001, 414, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.A.; Altman, J. Neocortical Development, 1st ed.; Raven Press: New York, NY, USA, 1999. [Google Scholar]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Klein-Flugge, M.C.; Howarth, C.; Attwell, D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Ochocki, J.D.; Simon, M.C. Nutrient-sensing pathways and metabolic regulation in stem cells. J. Cell Biol. 2013, 203, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Turrero Garcia, M.; Decimo, I.; Bifari, F.; Eelen, G.; Quaegebeur, A.; Boon, R.; Zhao, H.; Boeckx, B.; Chang, J.; et al. Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. EMBO J. 2016, 35, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Rafalski, V.A.; Brunet, A. Energy metabolism in adult neural stem cell fate. Prog. Neurobiol. 2011, 93, 182–203. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLIFE 2016, 10, e13374. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.C.; Keeney, P.M.; Bennett, J.P., Jr. Differentiation of Human Neural Stem Cells into Motor Neurons Stimulates Mitochondrial Biogenesis and Decreases Glycolytic Flux. Stem Cells Dev. 2015, 24, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Qing, Y.; Yan, S.; Chen, D.; Yan, S.S. Development and Dynamic Regulation of Mitochondrial Network in Human Midbrain Dopaminergic Neurons Differentiated from iPSCs. Stem Cell Rep. 2016, 7, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.Q.; Pan, Z.F.; Chen, W.T.; Xu, M.H.; Zhu, D.Y.; Yu, Y.P.; Lou, Y.J. A Flavonoid Compound Promotes Neuronal Differentiation of Embryonic Stem Cells via PPAR-β Modulating Mitochondrial Energy Metabolism. PLoS ONE 2016, 11, e0157747. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yin, R.; Liu, J.; Wang, P.; Wu, S.; Luo, J.; Zhelyabovska, O.; Yang, Q. Peroxisome proliferator-activated receptor delta regulates mitofusin 2 expression in the heart. J. Mol. Cell. Cardiol. 2009, 46, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Galloway, C.A.; Yoon, Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS ONE 2011, 6, e20655. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Langer, T. Mitofusin 2 builds a bridge between ER and mitochondria. Cell 2008, 135, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Furutachi, S.; Miya, H.; Watanabe, T.; Kawai, H.; Yamasaki, N.; Harada, Y.; Imayoshi, I.; Nelson, M.; Nakayama, K.I.; Hirabayashi, Y.; et al. Slowly dividing neural progenitors are an embryonic origin of adult neural stem cells. Nat. Neurosci. 2015, 18, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- Prozorovski, T.; Schneider, R.; Berndt, C.; Hartung, H.P.; Aktas, O. Redox-regulated fate of neural stem progenitor cells. Biochim. Biophys. Acta 2015, 1850, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.A.D.; Karlsson, A.; Martins, L.M.; Capasso, M.; et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Stoll, E.A.; Makin, R.; Sweet, I.R.; Trevelyan, A.J.; Miwa, S.; Horner, P.J.; Turnbull, D.M. Neural Stem Cells in the Adult Subventricular Zone Oxidize Fatty Acids to Produce Energy and Support Neurogenic Activity. Stem Cells 2015, 33, 2306–2319. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Braun, S.M.; Zurkirchen, L.; von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.J.; Karalay, O.; Suter, U.; Machado, R.A.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2013, 493, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Berg, D.A.; Zhu, Y.; Shin, J.Y.; Song, J.; Bonaguidi, M.A.; Enikolopov, G.; Nauen, D.W.; Christian, K.M.; Ming, G.L.; et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell 2015, 17, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Jones, A.; Deeney, J.T.; Hur, S.K.; Bankaitis, V.A. Inborn Errors of Long-Chain Fatty Acid β-Oxidation Link Neural Stem Cell Self-Renewal to Autism. Cell Rep. 2016, 14, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Brain insulin signalling, glucose metabolism and females’ reproductive aging: A dangerous triad in Alzheimer’s disease. Neuropharmacology 2018. [Google Scholar] [CrossRef] [PubMed]
- Hihi, A.K.; Michalik, L.; Wahli, W. PPARs: Transcriptional effectors of fatty acids and their derivatives. Cell. Mol. Life Sci. 2002, 59, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Diradourian, C.; Girard, J.; Pégorier, J.P. Phosphorylation of PPARs: From molecular characterization to physiological relevance. Biochimie 2005, 87, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Anbalagan, M.; Huderson, B.; Murphy, L.; Rowan, B.G. Post-translational modifications of nuclear receptors and human disease. Nucl. Recept. Signal. 2012, 10, e001. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Sahebkar, A.; Maffioli, P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J. Cell. Physiol. 2018, 233, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Cerù, M.P. Emerging roles of peroxisome proliferator-activated receptors (PPARs) in the regulation of neural stem cells proliferation and differentiation. Stem Cell Rev. 2008, 4, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hon, M.; Evans, R.M. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ). Mol. Cell. Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef] [PubMed]
- Barroso, E.; del Valle, J.; Porquet, D.; Vieira Santos, A.M.; Salvadó, L.; Rodríguez-Rodríguez, R.; Gutiérrez, P.; Anglada-Huguet, M.; Alberch, J.; Camins, A.; et al. Tau hyperphosphorylation and increased BACE1 and RAGE levels in the cortex of PPARβ/δ-null mice. Biochim. Biophys. Acta 2013, 1832, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Di Loreto, S.; D’Angelo, B.; D’Amico, M.A.; Benedetti, E.; Cristiano, L.; Cinque, B.; Cifone, M.G.; Cerù, M.P.; Festuccia, C.; Cimini, A. PPARbeta agonists trigger neuronal differentiation in the human neuroblastoma cell line SH-SY5Y. J. Cell. Physiol. 2007, 211, 837–847. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, B.; Benedetti, E.; Di Loreto, S.; Cristiano, L.; Laurenti, G.; Cerù, M.P.; Cimini, A. Signal transduction pathways involved in PPARβ/δ-induced neuronal differentiation. J. Cell. Physiol. 2011, 226, 2170–2180. [Google Scholar] [CrossRef] [PubMed]
- Beaven, S.W.; Tontonoz, P. Nuclear receptors in lipid metabolism: Targeting the heart of dyslipidemia. Annu. Rev. Med. 2006, 57, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.D.; Prabhu, K.S.; Thompson, J.T.; Reddy, P.S.; Peters, J.M.; Peterson, B.R.; Reddy, C.C.; Vanden Heuvel, J.P. The oxidative stress mediator 4-hydroxynonenal is anintracellular agonist of the nuclear receptor peroxisome proliferator-activatedreceptor-β/δ (PPARβ/δ). Free Radic. Biol. Med. 2007, 42, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Stump, M.; Guo, D.F.; Lu, K.T.; Mukohda, M.; Cassell, M.D.; Norris, A.W.; Rahmouni, K.; Sigmund, C.D. Nervous System Expression of PPARγ and Mutant PPARγ Has Profound Effects on Metabolic Regulation and Brain Development. Endocrinology 2016, 157, 4266–4275. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, E.; Benedetti, E.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Fidoamore, A.; Barone, D.; Moreno, S.; Ippoliti, R.; Cerù, M.P.; et al. Roles of PPAR transcription factors in the energetic metabolic switch occurring during adult neurogenesis. Cell Cycle 2017, 16, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Thambisetty, M.; Beason-Held, L.L.; An, Y.; Kraut, M.; Metter, J.; Egan, J.; Ferrucci, L.; O’Brien, R.; Resnick, S.M. Impaired glucose tolerance in midlife and longitudinal changes in brain function during aging. Neurobiol. Aging 2013, 34, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Pang, Z.; Geddes, J.W.; Uchida, K.; Mattson, M.P. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: Involvement of membrane lipid peroxidation. J. Neurosci. 1997, 17, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp. Gerontol. 2009, 44, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Denis, I.; Potier, B.; Vancassel, S.; Heberden, C.; Lavialle, M. Omega-3 fatty acids and brain resistance to ageing and stress: Body of evidence and possible mechanisms. Ageing Res. Rev. 2013, 12, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J.P.; Scutt, P.; Sprigg, N.; Bath, P.M. Hypercholesterolaemia and vascular dementia. Clin. Sci. 2017, 131, 1561–1578. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Philips, G.T.; Herz, J. More than cholesterol transporters: Lipoprotein receptors in CNS function and neurodegeneration. Neuron 2014, 83, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal Dysfunction in Age-Related Diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.W.; Tekirian, T.L.; Soultanian, N.S.; Ashford, J.W.; Davis, D.G.; Markesbery, W.R. Comparison of neuropathologic criteria for the diagnosis of Alzheimer’s disease. Neurobiol. Aging 1997, 18, 99–105. [Google Scholar] [CrossRef]
- Rodriguez-Oroz, M.C.; Jahanshahi, M.; Krack, P.; Litvan, I.; Macias, R.; Bezard, E.; Obeso, J.A. Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. Lancet Neurol. 2009, 8, 1128–1139. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson disease–the gut-brain axis and environmental factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Morsci, N.S.; Hall, D.H.; Driscoll, M.; Sheng, Z.H. Age-Related Phasic Patterns of Mitochondrial Maintenance in Adult Caenorhabditis elegans Neurons. J. Neurosci. 2016, 36, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 47, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Rcom-H’cheo-Gauthier, A.; Goodwin, J.; Pountney, D.L. Interactions between calcium and alpha synuclein in neurodegeneration. Biomolecules 2014, 4, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; Rutter, G.A.; Midgley, P.J.; McCormack, J.G. Effects of Ca2+ on the activities of the calcium-sensitive dehydrogenases within the mitochondria of mammalian tissues. J. Cardiovasc. Pharmacol. 1988, 12, S69–S72. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb. Perspect. Med. 2012, 2, a009415. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear gene encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Uldry, M.; Yang, W.; St-Pierre, J.; Lin, J.; Seale, P.; Spiegelman, B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006, 3, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Porcellotti, S.; Fanelli, F.; Fracassi, A.; Sepe, S.; Cecconi, F.; Bernardi, C.; Cimini, A.; Cerù, M.P.; Moreno, S. Oxidative Stress during the Progression of β-Amyloid Pathology in the Neocortex of the Tg2576 Mouse Model of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2015, 2015, 967203. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Sepe, S.; D’Amelio, M.; Bernardi, C.; Cristiano, L.; Cimini, A.; Cecconi, F.; Ceru, M.P.; Moreno, S. Age-dependent roles of peroxisomes in the hippocampus of a transgenic mouse model of Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Moreno, S.; D’Amelio, M.; Cristiano, L.; D’Angelo, B.; Falone, S.; Benedetti, E.; Carrara, P.; Fanelli, F.; Cecconi, F.; et al. Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: A role for peroxisomes. J. Alzheimers Dis. 2009, 18, 935–952. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, age-related diseases and peroxisomes. Subcell. Biochem. 2013, 69, 45–65. [Google Scholar] [PubMed]
- Terlecky, S.R.; Terlecky, L.J.; Giordano, C.R. Peroxisomes, oxidative stress, and inflammation. World J. Biol. Chem. 2012, 3, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martín, V.; Santpere, G.; Marín, R.; Torrent, L.; Ferrer, I.; Díaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Kovacs, G.G.; Höftberger, R.; Kulik, W.; Brodde, A.; Forss-Petter, S.; Hönigschnabl, S.; Gleiss, A.; Brügger, B.; Wanders, R.; et al. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, E.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; D’Angelo, B.; Selli, S.; Castelli, V.; Ippoliti, R.; Giordano, A.; Cimini, A. PPARs in Neurodegenerative and Neuroinflammatory Pathways. Curr. Alzheimer Res. 2018, 15, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Searcy, J.L.; Phelps, J.T.; Pancani, T.; Kadish, I.; Popovic, J.; Anderson, K.L.; Beckett, T.L.; Murphy, M.P.; Chen, K.C.; Blalock, E.M.; et al. Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2012, 30, 943–961. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Cholerton, B.; Baker, L.D. Insulin and Alzheimer’s disease: Untangling the web. J. Alzheimers Dis. 2013, 33, S263–S275. [Google Scholar] [CrossRef] [PubMed]
- Escribano, L.; Simón, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; López de Maturana, R.; García-Osta, A.; Ricobaraza, A.; Pérez-Mediavilla, A.; Del Río, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Rosenblat, J.D.; Brietzke, E.; Park, C.; Lee, Y.; Musial, N.; Pan, Z.; Mansur, R.B.; McIntyre, R.S. Comparative Efficacy and Acceptability of Anti-Diabetic Agents for Alzheimer’s Disease and Mild Cognitive Impairment: A Systematic Review and Network Meta-analysis. Diabetes Obes. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Gottschalk, W.K.; Kim, S.Y.; Roses, A.D.; Chiba-Falek, O. The effects of PPARγ on the regulation of the TOMM40-APOE-C1 genes cluster. Biochim. Biophys. Acta 2017, 1863, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.H.; Lai, S.W.; Chen, P.Y.; Chang, J.H.; Chang, N.W. PPARα activation attenuates amyloid-β-dependent neurodegeneration by modulating Endo G and AIF translocation. Neurotoxic. Res. 2015, 27, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Fanelli, F.; Cerù, M.P.; Moreno, S. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARα) and its lipid ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef] [PubMed]
- Brune, S.; Kölsch, H.; Ptok, U.; Majores, M.; Schulz, A.; Schlosser, R.; Rao, M.L.; Maier, W.; Heun, R. Polymorphism in the peroxisome proliferator-activated receptor alpha gene influences the risk for Alzheimer’s disease. J. Neural Transm. 2003, 110, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Kölsch, H.; Lehmann, D.J.; Ibrahim-Verbaas, C.A.; Combarros, O.; van Duijn, C.M.; Hammond, N.; Belbin, O.; Cortina-Borja, M.; Lehmann, M.G.; Aulchenko, Y.S.; et al. Interaction of insulin and PPAR-α genes in Alzheimer’s disease: The Epistasis Project. J. Neural Transm. 2012, 119, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Jana, M.; Kundu, M.; Corbett, G.T.; Rangaswamy, S.B.; Mishra, R.K.; Luan, C.H.; Gonzalez, F.J.; Pahan, K. HMG-CoA Reductase Inhibitors Bind to PPARα to Upregulate Neurotrophin Expression in the Brain and Improve Memory in Mice. Cell Metab. 2015, 22, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Kreisler, A.; Gelé, P.; Wiart, J.F.; Lhermitte, M.; Destée, A.; Bordet, R. Lipid-lowering drugs in the MPTP mouse model of Parkinson’s disease: Fenofibrate has a neuroprotective effect, whereas bezafibrate and HMG-CoA reductase inhibitors do not. Brain Res. 2007, 1135, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Giordano Attianese, G.M.; Desvergne, B. Integrative and systemic approaches for evaluating PPARβ/δ (PPARD) function. Nucl. Recept. Signal. 2015, 13, e001. [Google Scholar] [CrossRef] [PubMed]
- Zolezzi, J.M.; Bastías-Candia, S.; Santos, M.J.; Inestrosa, N.C. Alzheimer’s disease: Relevant molecular and physiopathological events affecting amyloid-β brain balance and the putative role of PPARs. Front. Aging Neurosci. 2014, 6, 176. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Neurodegenerative Diseases i.e. | ||
|---|---|---|
| AD | and | PD |
| Ref. Energy Metabolism Imbalance | Ref. PPARs and Their Ligands | |
| [47,64,65,66,67,68] | Insulin Resistance | [102,103,104,109,110] |
| [78,79,80,81,82,83,84,85] | Mitochondrial Dysregulation | [73,105,106,113] |
| [89,90,91,92,93,94,95,96,97,98] | Peroxisomal Dysregulation | [90,91,92] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Angelo, M.; Antonosante, A.; Castelli, V.; Catanesi, M.; Moorthy, N.; Iannotta, D.; Cimini, A.; Benedetti, E. PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation. Int. J. Mol. Sci. 2018, 19, 1869. https://doi.org/10.3390/ijms19071869
D’Angelo M, Antonosante A, Castelli V, Catanesi M, Moorthy N, Iannotta D, Cimini A, Benedetti E. PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation. International Journal of Molecular Sciences. 2018; 19(7):1869. https://doi.org/10.3390/ijms19071869
Chicago/Turabian StyleD’Angelo, Michele, Andrea Antonosante, Vanessa Castelli, Mariano Catanesi, NandhaKumar Moorthy, Dalila Iannotta, Annamaria Cimini, and Elisabetta Benedetti. 2018. "PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation" International Journal of Molecular Sciences 19, no. 7: 1869. https://doi.org/10.3390/ijms19071869
APA StyleD’Angelo, M., Antonosante, A., Castelli, V., Catanesi, M., Moorthy, N., Iannotta, D., Cimini, A., & Benedetti, E. (2018). PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation. International Journal of Molecular Sciences, 19(7), 1869. https://doi.org/10.3390/ijms19071869