Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression


 ,
, 
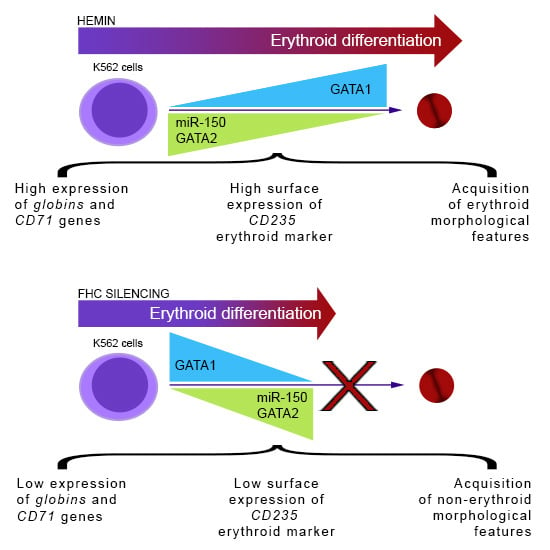
Abstract
:
1. Introduction
2. Results and Discussion
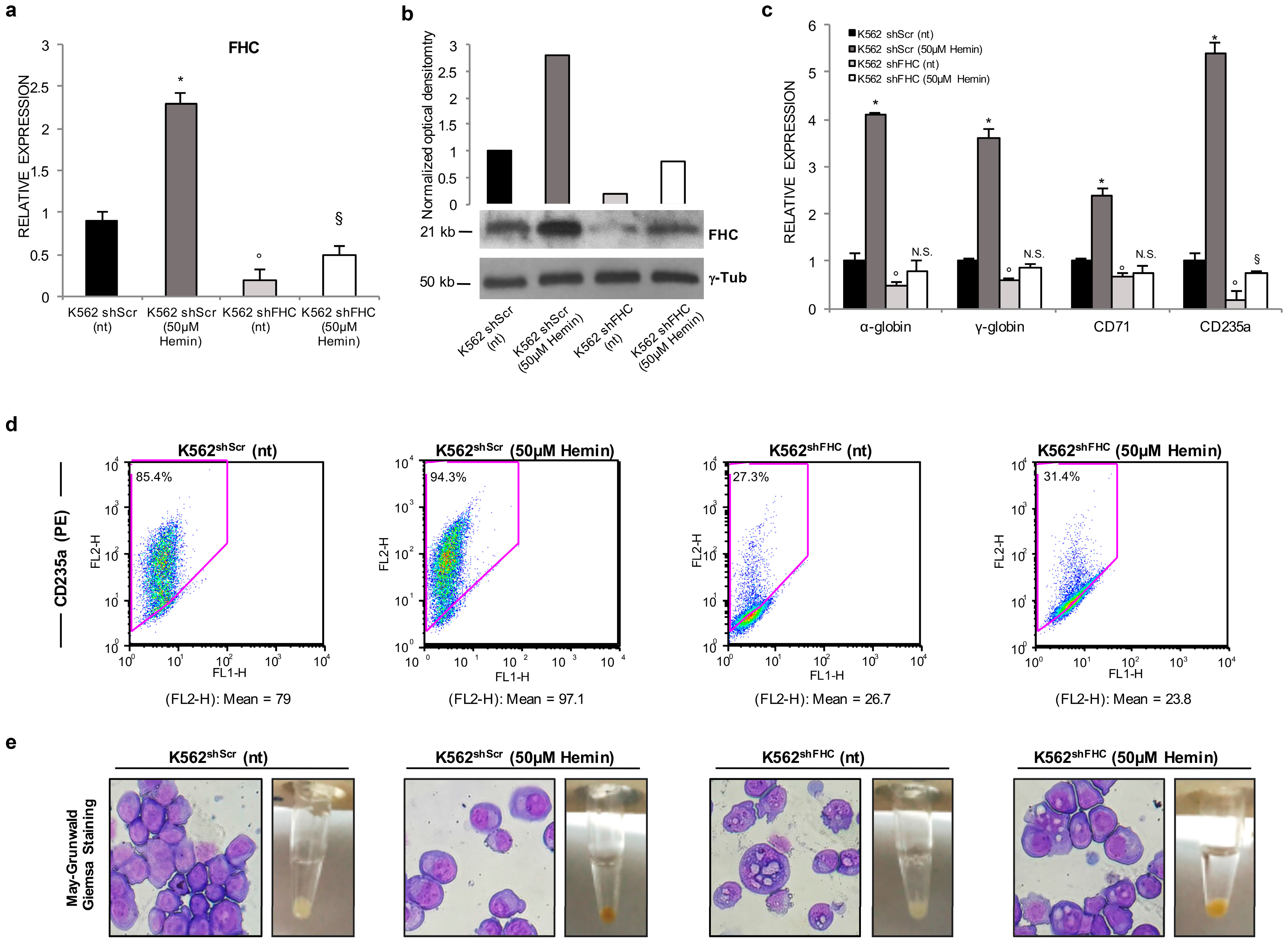
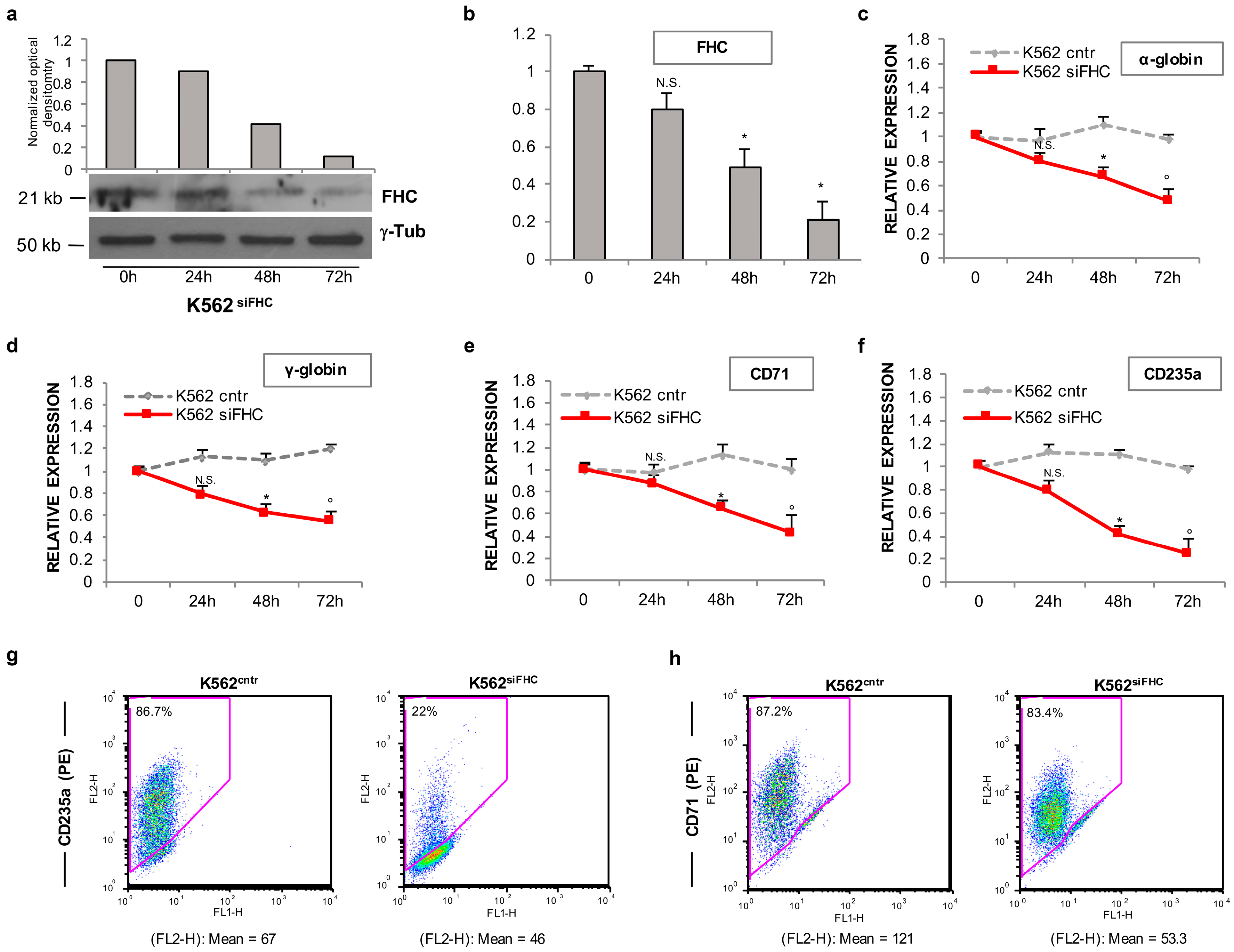
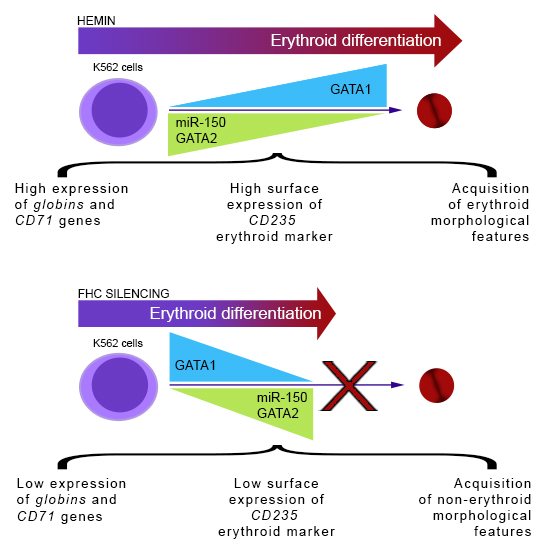
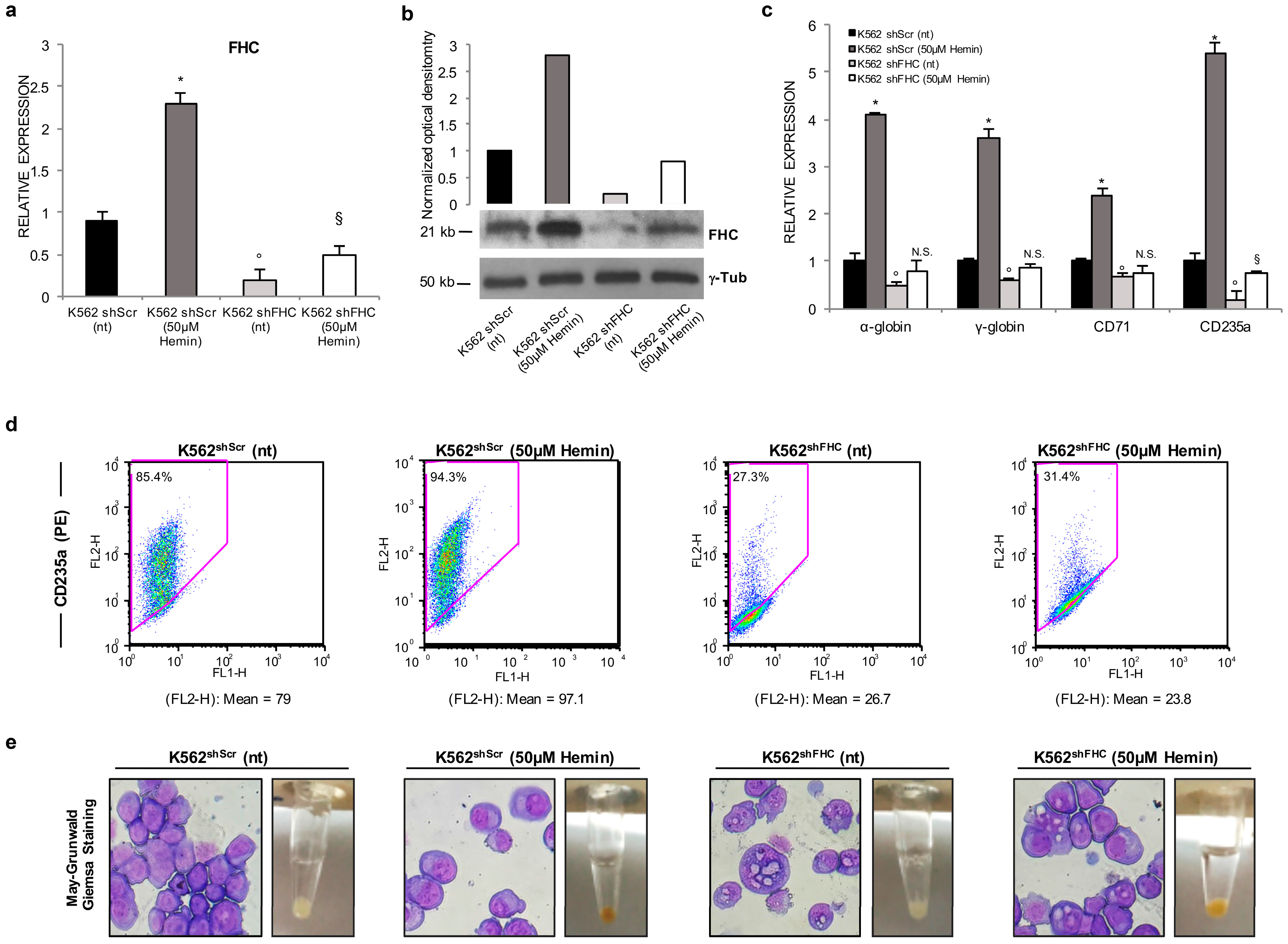
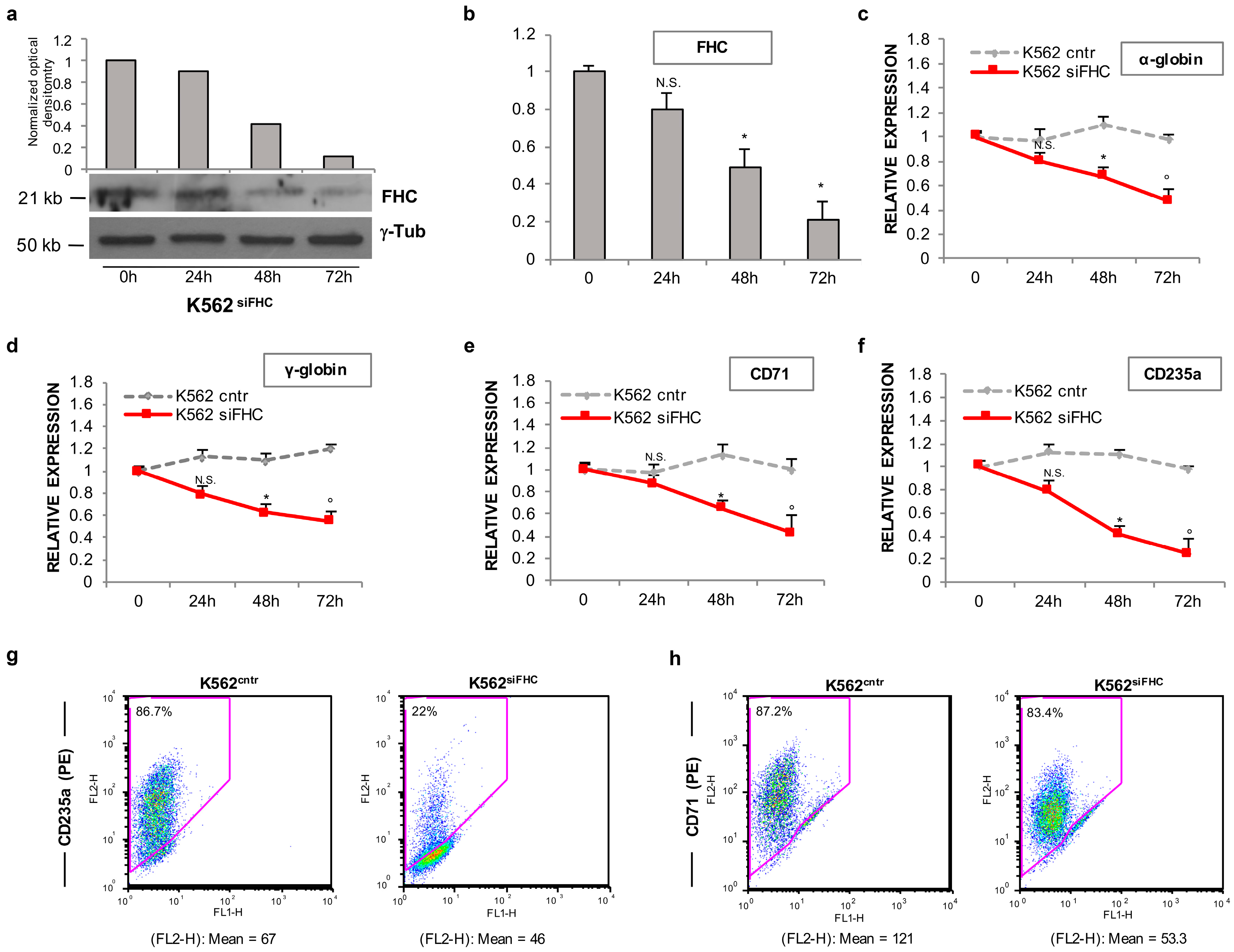
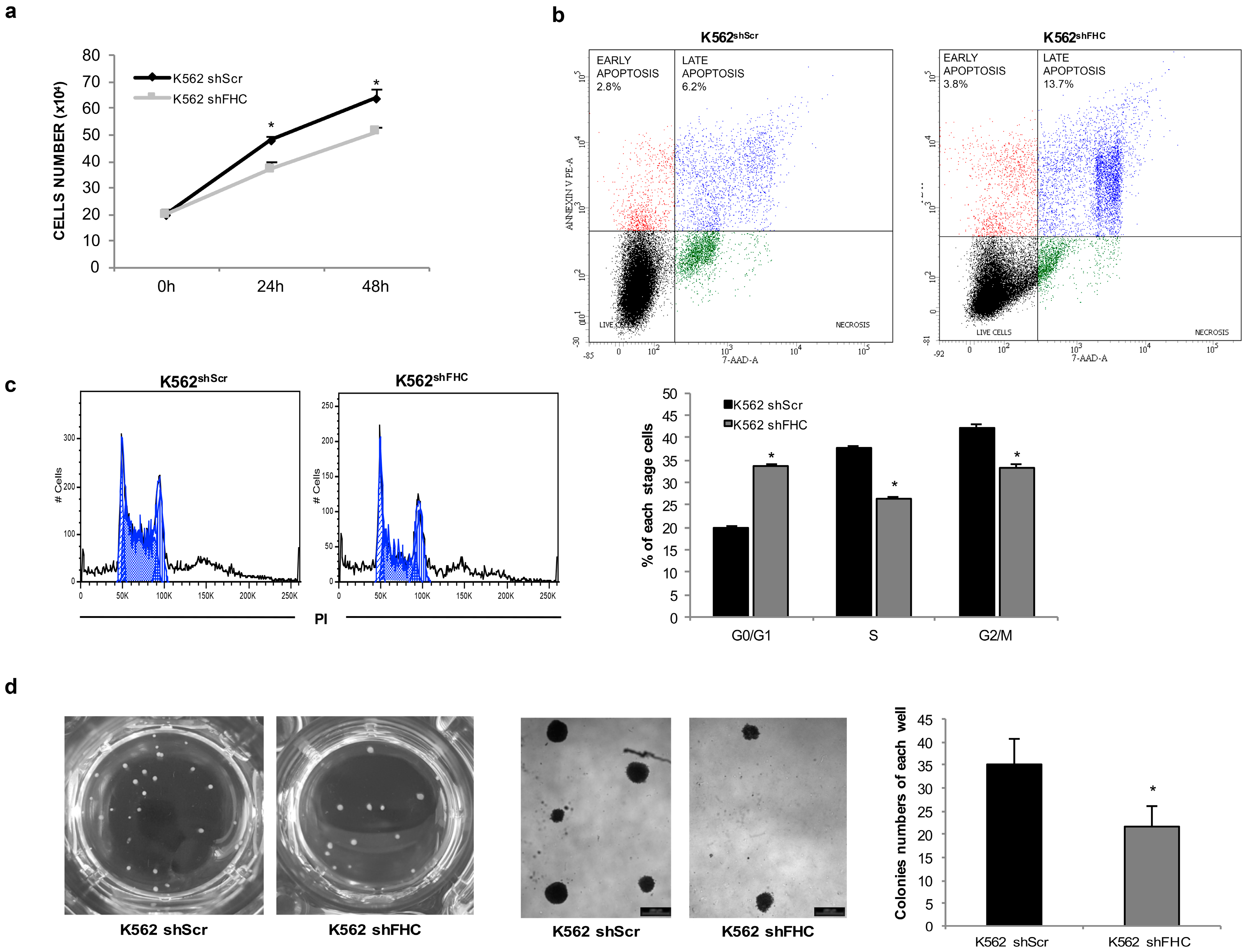
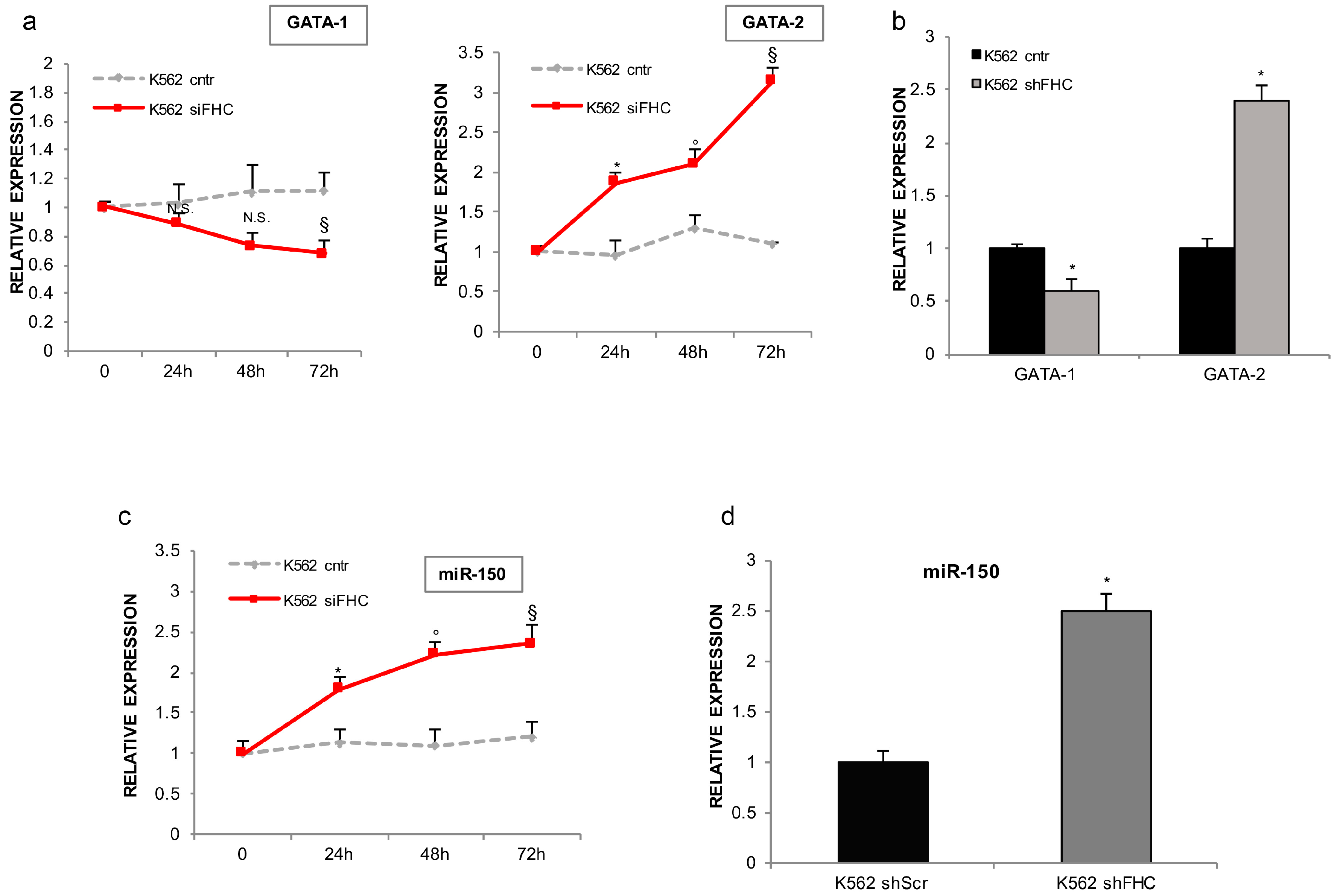
2.1. Ferritin Heavy Subunit (FHC) Knockdown Negatively Regulates the Erythroid Fate of K562 Cells
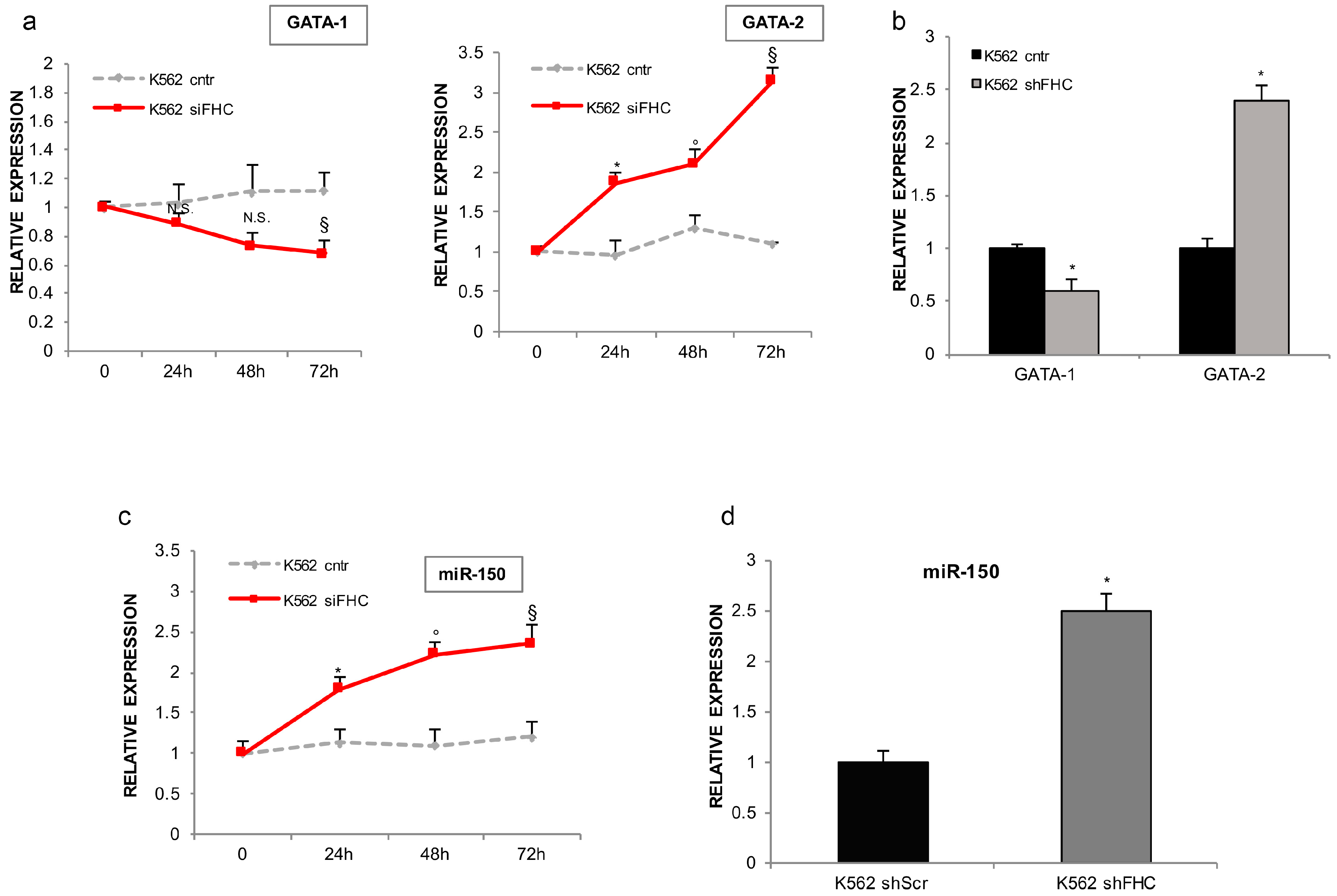
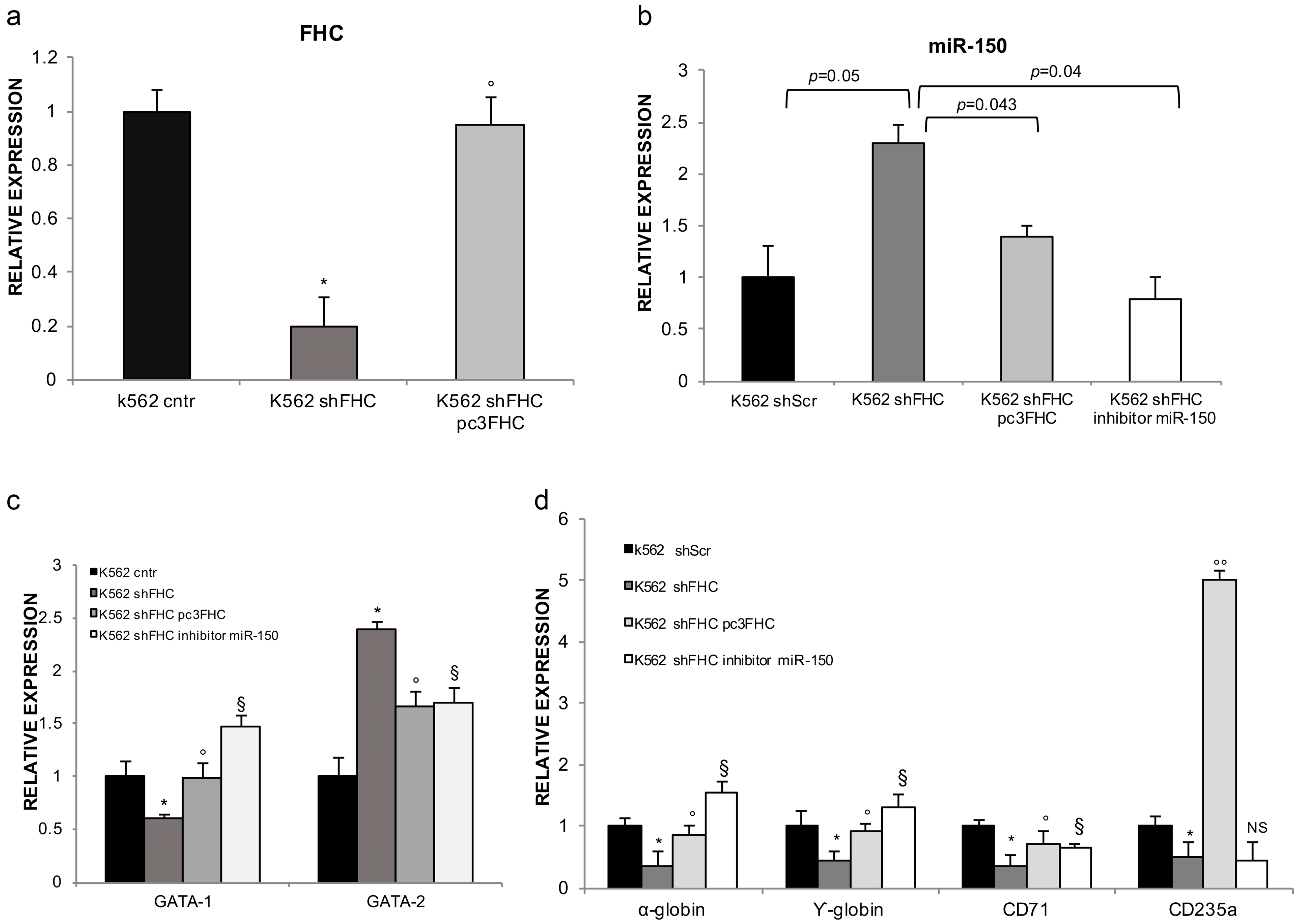
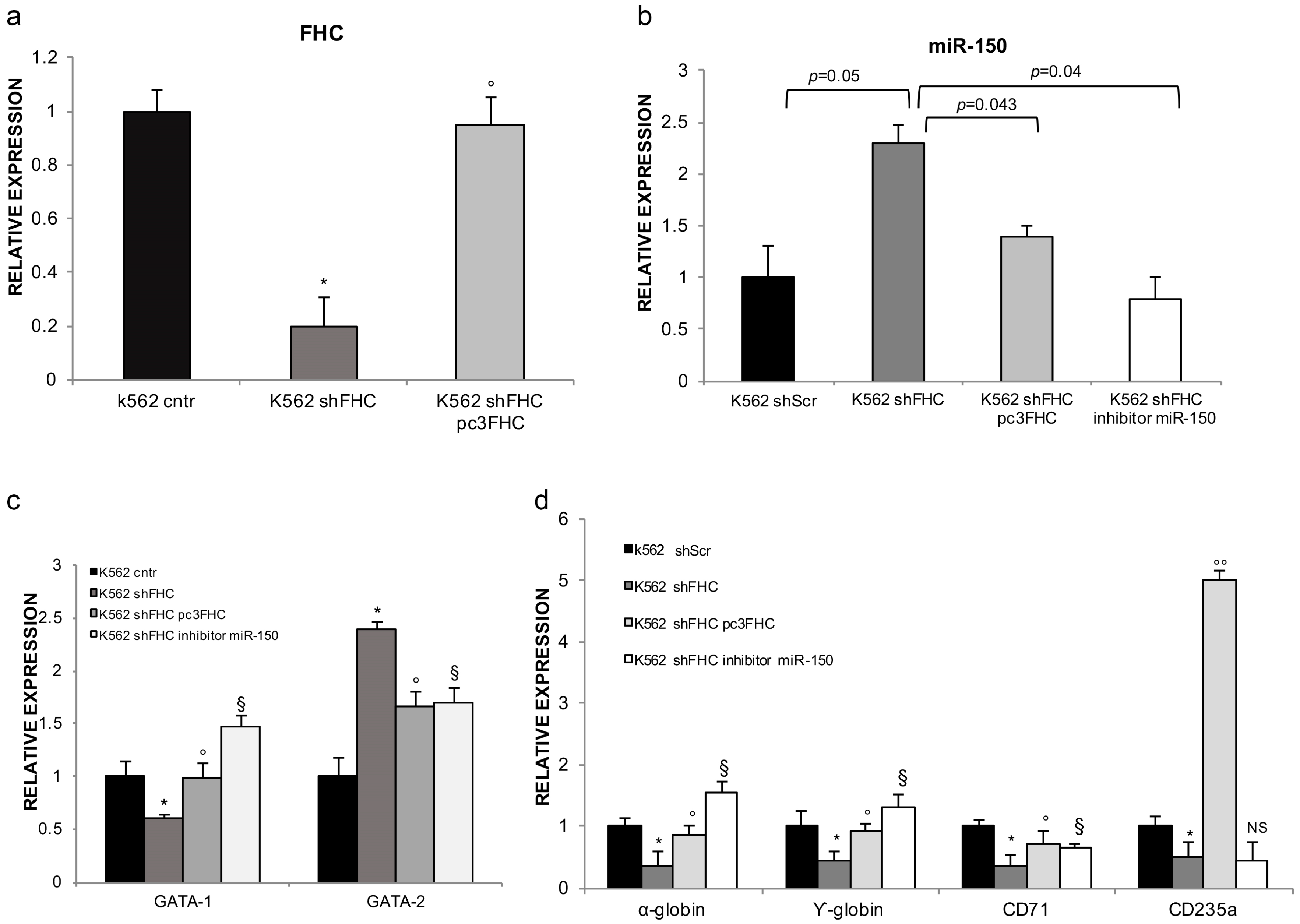
2.2. FHC Effects on K562 Erythroid Fate Are Mediated by miR-150
3. Materials and Methods
3.1. Cell Culture
3.2. K562 Cells Transfection and Transduction
3.3. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
3.4. TaqMan miR-150 Analysis
3.5. Western Blot Analysis
3.6. Flow Cytometry Analyses
3.7. Direct Cell Counting
3.8. Haematopoietic Colony Formation Assay
3.9. Cell Morphology Assay
3.10. Statistical Analysis
3.11. Bioinformatic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Weissman, I.L.; Anderson, D.J. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, G. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 2005, 33, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Fajtova, M.; Kovarikova, A.; Svec, P.; Kankuri, E.; Sedlak, J. Immunophenotypic profile of nucleated erythroid progenitors during maturation in regenerating bone marrow. Leuk. Lymphoma 2013, 54, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo. Blood 2013, 121, 3246–3253. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Kobayashi-Osaki, M.; Tsutsumi, S.; Pan, X.; Ohmori, S.; Takai, J.; Moriguchi, T.; Ohneda, O.; Ohneda, K.; Shimizu, R.; et al. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells 2013, 18, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Marr, C.; Schroeder, T.; Theis, F.J. Hierarchical differentiation of myeloid progenitors is encoded in the transcription factor network. PLoS ONE 2011, 6, e22649. [Google Scholar] [CrossRef] [PubMed]
- Teruel-Montoya, R.; Kong, X.; Abraham, S.; Ma, L.; Kunapuli, S.P.; Holinstat, M.; Shaw, C.A.; McKenzie, S.E.; Edelstein, L.C.; Bray, P.F. MicroRNA expression differences in human hematopoietic cell lineages enable regulated transgene expression. PLoS ONE 2014, 9, e102259. [Google Scholar] [CrossRef] [PubMed]
- Lazare, S.S.; Wojtowicz, E.E.; Bystrykh, L.V.; de Haan, G. MicroRNAs in hematopoiesis. Exp. Cell Res 2014, 329, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Dasaradhi, P.V.; Mohmmed, A.; Malhotra, P.; Bhatnagar, R.K.; Mukherjee, S.K. RNA interference: Biology, Mechanisms, and Applications. Microbiol. Mol. Biol. 2003, 67, 657–685. [Google Scholar] [CrossRef]
- Bissels, U.; Bosio, A.; Wagner, W. MicroRNAs are shaping the hematopoietic landscape. Haematologica 2012, 97, 160–167. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jiang, X.; Chen, J. The role of miR-150 in normal and malignant hematopoiesis. Oncogene 2014, 33, 3887–3893. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar] [CrossRef] [PubMed]
- Doré, L.C.; Crispino, J.D. Transcription factor networks in erythroid cell and megakaryocyte development. Blood 2011, 118, 231–239. [Google Scholar] [CrossRef] [PubMed]
- García, P.; Frampton, J. Hematopoietic lineage commitment: miRNAs add specificity to a widely expressed transcription factor. Dev. Cell 2008, 14, 815–816. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Guo, S.; Ebert, B.L.; Zhang, H.; Peng, X.; Bosco, J.; Pretz, J.; Schlanger, R.; Wang, J.Y.; Mak, R.H.; et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev. Cell 2008, 14, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sankaran, V.G.; Lodish, H.F. MicroRNAs in erythroid and megakaryocytic differentiation and megakaryocyte–erythroid progenitor lineage commitment. Leukemia 2012, 26, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, Y.; Han, X.; Zhao, X.; Peng, Y.; Li, Y.; Peng, M.; Song, J.; Wu, K.; Sun, S.; et al. miR-150 inhibits terminal erythroid proliferation and differentiation. Oncotarget 2015, 6, 43033–43047. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ginzburg, Y.Z. Crosstalk between Iron Metabolism and Erythropoiesis. Adv. Hematol. 2010, 605435. [Google Scholar] [CrossRef] [PubMed]
- Goodnough, L.T.; Skikne, B.; Brugnara, C. Erythropoietin, iron, and erythropoiesis. Blood 2000, 96, 823–833. [Google Scholar] [PubMed]
- Eliades, A.; Matsuura, S.; Ravid, K. Oxidases and reactive oxygen species during hematopoiesis: A focus on megakaryocytes. J. Cell. Physiol. 2012, 227, 3355–3362. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Q.; Zhao, Y.S.; Guo, J.; Zhao, S.D.; Song, L.X.; Fei, C.M.; Zhang, Z.; Li, X.; Chang, C.K. Iron overload promotes erythroid apoptosis through regulating HIF-1a/ROS signaling pathway in patients with myelodysplastic syndrome. Leuk. Res. 2017, 58, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; MacKenzie, E.L.; Hailemariam, K.; Sakamoto, K.; Tsuji, Y. Hemin-Mediated Regulation of an Antioxidant-Responsive Element of the Human Ferritin H Gene and Role of Ref-1 during Erythroid Differentiation of K562 Cells. Mol. Cell. Biol. 2006, 26, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, F.; Zolea, F.; Bisognin, A.; Di Sanzo, M.; Saccoman, C.; Scumaci, D.; Aversa, I.; Panebianco, M.; Faniello, M.C.; Bortoluzzi, S.; et al. H-ferritin-regulated microRNAs modulate gene expression in K562 cells. PLoS ONE 2015, 10, e0122105. [Google Scholar] [CrossRef] [PubMed]
- Zolea, F.; Biamonte, F.; Candeloro, P.; Di Sanzo, M.; Cozzi, A.; Di Vito, A.; Quaresima, B.; Lobello, N.; Trecroci, F.; Di Fabrizio, E.; et al. H ferritin silencing induces protein misfolding in K562 cells: A Raman analysis. Free Radic. Biol. Med. 2015, 89, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Zolea, F.; Biamonte, F.; Battaglia, A.M.; Faniello, M.C.; Cuda, G.; Costanzo, F. Caffeine Positively Modulates Ferritin Heavy Chain Expression in H460 Cells: Effects on Cell Proliferation. PLoS ONE 2016, 11, e0163078. [Google Scholar] [CrossRef] [PubMed]
- Lobello, N.; Biamonte, F.; Pisanu, M.E.; Faniello, M.C.; Jakopin, Ž.; Chiarella, E.; Giovannone, E.D.; Mancini, R.; Ciliberto, G.; Cuda, G.; et al. Ferritin heavy chain is a negative regulator of ovarian cancer stem cell expansion and epithelial to mesenchymal transition. Oncotarget 2016, 7, 62019–62033. [Google Scholar] [CrossRef] [PubMed]
- Misaggi, R.; Di Sanzo, M.; Cosentino, C.; Bond, H.M.; Scumaci, D.; Romeo, F.; Stellato, C.; Giurato, G.; Weisz, A.; Quaresima, B.; et al. Identification of H ferritin-dependent and independent genes in K562 differentiating cells by targeted gene silencing and expression profiling. Gene 2014, 535, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.F.; Yu, J.; Peng, H.; Du, Z.W.; Liu, X.L.; Ma, Y.N.; Zhang, X.; Zhang, Y.; Zhao, H.L.; Zhang, J.W. Differential expression changes in K562 cells during the hemin-induced erythroid differentiation and the phorbolmyristate acetate (PMA)-induced megakaryocytic differentiation. Mol. Cell. Biochem. 2006, 292, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Broyles, R.H.; Belegu, V.; DeWitt, C.R.; Shah, S.N.; Stewart, C.A.; Pye, Q.N.; Floyd, R.A. Specific repression of β-globin promoter activity by nuclear ferritin. Proc. Natl. Acad. Sci. USA 2001, 98, 9145–9150. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yu, J.; Yang, G.H.; Wang, X.S.; Zhang, J.W. Regulation of erythroid differentiation by miR-376a and its targets. Cell Res. 2011, 21, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H. Role of transcription factors in differentiation and reprogramming of hematopoietic cells. Keio J. Med. 2011, 60, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.J.; Watts, J.A.; Vakoc, C.R.; Yao, Y.; Wang, H.; Hardison, R.C.; Blobel, G.A.; Chodosh, L.A.; Weiss, M.J. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 2004, 104, 3136–3147. [Google Scholar] [CrossRef] [PubMed]
- Whitelaw, E.; Tsai, S.F.; Hogben, P.; Orkin, S.H. Regulated expression of globin chains and the erythroid transcription factor GATA-1 during erythropoiesis in the developing mouse. Mol. Cell. Biol. 1990, 10, 6596–6606. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, L.C.; McKenzie, S.E.; Shaw, C.; Holinstat, M.A.; Kunapuli, S.P.; Bray, P.F. MicroRNAs in platelet production and activation. J. Thromb. Haemost. 2013, 1, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z. Long non-coding RNA FTH1P3 facilitates oral squamous cell carcinoma progression by acting as a molecular sponge of miR-224–5p to modulate fizzled 5 expression. Gene 2017, 607, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, E.; Carrà, G.; Scicchitano, S.; Codispoti, B.; Mega, T.; Lupia, M.; Pelaggi, D.; Marafioti, M.G.; Aloisio, A.; Giordano, M.; et al. UMG Lenti: Novel lentiviral vectors for efficient transgene- and reporter gene expression in human early hematopoietic progenitors. PLoS ONE 2014, 12, e114795. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | 5′-TGA TGA CAT CAA GAA GGT GGT GAA G-3′ | 5′-TCC TTG GAG GCC ATG TGG GCC AT-3′ |
| FHC | 5′-CAT CAA CCG CCA GAT CAA C-3′ | 5′-GAT GGC TTT CAC CTG CTC AT-3′ |
| α-globin | 5′-GTG GAC GAC ATG CCC AAC-3′ | 5′-TAT TTG GAG GTC AGC ACG GT-3′ |
| γ-globin | 5′-CAG AAA TAC ACA TAC ACA CTT CC-3′ | 5′-GAG AGA TCA CAC ATG ATT TTC TT-3′ |
| CD71 | 5′-ACT GGT CCA TGC TAA TTT TGG T-3′ | 5′-AGT TCT GCG TTA ACA ATG GGA-3′ |
| CD235a | 5′-GAG AAA GGG TAC AAC TTG CC-3′ | 5′-CAT TGA TCA CTT GTC TCT GG-3′ |
| GATA-1 | 5′-GAT GAA TGG GCA GAA CAG GC-3′ | 5′-TAG CTT GTA GTA GAG GCC GC-3′ |
| GATA-2 | 5′-GAA CCG ACC ACT CAT CAA GC-3′ | 5′-GCA GCT TGT AGT AGA GGC CA-3′ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolea, F.; Battaglia, A.M.; Chiarella, E.; Malanga, D.; Marco, C.D.; Bond, H.M.; Morrone, G.; Costanzo, F.; Biamonte, F. Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression. Int. J. Mol. Sci. 2017, 18, 2167. https://doi.org/10.3390/ijms18102167
Zolea F, Battaglia AM, Chiarella E, Malanga D, Marco CD, Bond HM, Morrone G, Costanzo F, Biamonte F. Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression. International Journal of Molecular Sciences. 2017; 18(10):2167. https://doi.org/10.3390/ijms18102167
Chicago/Turabian StyleZolea, Fabiana, Anna Martina Battaglia, Emanuela Chiarella, Donatella Malanga, Carmela De Marco, Heather Mandy Bond, Giovanni Morrone, Francesco Costanzo, and Flavia Biamonte. 2017. "Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression" International Journal of Molecular Sciences 18, no. 10: 2167. https://doi.org/10.3390/ijms18102167
APA StyleZolea, F., Battaglia, A. M., Chiarella, E., Malanga, D., Marco, C. D., Bond, H. M., Morrone, G., Costanzo, F., & Biamonte, F. (2017). Ferritin Heavy Subunit Silencing Blocks the Erythroid Commitment of K562 Cells via miR-150 up-Regulation and GATA-1 Repression. International Journal of Molecular Sciences, 18(10), 2167. https://doi.org/10.3390/ijms18102167