The Making of Leukemia

 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
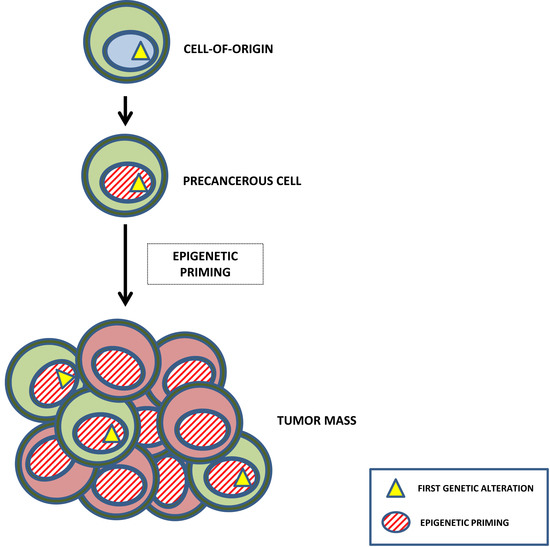
1. The Making of Leukemia: The Concept of Epigenetic Reprogramming
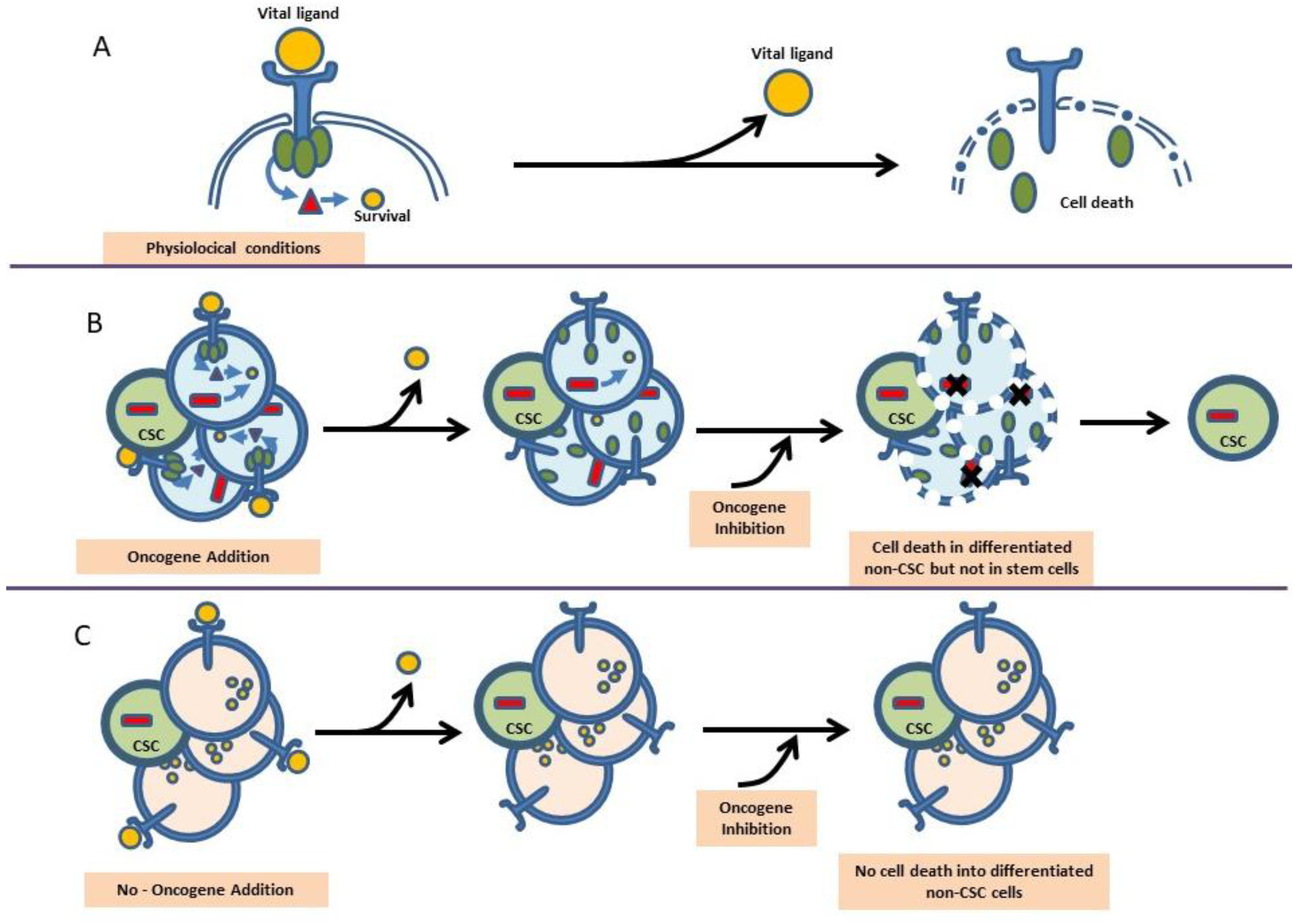
2. Oncogene Addition versus Reprogramming Leukemia Predisposition
3. Restriction of Lineage Options during the Making of Leukemia
4. Epigenetic Reprogramming in Non-Hematopoietic Tumors
5. Hematopoiesis and Leukemia Are Both Lineage Decision-Making Processes
6. The Importance of Environmental Signals in the Making of Leukemia
7. Therapeutic Intervention in Leukemia and the Prospect of Modifying the Making of Leukemia
8. Future Opportunities and Challenges
Acknowledgments
Conflicts of Interest
References
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, R.; Urban, N.; Ramsey, S.; McIntosh, M.; Schwartz, S.; Reid, B.; Radich, J.; Anderson, G.; Hartwell, L. The case for early detection. Nat. Rev. Cancer 2003, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Romero-Camarero, I.; Cobaleda, C.; Sanchez-Garcia, I. Function of oncogenes in cancer development: A changing paradigm. EMBO J. 2013, 32, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.; Lynch, H.T.; Neuhausen, S.L.; van’t Veer, L.; Garber, J.E.; Evans, G.R.; Narod, S.A.; Isaacs, C.; Matloff, E.; et al. Bilateral prophylactic mastectomy reduces breast cancer risk in brca1 and brca2 mutation carriers: The prose study group. J. Clin. Oncol. 2004, 22, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.M.; Del Paggio, J.C. Approvals in 2016: Questioning the clinical benefit of anticancer therapies. Nat. Rev. Clin. Oncol. 2017, 14, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Boxer, R.B.; Jang, J.W.; Sintasath, L.; Chodosh, L.A. Lack of sustained regression of c-myc-induced mammary adenocarcinomas following brief or prolonged myc inactivation. Cancer Cell 2004, 6, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic ras in tumour maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Huettner, C.S.; Zhang, P.; Van Etten, R.A.; Tenen, D.G. Reversibility of acute b-cell leukaemia induced by bcr-abl1. Nat. Genet. 2000, 24, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The achilles heel of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Hamburger, A.; Salmon, S.E. Primary bioassay of human myeloma stem cells. J. Clin. Investig. 1977, 60, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Hauer, J.; Ruiz-Roca, L.; Ingenhag, D.; Rodriguez-Meira, A.; Auer, F.; Borkhardt, A.; Sanchez-Garcia, I. Tumoral stem cell reprogramming as a driver of cancer: Theory, biological models, implications in cancer therapy. Semin. Cancer Biol. 2015, 32, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sanchez-Martin, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Gupta, R.; Ancliff, P.; Atzberger, A.; Brown, J.; Soneji, S.; Green, J.; Colman, S.; Piacibello, W.; Buckle, V.; et al. Initiating and cancer-propagating cells in tel-aml1-associated childhood leukemia. Science 2008, 319, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Dobbins, S.E.; Sherborne, A.L.; Chubb, D.; Galbiati, M.; Cazzaniga, G.; Micalizzi, C.; Tearle, R.; Lloyd, A.L.; Hain, R.; et al. Developmental timing of mutations revealed by whole-genome sequencing of twins with acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 7429–7433. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.J.; Melo, J.V. Primitive, quiescent and difficult to kill: The role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle 2006, 5, 2862–2866. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to sti571 In Vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Romero-Camarero, I.; Jiang, X.; Natkunam, Y.; Lu, X.; Vicente-Duenas, C.; Gonzalez-Herrero, I.; Flores, T.; Luis Garcia, J.; McNamara, G.; Kunder, C.; et al. Germinal centre protein hgal promotes lymphoid hyperplasia and amyloidosis via bcr-mediated syk activation. Nat. Commun. 2013, 4, 1338. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Fontan, L.; Gonzalez-Herrero, I.; Romero-Camarero, I.; Segura, V.; Aznar, M.A.; Alonso-Escudero, E.; Campos-Sanchez, E.; Ruiz-Roca, L.; Barajas-Diego, M.; et al. Expression of malt1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10534–10539. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Romero-Camarero, I.; Gonzalez-Herrero, I.; Alonso-Escudero, E.; Abollo-Jimenez, F.; Jiang, X.; Gutierrez, N.C.; Orfao, A.; Marin, N.; Villar, L.M.; et al. A novel molecular mechanism involved in multiple myeloma development revealed by targeting mafb to haematopoietic progenitors. EMBO J. 2012, 31, 3704–3717. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Vicente-Duenas, C.; Romero-Camarero, I.; Long Liu, C.; Dai, B.; Gonzalez-Herrero, I.; Garcia-Ramirez, I.; Alonso-Escudero, E.; Iqbal, J.; Chan, W.C.; et al. Transient expression of bcl6 is sufficient for oncogenic function and induction of mature b-cell lymphoma. Nat. Commun. 2014, 5, 3904. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, G.; Hauer, J.; Martin-Lorenzo, A.; Schafer, D.; Bartenhagen, C.; Garcia-Ramirez, I.; Auer, F.; Gonzalez-Herrero, I.; Ruiz-Roca, L.; Gombert, M.; et al. Infection exposure promotes etv6-runx1 precursor b-cell leukemia via impaired h3k4 demethylases. Cancer Res. 2017, 77, 4365–4377. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Romero-Camarero, I.; Garcia-Criado, F.J.; Cobaleda, C.; Sanchez-Garcia, I. The cellular architecture of multiple myeloma. Cell Cycle 2012, 11, 3715–3717. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vicente-Duenas, C.; Cobaleda, C.; Martinez-Climent, J.A.; Sanchez-Garcia, I. Malt lymphoma meets stem cells. Cell Cycle 2012, 11, 2961–2962. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riggi, N.; Suva, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suva, D.; et al. Ews-fli-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.B.; Shaffer, C.M.; Alfaro, M.P.; Smith, A.L.; Sun, J.; Zhao, Z.; Young, P.P.; VanSaun, M.N.; Eid, J.E. Reprogramming of mesenchymal stem cells by the synovial sarcoma-associated oncogene SYT-SSX2. Oncogene 2012, 31, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone h3.3k36m mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, S.; Mijimolle, N.; Francoz, S.; Vicente-Duenas, C.; Sanchez-Garcia, I.; Barbacid, M. Identification of cancer initiating cells in k-ras driven lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lim, A.; Odegaard, J.I.; Honeycutt, J.D.; Kawano, S.; Hsieh, M.H.; Beachy, P.A. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat. Cell Biol. 2014, 16, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Torres, D.; Marone, R.; Feng, X.; Martin, D.; Simaan, M.; Chen, M.; Weinstein, L.S.; Taylor, S.S.; Molinolo, A.A.; et al. Inactivation of a galpha(s)-pka tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat. Cell Biol. 2015, 17, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, T.E.; Chindera, K.; McDermott, J.; Breeze, C.E.; Cooke, W.R.; Jones, A.; Reisel, D.; Karegodar, S.T.; Arora, R.; Beck, S.; et al. Epigenetic reprogramming of fallopian tube fimbriae in brca mutation carriers defines early ovarian cancer evolution. Nat. Commun. 2016, 7, 11620. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kozono, D.; Li, J.; Nitta, M.; Sampetrean, O.; Gonda, D.; Kushwaha, D.S.; Merzon, D.; Ramakrishnan, V.; Zhu, S.; Zhu, K.; et al. Dynamic epigenetic regulation of glioblastoma tumorigenicity through lsd1 modulation of myc expression. Proc. Natl. Acad. Sci. USA 2015, 112, E4055–E4064. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Chesnelong, C.; Pankov, A.; Jalbert, L.E.; Hong, C.; Hayes, J.; Smirnov, I.V.; Marshall, R.; Souza, C.F.; Shen, Y.; et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant idh1. Proc. Natl. Acad. Sci. USA 2017, 114, 10743–10748. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ahmed, M.; Guo, H.; Soares, F.; Hua, J.T.; Gao, S.; Lu, C.; Poon, C.; Han, W.; Langstein, J.; et al. Lsd1-mediated epigenetic reprogramming drives cenpe expression and prostate cancer progression. Cancer Res. 2017, 77, 5479–5490. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Chomel, J.C.; Bonnet, M.L.; Sorel, N.; Bertrand, A.; Meunier, M.C.; Fichelson, S.; Melkus, M.; Bennaceur-Griscelli, A.; Guilhot, F.; Turhan, A.G. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood 2011, 118, 3657–3660. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; McDonald, T.; Lin, A.; Chakraborty, S.; Huang, Q.; Snyder, D.S.; Bhatia, R. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 2011, 118, 5565–5572. [Google Scholar] [CrossRef] [PubMed]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of bcr-abl activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Helgason, G.V.; Schemionek, M.; Zhang, B.; Myssina, S.; Allan, E.K.; Nicolini, F.E.; Muller-Tidow, C.; Bhatia, R.; Brunton, V.G.; et al. Chronic myeloid leukemia stem cells are not dependent on bcr-abl kinase activity for their survival. Blood 2012, 119, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Brendel, C.; Hochhaus, A.; Neubauer, A.; Burchert, A. Low bcr-abl expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood 2012, 119, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Trans. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Stricker, S.H.; Feber, A.; Engstrom, P.G.; Caren, H.; Kurian, K.M.; Takashima, Y.; Watts, C.; Way, M.; Dirks, P.; Bertone, P.; et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev. 2013, 27, 654–669. [Google Scholar] [CrossRef] [PubMed]
- McClellan, J.S.; Dove, C.; Gentles, A.J.; Ryan, C.E.; Majeti, R. Reprogramming of primary human philadelphia chromosome-positive B cell acute lymphoblastic leukemia cells into nonleukemic macrophages. Proc. Natl. Acad. Sci. USA 2015, 112, 4074–4079. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, I. How tumour cell identity is established? Semin. Cancer Biol. 2015, 32, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Hernandez, T.; Vicente-Duenas, C.; Sanchez-Garcia, I.; Martin-Zanca, D. P53 restoration kills primitive leukemia cells in vivo and increases survival of leukemic mice. Cell Cycle 2013, 12, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lorenzo, A.; Hauer, J.; Vicente-Duenas, C.; Auer, F.; Gonzalez-Herrero, I.; Garcia-Ramirez, I.; Ginzel, S.; Thiele, R.; Constantinescu, S.N.; Bartenhagen, C.; et al. Infection exposure is a causal factor in b-cell precursor acute lymphoblastic leukemia as a result of pax5-inherited susceptibility. Cancer Discov. 2015, 5, 1328–1343. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Klemm, L.; Park, E.; Papaemmanuil, E.; Ford, A.; Kweon, S.M.; Trageser, D.; Hasselfeld, B.; Henke, N.; Mooster, J.; et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat. Immunol. 2015, 16, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Pereira, H.M. A latitudinal gradient for genetic diversity. Science 2016, 353, 1494–1495. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Luo, R.T.; Ptasinska, A.; Kerry, J.; Assi, S.A.; Wunderlich, M.; Imamura, T.; Kaberlein, J.J.; Rayes, A.; Althoff, M.J.; et al. Instructive role of mll-fusion proteins revealed by a model of t(4;11) pro-b acute lymphoblastic leukemia. Cancer Cell 2016, 30, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. Cd19 car immune pressure induces b-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Bhatla, T.; Wang, J.; Morrison, D.J.; Raetz, E.A.; Burke, M.J.; Brown, P.; Carroll, W.L. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood b-lymphoblastic leukemia. Blood 2012, 119, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Connelly, M.C.; Wetmore, C.; Curran, T.; Morgan, J.I. Mouse embryos cloned from brain tumors. Cancer Res. 2003, 63, 2733–2736. [Google Scholar] [PubMed]
- Blelloch, R.H.; Hochedlinger, K.; Yamada, Y.; Brennan, C.; Kim, M.; Mintz, B.; Chin, L.; Jaenisch, R. Nuclear cloning of embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13985–13990. [Google Scholar] [PubMed]
- Hochedlinger, K.; Blelloch, R.; Brennan, C.; Yamada, Y.; Kim, M.; Chin, L.; Jaenisch, R. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004, 18, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic reprogramming sensitizes cml stem cells to combined ezh2 and tyrosine kinase inhibition. Cancer Discov. 2016, 6, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Chheda, M.G.; Gutmann, D.H. Using epigenetic reprogramming to treat pediatric brain cancer. Cancer Cell 2017, 31, 609–611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 2017, 31, 635–652 e636. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Grimm, S.A.; Chrysovergis, K.; Kosak, J.; Wang, X.; Du, Y.; Burkholder, A.; Janardhan, K.; Mav, D.; Shah, R.; et al. Obesity, rather than diet, drives epigenomic alterations in colonic epithelium resembling cancer progression. Cell Metab. 2014, 19, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Vaz, M.; Hwang, S.Y.; Kagiampakis, I.; Phallen, J.; Patil, A.; O’Hagan, H.M.; Murphy, L.; Zahnow, C.A.; Gabrielson, E.; Velculescu, V.E.; et al. Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by kras mutations. Cancer Cell 2017, 32, 360–376.e6. [Google Scholar] [CrossRef] [PubMed]
- Greathouse, K.L.; Bredfeldt, T.; Everitt, J.I.; Lin, K.; Berry, T.; Kannan, K.; Mittelstadt, M.L.; Ho, S.M.; Walker, C.L. Environmental estrogens differentially engage the histone methyltransferase ezh2 to increase risk of uterine tumorigenesis. Mol. Cancer Res. 2012, 10, 546–557. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Herrero, I.; Rodríguez-Hernández, G.; Luengas-Martínez, A.; Isidro-Hernández, M.; Jiménez, R.; García-Cenador, M.B.; García-Criado, F.J.; Sánchez-García, I.; Vicente-Dueñas, C. The Making of Leukemia. Int. J. Mol. Sci. 2018, 19, 1494. https://doi.org/10.3390/ijms19051494
González-Herrero I, Rodríguez-Hernández G, Luengas-Martínez A, Isidro-Hernández M, Jiménez R, García-Cenador MB, García-Criado FJ, Sánchez-García I, Vicente-Dueñas C. The Making of Leukemia. International Journal of Molecular Sciences. 2018; 19(5):1494. https://doi.org/10.3390/ijms19051494
Chicago/Turabian StyleGonzález-Herrero, Inés, Guillermo Rodríguez-Hernández, Andrea Luengas-Martínez, Marta Isidro-Hernández, Rafael Jiménez, Maria Begoña García-Cenador, Francisco Javier García-Criado, Isidro Sánchez-García, and Carolina Vicente-Dueñas. 2018. "The Making of Leukemia" International Journal of Molecular Sciences 19, no. 5: 1494. https://doi.org/10.3390/ijms19051494
APA StyleGonzález-Herrero, I., Rodríguez-Hernández, G., Luengas-Martínez, A., Isidro-Hernández, M., Jiménez, R., García-Cenador, M. B., García-Criado, F. J., Sánchez-García, I., & Vicente-Dueñas, C. (2018). The Making of Leukemia. International Journal of Molecular Sciences, 19(5), 1494. https://doi.org/10.3390/ijms19051494