Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings

 ,
,  and
and 
Abstract

1. Introduction
2. Results
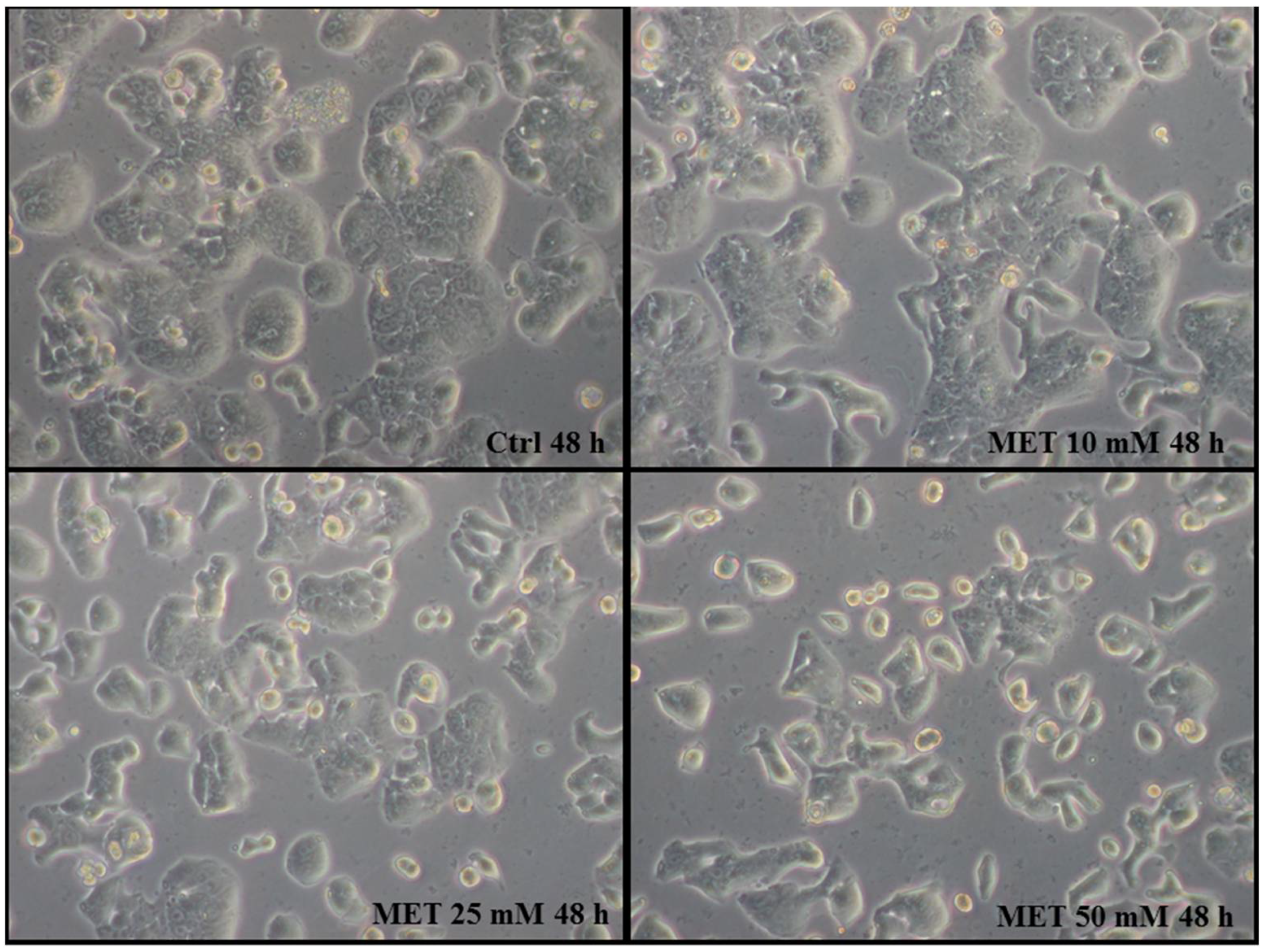
2.1. Metformin Suppresses the Proliferation of HT29 Cells in a Dose- and Time-Dependent Manner
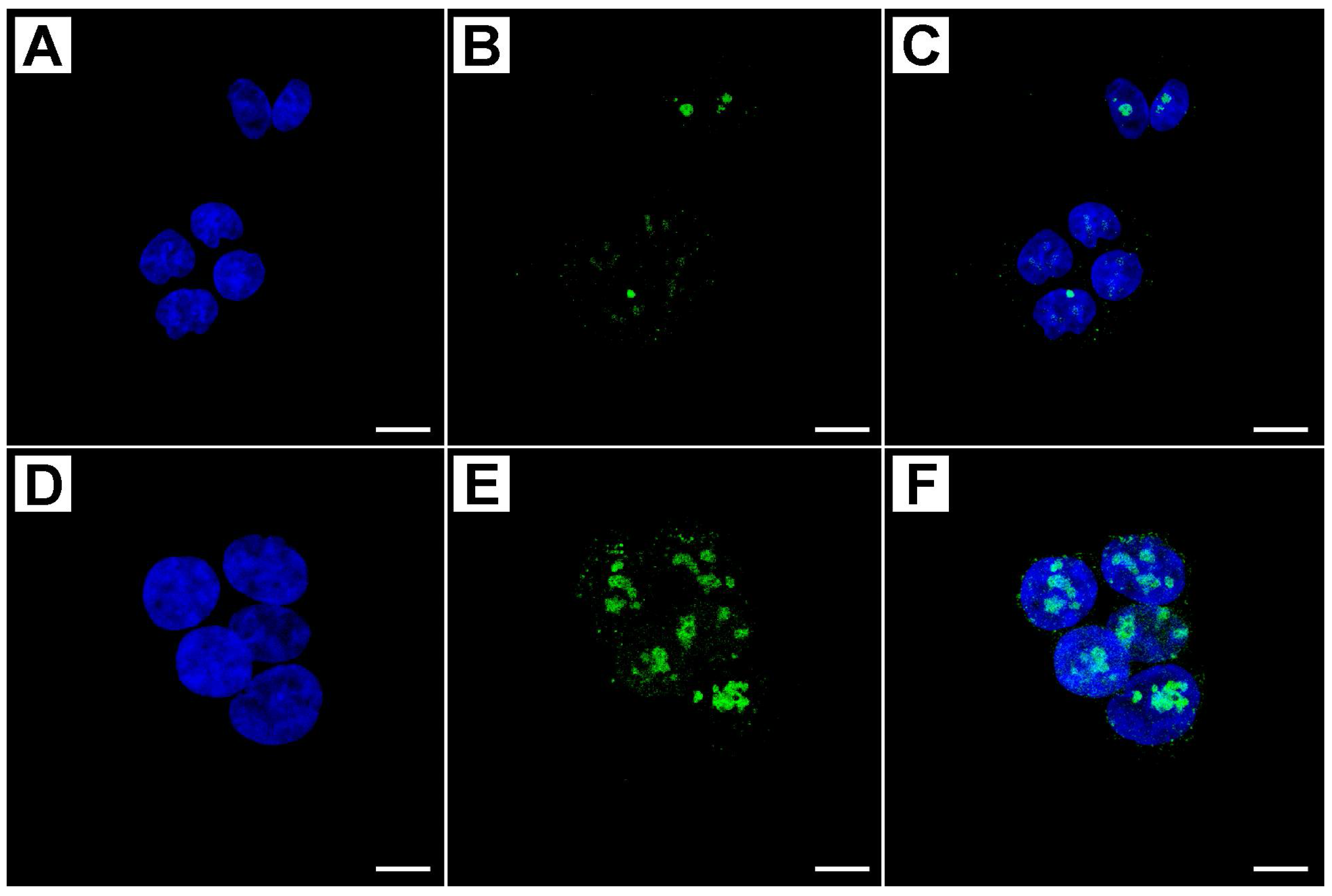
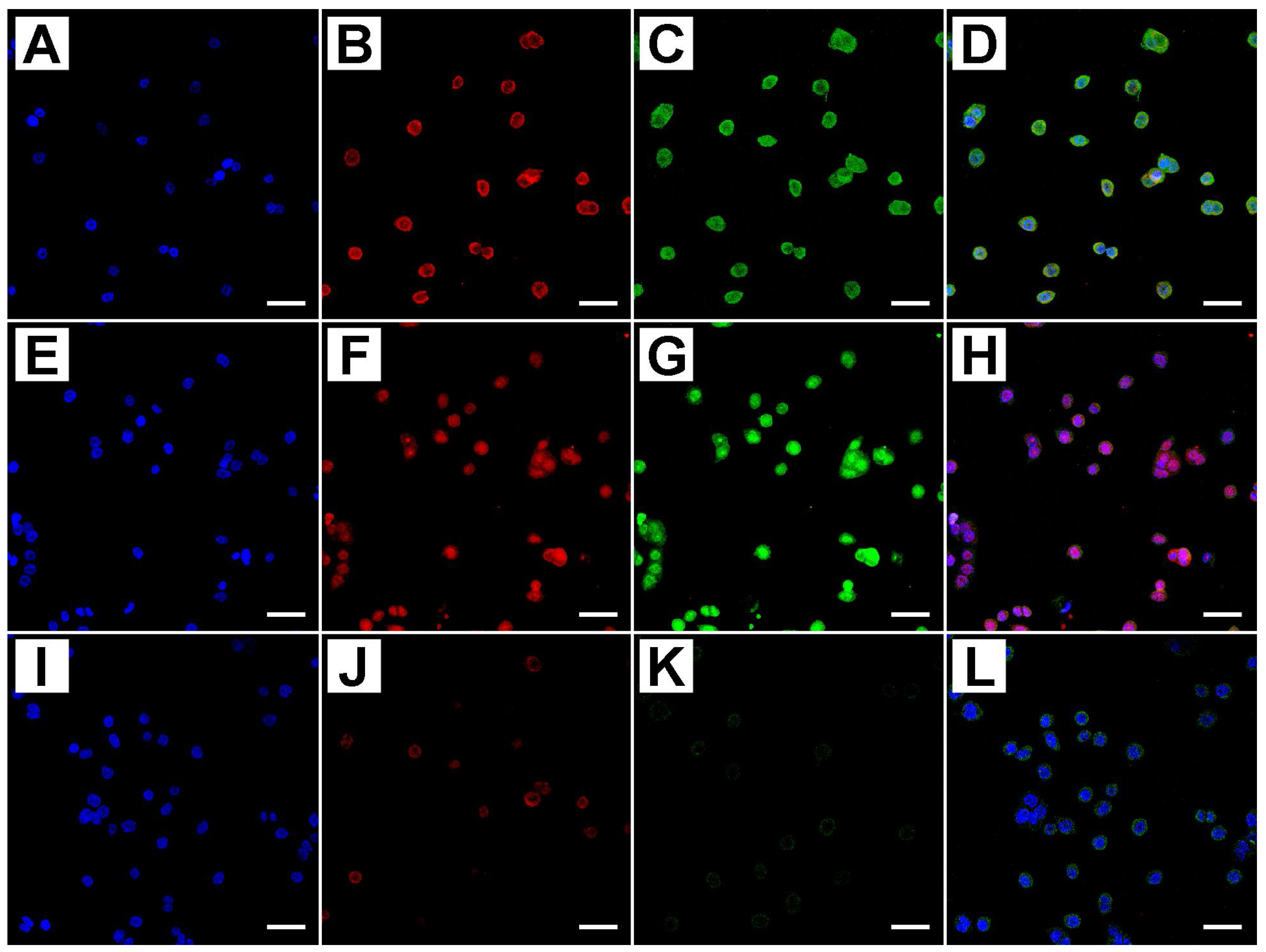
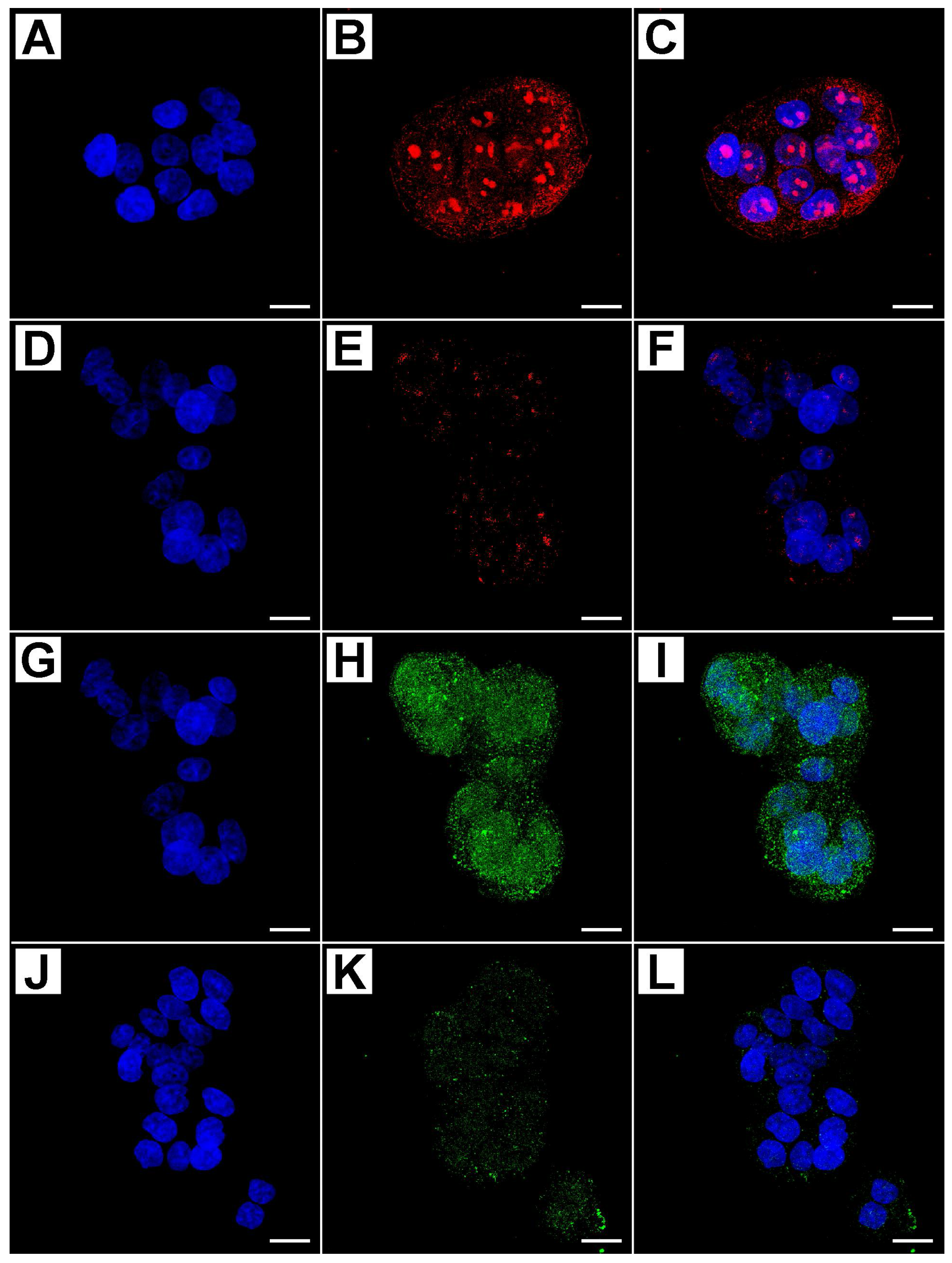
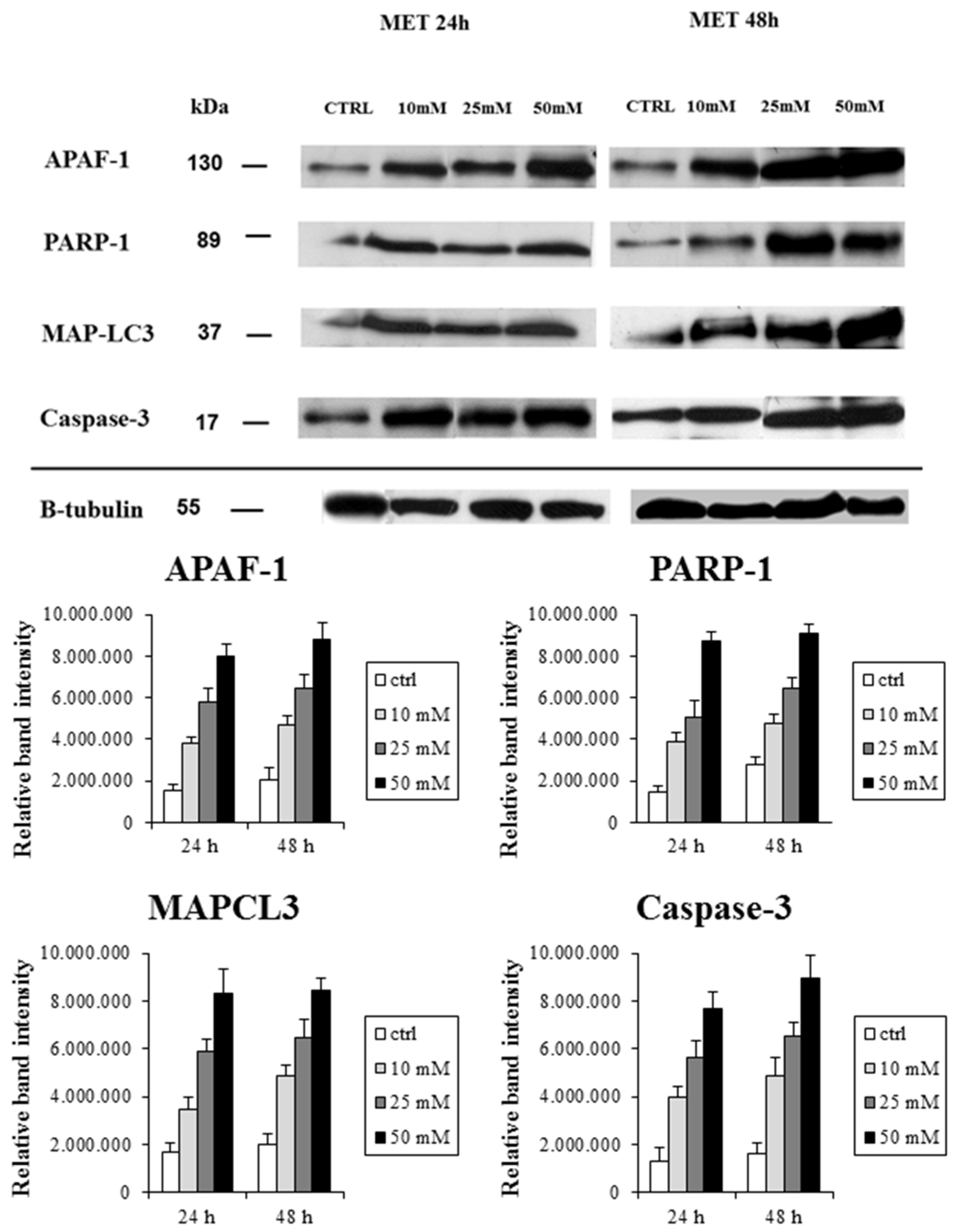
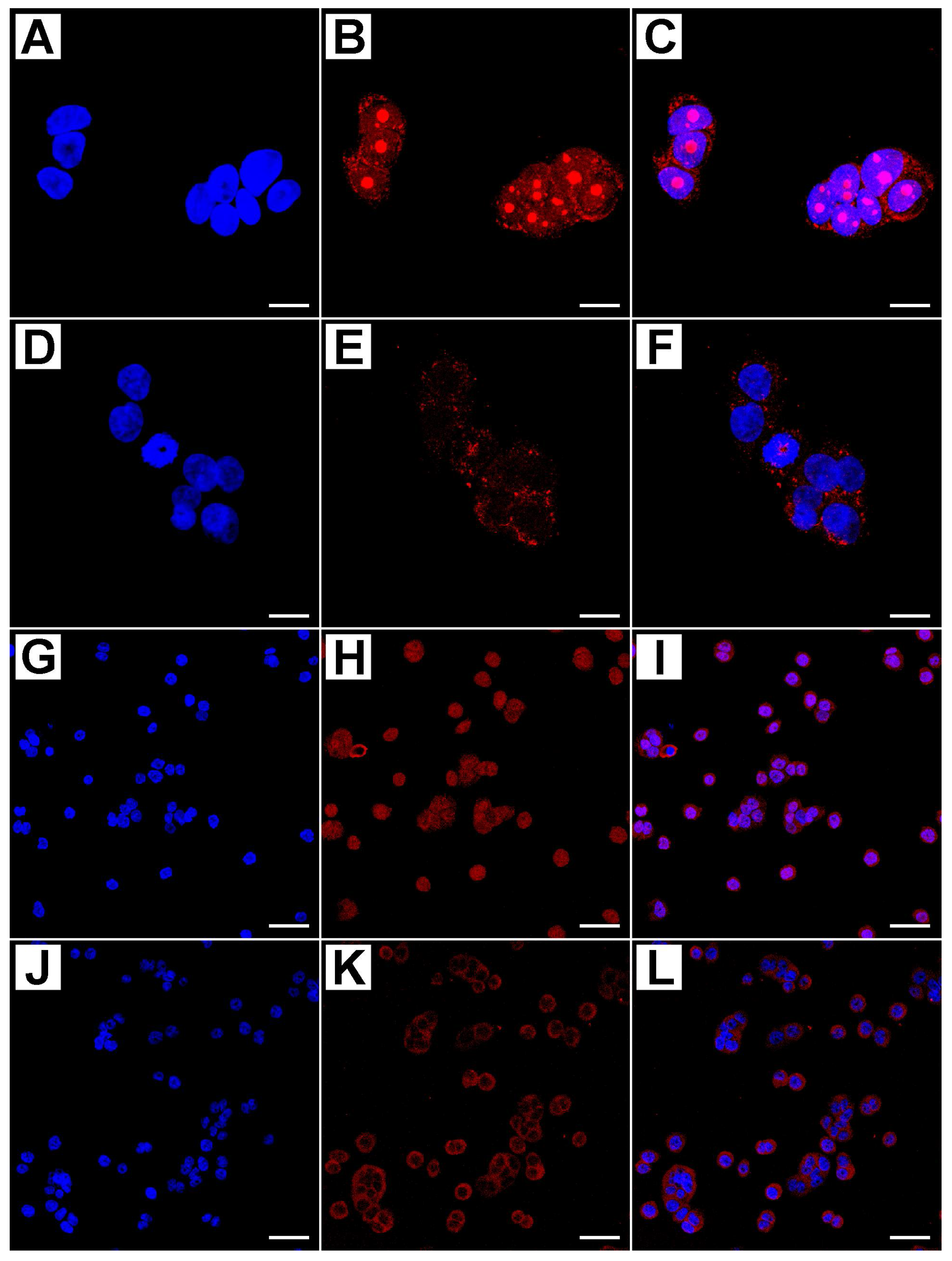
2.2. Metformin Alters NRF-2 and NF-κB Expression in HT29 Cells in a Dose- and Time-Dependent Manner
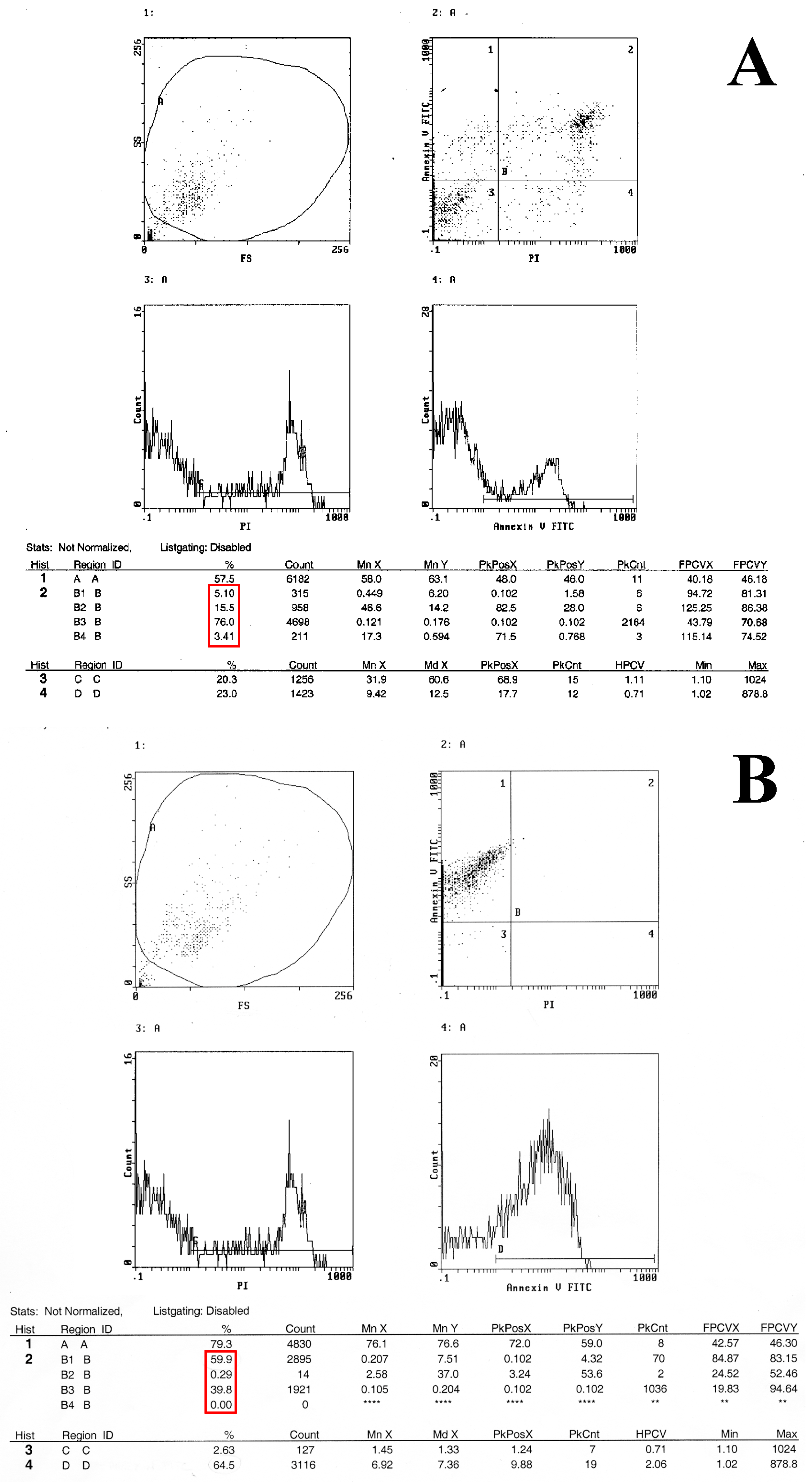
2.3. Flow Cytometry Analysis Demonstrates That Metformin Exerts an Inhibitory Effect on HT29 Cells Survival
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Counting
4.3. Western Blot Analysis
4.4. Evaluation of Immunofluorescence by Confocal Microscopy
4.5. Flow Cytometry Analysis
4.6. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Stacey, A.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, S.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Umar, A.; Brawley, O.W. Colorectal carcinoma in black and white race. Cancer Metastab. Rev. 2003, 22, 67–82. [Google Scholar] [CrossRef]
- Das, D.; Arber, N.; Jankowski, J.A. Chemoprevention of colorectal cancer. Digestion 2007, 76, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Dubois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Psaty, B.M.; Furberg, C.D. COX-2 inhibitors—Lessons in drug safety. N. Engl. J. Med. 2005, 352, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr.; McLaren, C.E.; Pelot, D.; Fujikawa-Brooks, S.; Carpenter, P.M.; Hawk, E.; Kelloff, G.; Lawson, M.J.; Kidao, J.; McCracken, J.; et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: A randomized placebo-controlled, double-blind trial. Cancer Prev. Res. 2008, 1, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Limburg, P.J.; Anderson, K.E.; Johnson, T.W; Jacobs, D.R., Jr; Lazovich, D.; Hong, C.P.; Nicodemus, K.K.; Folsom, A.R. Diabetes mellitus and subsite-specific colorectal cancer risks in the Iowa Women's Health Study. Cancer Epidemiol. Biomark. Prev. 2005, 14, 133–137. [Google Scholar] [CrossRef]
- Larsson, S.C.; Giovannucci, E.; Wolk, A. Diabetes and colorectal cancer incidence in the cohort of Swedish men. Diabetes Care 2005, 28, 1805–1807. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giovannucci, E.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Physical activity, obesity, and risk for colon cancer and adenoma in men. Ann. Intern. Med. 1995, 122, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Frezza, E.E.; Wachtel, M.S.; Chiriva-Internati, M. Influence of obesity on the risk of developing colon cancer. Gut 2006, 55, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Witters, L.A. The blooming of the French lilac. J. Clin. Investig. 2001, 108, 1105–1107. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Meier, C.; Krähenbühl, S.; Jick, S.S.; Meier, C.R. Metformin, sulfonylureas, or other antidiabetes drugs and the risk of lactic acidosis or hypoglycemia: A nested case-control analysis. Diabetes Care 2008, 31, 2086–2091. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Lamina, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 medited glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [PubMed]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Paraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bosi, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Sruhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Lee, H.; Dings, R.P.; Williams, B.; Powers, J.; Santos, T.D.; Cho, B.H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef] [PubMed]
- Jurjus, A.; Eid, A.; Al Kattar, S.; Zeenny, M.N.; Gerges-Geagea, A.; Haydar, H.; Hilal, A.; Oueidat, D.; Matar, M.; Tawilah, J.; et al. Inflammatory bowel disease, colorectal cancer and type 2 diabetes mellitus: The links. BBA Clin. 2016, 5, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Oudejans, J.J.; Muris, J.J.; Meijer, C.J. Inhibition of caspase 9 and notcaspase 8 mediated apoptosis may determine clinical response tochemotherapy in primary nodal diffuse large B-cell lymphomas. Cell Cycle 2005, 4, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Li, Z.S.; Zou, D.W.; Jin, Z.D.; Gao, J.; Xu, G.M. Metformin induces apoptosis of pancreatic cancer cells. World J. Gastroenterol. 2008, 14, 7192–7198. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Janjetovic, K.; Harhaji-Trajkovic, L.; Misirkic-Marjanovic, M.; Vucicevic, L.; Stevanovic, D.; Zogovic, N.; Sumarac-Dumanovic, M.; Micic, D.; Trajkovic, V. In vitro and in vivo anti-melanoma action of metformin. Eur. J. Pharmacol. 2011, 668, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Gong, J.; Iwama, H.; Kitanaka, A.; Tani, J.; Miyoshi, H.; Nomura, K.; Mimura, S.; Kobayashi, H.; Aritomo, Y.; et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentration matters. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Interactive effects of Nrf2 geno-type and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis 2003, 24, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Itoh, K.; Kumagai, Y.; Oyasu, R.; Hattori, K.; Kawai, K.; Shimazui, T.; Akaza, H.; Yamamoto, M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004, 64, 6424–6431. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T., Jr.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, G.; Sekhar, K.R.; Jowhar, D.; Samson, P.C.; Wikswo, J.P.; Beauchamp, R.D.; Datta, P.K.; Freeman, M.L. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene 2010, 29, 3703–3714. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Wang X, J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K. NRF2 and cancer: The good, the bad and the importance of context. Nat Rev Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkowa-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1–Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Akhdar, H.; Loyer, P.; Rauch, C.; Corlu, A.; Guillouzo, A.; Morel, F. Involvement of Nrf2 activation in resistance to 5-fluorouracil in human colon cancer HT-29 cells. Eur. J. Cancer 2009, 45, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Caamaño, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Viennois, E.; Chen, F.; Merlin, D. NF-κB pathway in colitis-associated cancers. Transl. Gastrointest. Cancer 2013, 2, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation Meets Cancer, with NF-[Kappa] B as the Matchmaker. Nat. Immunol. 2011, 19, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-κB and Epithelial to Mesenchymal Transition of Cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Sena, P.; Mariani, F.; Mancini, S.; Benincasa, M.; Magnani, G.; Pedroni, M.; Palumbo, C.; Roncucci, L. Autophagy is upregulated during colorectal carcinogenesis, and in DNA microsatellite stable carcinomas. Oncol. Rep. 2015, 34, 3222–3230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eray M, M.; Mättö, N.; Kaartinen, L.; Andersson, L.; Pelkonen, J. Flow cytometric analysis of apoptotic subpopulations with a combination of Annexin V-FITC, propidium iodide, and SYTO 17. Cytometry 2001, 43, 134–142. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | IFIS (Mean ± SD)-24 h | IFIS (mean ± SD)-48 h | ||||||
|---|---|---|---|---|---|---|---|---|
| CTRL | 10 mM | 25 mM | 50 mM | CTRL | 10 mM | 25 mM | 50 mM | |
| PARP-1 | 20.0 ± 4.3 | 47.0 ± 11.0 | 63.0 ± 9.8 | 76.0 ± 15.0 | 23.0 ± 12.0 | 55.0 ± 6.7 | 74.0 ± 13.0 | 89.0 ± 22.0 |
| Caspase-3 | 36.0 ± 9.5 | 49.0 ± 23.0 | 69.0 ± 15.0 | 91.0 ± 7.4 | 32.0 ± 5.7 | 64.0 ± 17.0 | 97.0 ± 20.0 | 101.0 ± 29.0 |
| APAF-1 | 31.0 ± 7.5 | 54.0 ± 3.9 | 66.0 ± 19.0 | 85.0 ± 9.5 | 27.0 ± 7.0 | 72.0 ± 17.0 | 72.0 ± 17.0 | 93.0 ± 12.0 |
| MAP-LC3 | 26.0 ±4.7 | 45.0 ± 7.2 | 71.0 ± 7.6 | 93.0 ± 15.0 | 28.0 ± 8.3 | 57.0 ± 6.3 | 84.0 ± 5.4 | 103.0 ± 32.0 |
| NRF-2 | 55.0 ± 7.2 | 43.0 ± 6.3 | 31.0 ± 4.8 | 19.0 ± 3.7 | 63.0 ± 12.0 | 47.0 ± 5.4 | 30.0 ± 7.5 | 23.0 ± 3.0 |
| NF-κB | 60.0 ± 9.5 | 54.0 ± 8.3 | 47.0 ± 15.0 | 32.0 ± 7.4 | 67.0 ± 5.7 | 50.0 ± 17.0 | 33.0 ± 2.0 | 25.0 ± 2.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sena, P.; Mancini, S.; Benincasa, M.; Mariani, F.; Palumbo, C.; Roncucci, L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. Int. J. Mol. Sci. 2018, 19, 1478. https://doi.org/10.3390/ijms19051478
Sena P, Mancini S, Benincasa M, Mariani F, Palumbo C, Roncucci L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. International Journal of Molecular Sciences. 2018; 19(5):1478. https://doi.org/10.3390/ijms19051478
Chicago/Turabian StyleSena, Paola, Stefano Mancini, Marta Benincasa, Francesco Mariani, Carla Palumbo, and Luca Roncucci. 2018. "Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings" International Journal of Molecular Sciences 19, no. 5: 1478. https://doi.org/10.3390/ijms19051478
APA StyleSena, P., Mancini, S., Benincasa, M., Mariani, F., Palumbo, C., & Roncucci, L. (2018). Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. International Journal of Molecular Sciences, 19(5), 1478. https://doi.org/10.3390/ijms19051478