CREBH Regulates Systemic Glucose and Lipid Metabolism


Abstract
1. Introduction
2. The Gene Regulation of CREB3L3 in Response to Nutrient Condition
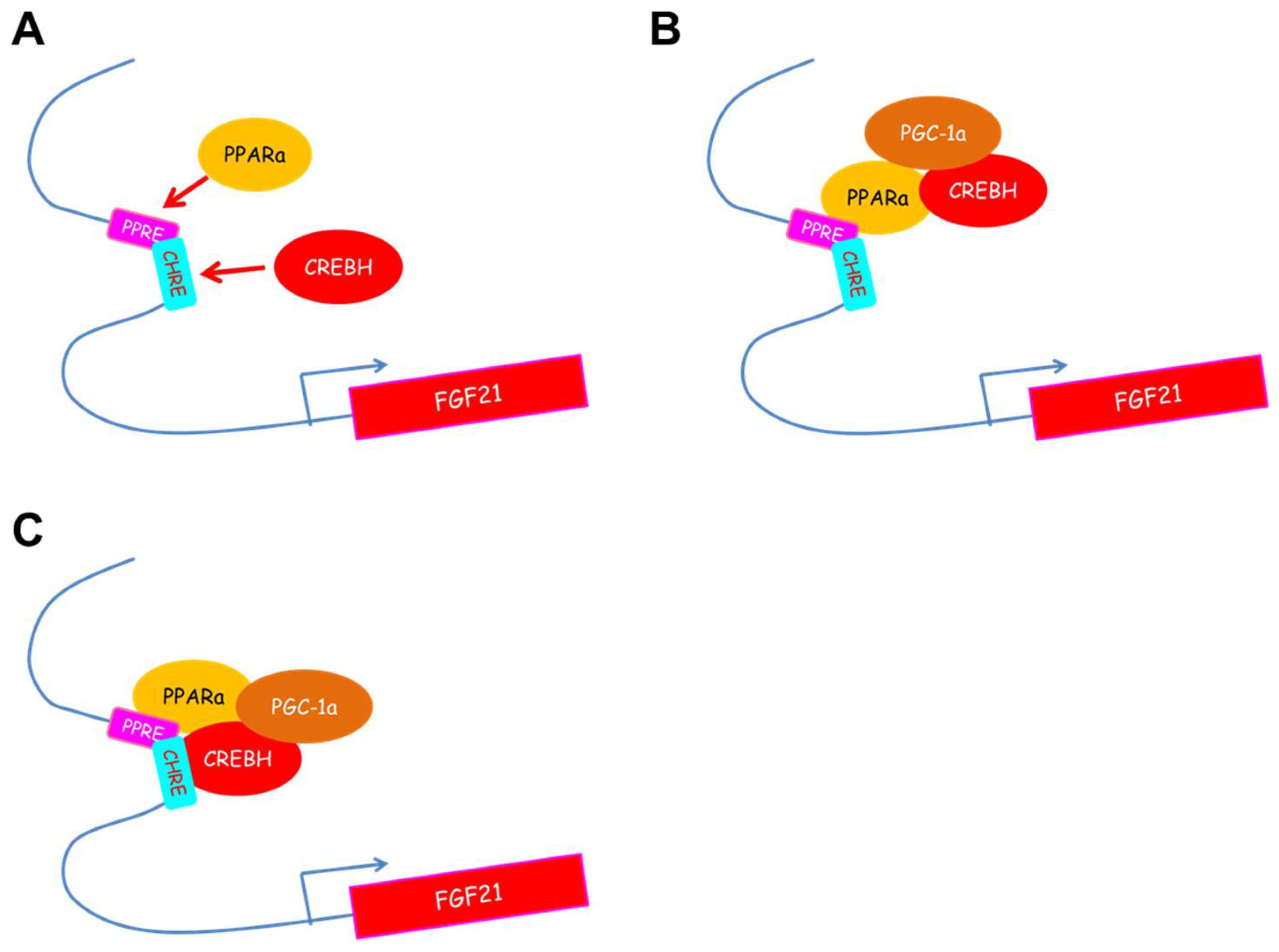
3. CREBH Regulates Fgf21 Expression in the Liver and Subsequently Regulates Glucose and Lipid Metabolism
4. CREBH Regulates Gluconeogenesis Gene Expression
5. CREBH Regulates Lipid Metabolism in Fatty Liver
5.1. Deficiency of CREBH Exacerbates Non-Alcoholic Fatty Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH)
5.2. CREBH Regulates Very Low-density Lipoprotein( VLDL) Particle Metabolism in Fatty Liver
6. CREBH Regulates Lipoprotein Metabolism
6.1. CREBH Regulates the Expression of Apoa4, a Multitasking Apolipoprotein, in the Liver and Small Intestine
6.2. CREBH Regulates Lipoprotein Metabolism in Response to Endotoxemia
7. Intestinal CREBH Overexpression Controls Intestinal Cholesterol Absorption
8. CREBH Regulates the Progression of Atherosclerosis
9. CREBH Rhythmically Interacts with the Transcription Factors for Lipid Metabolism
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Despres, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Mertens, I.L.; de Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell Biol. 2015, 208, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G. The role of the liver in metabolic homeostasis: Implications for inborn errors of metabolism. J. Inherit. Metab. Dis. 1991, 14, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Imai, J.; Watanabe, M.; Komatsu, T.; Suzuki, Y.; Kataoka, K.; Watanabe, S.; Tanigami, A.; Sugano, S. CREB-H: A novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the box-B element with a liver-specific expression. Nucleic Acids Res. 2001, 29, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Satoh, A.; Tezuka, H.; Han, S.I.; Takei, K.; Iwasaki, H.; Yatoh, S.; Yahagi, N.; Suzuki, H.; Iwasaki, Y.; et al. CREB3L3 controls fatty acid oxidation and ketogenesis in synergy with PPARα. Sci. Rep. 2016, 6, 39182. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Mendez, R.; Zheng, Z.; Chang, L.; Cai, J.; Zhang, R.; Zhang, K. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor alpha to regulate metabolic hormone FGF21. Endocrinology 2014, 155, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Giannikopoulos, P.; Duncan, S.A.; Wang, J.; Johansen, C.T.; Brown, J.D.; Plutzky, J.; Hegele, R.A.; Glimcher, L.H.; Lee, A.H. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat. Med. 2011, 17, 812–815. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Satoh, A.; Yabe, S.; Furusawa, M.; Tokushige, N.; Tezuka, H.; Mikami, M.; Iwata, W.; Shingyouchi, A.; Matsuzaka, T.; et al. Hepatic CREB3L3 Controls Whole-Body Energy Homeostasis and Improves Obesity and Diabetes. Endocrinology 2014, 155, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Chanda, D.; Yang, J.; Oh, H.; Kim, S.S.; Yoon, Y.S.; Hong, S.; Park, K.G.; Lee, I.K.; Choi, C.S.; et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010, 11, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.T.; Zhou, H.J.; Wong, C.M.; Lee, J.M.; Chan, C.P.; Qiang, B.Q.; Yuan, J.G.; Ng, I.O.; Jin, D.Y. The liver-enriched transcription factor CREB-H is a growth suppressor protein underexpressed in hepatocellular carcinoma. Nucleic Acids Res. 2005, 33, 1859–1873. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Xu, X.; Cho, S.; Lee, A.H. Loss of Transcription Factor CREBH Accelerates Diet-Induced Atherosclerosis in Ldlr−/− Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1772–1781. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, X.; Park, J.G.; So, J.S.; Hur, K.Y.; Lee, A.H. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J. Lipid Res. 2014, 55, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, M.; Cheng, X.; Shen, J.; Khound, R.; Zhang, K.; Su, Q. CREBH mediates metabolic inflammation to hepatic VLDL overproduction and hyperlipoproteinemia. J. Mol. Med. 2017, 95, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, M.; Sud, N.; Christian, P.; Shen, J.; Song, Y.; Pashaj, A.; Zhang, K.; Carr, T.; Su, Q. Glucagon regulates hepatic lipid metabolism via cAMP and Insig-2 signaling: Implication for the pathogenesis of hypertriglyceridemia and hepatic steatosis. Sci. Rep. 2016, 6, 32246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Park, J.G.; So, J.S.; Lee, A.H. Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology 2015, 61, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Orihara, K.; Oikawa, F.; Han, S.I.; Kuba, M.; Okuda, K.; Satoh, A.; Osaki, Y.; Takeuchi, Y.; Aita, Y.; et al. Intestinal CREBH overexpression prevents high-cholesterol diet-induced hypercholesterolemia by reducing Npc1l1 expression. Mol. Metab. 2016, 5, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Danno, H.; Ishii, K.A.; Nakagawa, Y.; Mikami, M.; Yamamoto, T.; Yabe, S.; Furusawa, M.; Kumadaki, S.; Watanabe, K.; Shimizu, H.; et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARα. Biochem. Biophys. Res. Commun. 2010, 391, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Han, S.I.; Murayama, Y.; Satoh, A.; Oikawa, F.; Ohno, H.; Osaki, Y.; Matsuzaka, T.; Sekiya, M.; Iwasaki, H.; et al. The selective PPARα modulator K-877 efficiently activates the PPARα pathway and improves lipid metabolism in mice. J. Diabetes Investig. 2017. [Google Scholar] [CrossRef] [PubMed]
- Luebke-Wheeler, J.; Zhang, K.; Battle, M.; Si-Tayeb, K.; Garrison, W.; Chhinder, S.; Li, J.; Kaufman, R.J.; Duncan, S.A. Hepatocyte nuclear factor 4α is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology 2008, 48, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Min, A.K.; Jeong, J.Y.; Go, Y.; Choi, Y.K.; Kim, Y.D.; Lee, I.K.; Park, K.G. cAMP response element binding protein H mediates fenofibrate-induced suppression of hepatic lipogenesis. Diabetologia 2013, 56, 412–422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kharitonenkov, A. FGFs and metabolism. Curr. Opin. Pharmacol. 2009, 9, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Wroblewski, V.J.; Koester, A.; Chen, Y.F.; Clutinger, C.K.; Tigno, X.T.; Hansen, B.C.; Shanafelt, A.B.; Etgen, G.J. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology 2007, 148, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, C.; Fang, Q.C.; Shao, M.L.; Zhang, Y.L.; Liu, Y.; Deng, Y.P.; Shan, B.; Liu, J.Q.; Li, H.T.; et al. Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J. Biol. Chem. 2014, 289, 29751–29765. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.S.; Lu, X.H.; Xiao, Y.C.; Lin, Y.; Zhu, H.; Ding, T.; Yang, Y.; Huang, Y.; Zhang, Y.; Liu, Y.L.; et al. ATF4- and CHOP-dependent induction of FGF21 through endoplasmic reticulum stress. BioMed Res. Int. 2014, 2014, 807874. [Google Scholar] [CrossRef] [PubMed]
- De Sousa-Coelho, A.L.; Marrero, P.F.; Haro, D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem. J. 2012, 443, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Shan, B.; Liu, Y.; Deng, Y.; Yan, C.; Wu, Y.; Mao, T.; Qiu, Y.; Zhou, Y.; Jiang, S.; et al. Hepatic IRE1α regulates fasting-induced metabolic adaptive programs through the XBP1s-PPARα axis signalling. Nat. Commun. 2014, 5, 3528. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Regnier, M.; Fouche, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Uebanso, T.; Taketani, Y.; Yamamoto, H.; Amo, K.; Tanaka, S.; Arai, H.; Takei, Y.; Masuda, M.; Yamanaka-Okumura, H.; Takeda, E. Liver X receptor negatively regulates fibroblast growth factor 21 in the fatty liver induced by cholesterol-enriched diet. J. Nutr. Biochem. 2012, 23, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, Z.; Walker, P.D.; Kapatos, G.; Zhang, K. CREBH Maintains Circadian Glucose Homeostasis by Regulating Hepatic Glycogenolysis and Gluconeogenesis. Mol. Cell. Biol. 2017, 37, e00048-17. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Oikawa, F.; Mizuno, S.; Ohno, H.; Yagishita, Y.; Satoh, A.; Osaki, Y.; Takei, K.; Kikuchi, T.; Han, S.I.; et al. Hyperlipidemia and hepatitis in liver-specific CREB3L3 knockout mice generated using a one-step CRISPR/Cas9 system. Sci. Rep. 2016, 6, 27857. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Xu, X.; Cho, S.; Hur, K.Y.; Lee, M.S.; Kersten, S.; Lee, A.H. CREBH-FGF21 axis improves hepatic steatosis by suppressing adipose tissue lipolysis. Sci. Rep. 2016, 6, 27938. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Yabe, D.; Komuro, R.; Liang, G.; Goldstein, J.L.; Brown, M.S. Liver-specific mRNA for Insig-2 down-regulated by insulin: Implications for fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Sozio, M.S.; Liangpunsakul, S.; Crabb, D. The role of lipid metabolism in the pathogenesis of alcoholic and nonalcoholic hepatic steatosis. Semin. Liver Dis. 2010, 30, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Ginsberg, H.N. Complexity in the secretory pathway: The assembly and secretion of apolipoprotein B-containing lipoproteins. J. Biol. Chem. 2002, 277, 17377–17380. [Google Scholar] [CrossRef] [PubMed]
- Shelness, G.S.; Ledford, A.S. Evolution and mechanism of apolipoprotein B-containing lipoprotein assembly. Curr. Opin. Lipidol. 2005, 16, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Jadhav, A.B.; Hassan, A.; Meng, Q.H. Acute phase reactants as novel predictors of cardiovascular disease. ISRN Inflamm. 2012, 2012, 953461. [Google Scholar] [CrossRef] [PubMed]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.J.; Rodriguez, M.D.; Tso, P. Control of synthesis and secretion of intestinal apolipoprotein A-IV by lipid. J. Nutr. 1997, 127, 537S–543S. [Google Scholar] [CrossRef] [PubMed]
- Black, D.D. Development and physiological regulation of intestinal lipid absorption. I. Development of intestinal lipid absorption: Cellular events in chylomicron assembly and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G519–G524. [Google Scholar] [CrossRef] [PubMed]
- VerHague, M.A.; Cheng, D.; Weinberg, R.B.; Shelness, G.S. Apolipoprotein A-IV expression in mouse liver enhances triglyceride secretion and reduces hepatic lipid content by promoting very low density lipoprotein particle expansion. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.W.; Weinberg, R.B.; Shelness, G.S. apoA-IV tagged with the ER retention signal KDEL perturbs the intracellular trafficking and secretion of apoB. J. Lipid Res. 2004, 45, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.B.; Gallagher, J.W.; Fabritius, M.A.; Shelness, G.S. ApoA-IV modulates the secretory trafficking of apoB and the size of triglyceride-rich lipoproteins. J. Lipid Res. 2012, 53, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Yao, Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr. Metab. 2010, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Hanniman, E.A.; Lambert, G.; Inoue, Y.; Gonzalez, F.J.; Sinal, C.J. Apolipoprotein A-IV is regulated by nutritional and metabolic stress: Involvement of glucocorticoids, HNF-4α, and PGC-1α. J. Lipid Res. 2006, 47, 2503–2514. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Muller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/Δ in regulation of gene expression in mouse liver. Physiol. Genom. 2010, 41, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Elshourbagy, N.A.; Walker, D.W.; Boguski, M.S.; Gordon, J.I.; Taylor, J.M. The nucleotide and derived amino acid sequence of human apolipoprotein A-IV mRNA and the close linkage of its gene to the genes of apolipoproteins A-I and C-III. J. Biol. Chem. 1986, 261, 1998–2002. [Google Scholar] [PubMed]
- Karathanasis, S.K.; Yunis, I.; Zannis, V.I. Structure, evolution, and tissue-specific synthesis of human apolipoprotein AIV. Biochemistry 1986, 25, 3962–3970. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.B.; Spector, M.S. Human apolipoprotein A-IV: Displacement from the surface of triglyceride-rich particles by HDL2-associated C-apoproteins. J. Lipid Res. 1985, 26, 26–37. [Google Scholar] [PubMed]
- Goldberg, I.J.; Scheraldi, C.A.; Yacoub, L.K.; Saxena, U.; Bisgaier, C.L. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J. Biol. Chem. 1990, 265, 4266–4272. [Google Scholar] [PubMed]
- Duverger, N.; Tremp, G.; Caillaud, J.M.; Emmanuel, F.; Castro, G.; Fruchart, J.C.; Steinmetz, A.; Denefle, P. Protection against atherogenesis in mice mediated by human apolipoprotein A-IV. Science 1996, 273, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Yao, Y.; Meng, S.; Cheng, X.; Black, D.D. Overexpression of apolipoprotein A-IV enhances lipid transport in newborn swine intestinal epithelial cells. J. Biol. Chem. 2002, 277, 31929–31937. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Lu, S.; Huang, Y.; Beeman-Black, C.C.; Lu, R.; Pan, X.; Hussain, M.M.; Black, D.D. Regulation of microsomal triglyceride transfer protein by apolipoprotein A-IV in newborn swine intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G357–G363. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Munshi, M.K.; Iqbal, J.; Queiroz, J.; Sirwi, A.A.; Shah, S.; Younus, A.; Hussain, M.M. Circadian regulation of intestinal lipid absorption by apolipoprotein AIV involves forkhead transcription factors A2 and O1 and microsomal triglyceride transfer protein. J. Biol. Chem. 2013, 288, 20464–20476. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Fukagawa, K.; Sakata, T.; Tso, P. Suppression of food intake by apolipoprotein A-IV is mediated through the central nervous system in rats. J. Clin. Investig. 1993, 91, 1830–1833. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kohan, A.B.; Kindel, T.L.; Corbin, K.L.; Nunemaker, C.S.; Obici, S.; Woods, S.C.; Davidson, W.S.; Tso, P. Apolipoprotein A-IV improves glucose homeostasis by enhancing insulin secretion. Proc. Natl. Acad. Sci. USA 2012, 109, 9641–9646. [Google Scholar] [CrossRef] [PubMed]
- Raggo, C.; Rapin, N.; Stirling, J.; Gobeil, P.; Smith-Windsor, E.; O’Hare, P.; Misra, V. Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Mol. Cell. Biol. 2002, 22, 5639–5649. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Kim, J.; An, H.T.; Ko, J. Human leucine zipper protein promotes hepatic steatosis via induction of apolipoprotein A-IV. FASEB J. 2017, 31, 2548–2561. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Wang, Y.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. LPS decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J. Lipid Res. 2008, 49, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Grunfeld, C. The role of HDL in innate immunity. J. Lipid Res. 2011, 52, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Peng, Y.S.; Chen, Y.C.; Lien, J.M.; Tian, Y.C.; Fang, J.T.; Weng, H.H.; Chen, P.C.; Yang, C.W.; Wu, C.S. Low serum concentration of apolipoprotein A-I is an indicator of poor prognosis in cirrhotic patients with severe sepsis. J. Hepatol. 2009, 50, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.Y.; Jerng, J.S.; Yu, C.J.; Yang, P.C. Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis. Crit. Care Med. 2005, 33, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, H.J.; van Beek, A.P.; Dallinga-Thie, G.M.; van Strijp, J.A.; Verhoef, J.; van Kessel, K.P. The role of high density lipoprotein in sepsis. Neth. J. Med. 2001, 59, 102–110. [Google Scholar] [CrossRef]
- Saemann, M.D.; Poglitsch, M.; Kopecky, C.; Haidinger, M.; Horl, W.H.; Weichhart, T. The versatility of HDL: A crucial anti-inflammatory regulator. Eur. J. Clin. Investig. 2010, 40, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Dandekar, A.; Qiu, Y.; Kim, H.; Wang, J.; Hou, X.; Zhang, X.; Zheng, Z.; Mendez, R.; Yu, F.S.; Kumar, A.; et al. Toll-like Receptor (TLR) Signaling Interacts with CREBH to Modulate High-density Lipoprotein (HDL) in Response to Bacterial Endotoxin. J. Biol. Chem. 2016, 291, 23149–23158. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Yamamoto, H.; Onishi, M.; Miyamoto, N.; Oki, R.; Ueda, H.; Ishigami, M.; Hiraoka, H.; Matsuzawa, Y.; Kihara, S. Novel combined GPIHBP1 mutations in a patient with hypertriglyceridemia associated with CAD. J. Atheroscler. Thromb. 2013, 20, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, B.S.; Goldberg, I.J.; Merab, J.; Vanni, T.M.; Ramakrishnan, R.; Ginsberg, H.N. Increased plasma and renal clearance of an exchangeable pool of apolipoprotein A-I in subjects with low levels of high density lipoprotein cholesterol. J. Clin. Investig. 1993, 91, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Parks, J.S. ATP-binding cassette transporter AI and its role in HDL formation. Curr. Opin. Lipidol. 2005, 16, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E.; Kawashiri, M.A.; Kitajima, K.; Secreto, A.; Millar, J.S.; Pratico, D.; Rader, D.J. Apolipoprotein A-I deficiency results in markedly increased atherosclerosis in mice lacking the LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Tzameli, I.; Zannis, V.I. Binding specificity and modulation of the ApoA-I promoter activity by homo- and heterodimers of nuclear receptors. J. Biol. Chem. 1996, 271, 8402–8415. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ning, G.; Duncan, S.A. Mammalian hepatocyte differentiation requires the transcription factor HNF-4α. Genes Dev. 2000, 14, 464–474. [Google Scholar] [PubMed]
- Sladek, F.M.; Zhong, W.M.; Lai, E.; Darnell, J.E., Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990, 4, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Harnish, D.C.; Malik, S.; Karathanasis, S.K. Activation of apolipoprotein AI gene transcription by the liver-enriched factor HNF-3. J. Biol. Chem. 1994, 269, 28220–28226. [Google Scholar] [PubMed]
- Chen, C.H.; Albers, J.J. Activation of lecithin: Cholesterol acyltransferase by apolipoproteins E-2, E-3, and A-IV isolated from human plasma. Biochim. Biophys. Acta 1985, 836, 279–285. [Google Scholar] [CrossRef]
- Steinmetz, A.; Utermann, G. Activation of lecithin: Cholesterol acyltransferase by human apolipoprotein A-IV. J. Biol. Chem. 1985, 260, 2258–2264. [Google Scholar] [PubMed]
- Fournier, N.; Atger, V.; Paul, J.L.; Sturm, M.; Duverger, N.; Rothblat, G.H.; Moatti, N. Human ApoA-IV overexpression in transgenic mice induces cAMP-stimulated cholesterol efflux from J774 macrophages to whole serum. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, A.; Barbaras, R.; Ghalim, N.; Clavey, V.; Fruchart, J.C.; Ailhaud, G. Human apolipoprotein A-IV binds to apolipoprotein A-I/A-II receptor sites and promotes cholesterol efflux from adipose cells. J. Biol. Chem. 1990, 265, 7859–7863. [Google Scholar] [PubMed]
- Ostos, M.A.; Conconi, M.; Vergnes, L.; Baroukh, N.; Ribalta, J.; Girona, J.; Caillaud, J.M.; Ochoa, A.; Zakin, M.M. Antioxidative and antiatherosclerotic effects of human apolipoprotein A-IV in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.D.; Castellani, L.W.; Qiao, J.H.; Van Lenten, B.J.; Lusis, A.J.; Reue, K. Reduced aortic lesions and elevated high density lipoprotein levels in transgenic mice overexpressing mouse apolipoprotein A-IV. J. Clin. Investig. 1997, 99, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Schlein, C.; Talukdar, S.; Heine, M.; Fischer, A.W.; Krott, L.M.; Nilsson, S.K.; Brenner, M.B.; Heeren, J.; Scheja, L. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016, 23, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Pan, X.; Wu, F.; Ye, D.; Zhang, Y.; Wang, Y.; Jin, L.; Lian, Q.; Huang, Y.; Ding, H.; et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation 2015, 131, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qi, Y.F.; Chang, J.R.; Lu, W.W.; Zhang, J.S.; Wang, S.P.; Cheng, S.J.; Zhang, M.; Fan, Q.; Lv, Y.; et al. Possible role of fibroblast growth factor 21 on atherosclerosis via amelioration of endoplasmic reticulum stress-mediated apoptosis in apoE−/− mice. Heart Vessels 2015, 30, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Kim, H.; Qiu, Y.; Chen, X.; Mendez, R.; Dandekar, A.; Zhang, X.; Zhang, C.; Liu, A.C.; Yin, L.; et al. CREBH Couples Circadian Clock With Hepatic Lipid Metabolism. Diabetes 2016, 65, 3369–3383. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Touche, V.; Tailleux, A.; Fruchart, J.C.; Fievet, C.; Clavey, V.; Staels, B.; Lestavel, S. Niemann-Pick C1 like 1 gene expression is down-regulated by LXR activators in the intestine. Biochem. Biophys. Res. Commun. 2006, 340, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Qian, K.; Jiang, N.; Zheng, X.L.; Cayabyab, F.S.; Tang, C.K. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clin. Chim. Acta 2014, 428, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors αand β. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]


{kind=link}
| Metabolic Function | Target Gene | Co-Factor | Reference |
|---|---|---|---|
| Metabolic hormone | Fgf21 | PPARα | [8,10] |
| Gluconeogenesis | Pck1, G6pc | CRTC2 | [11,12] |
| Fatty acid oxidation | Ppara | – | [8,10] |
| Cpt1a | – | [7] | |
| Ketogenesis | Bdh1 | – | [7] |
| Apolipoprotein | Apoa1 | HNF4α | [13] |
| Apoa4, Apoa5, Apoc2, Apoc3 | – | [9,14] | |
| Apob | – | [15,16] | |
| SREBP suppressor | Insig2a | – | [17] |
| Fatty acid elongation | Elovl2, Elovl5, Elovl6 | – | [18] |
| Lipid droplet formation | Fsp27β | – | [19] |
| Cholesterol absorption | Npc1l1 | – | [20] |
| Transcription Factor | Inducer | Reference |
|---|---|---|
| Up-Regulation | ||
| CREBH | Fasting | [8,10] |
| PPARα | Fasting, Fibrates | [8,10] |
| Activating transcription factor 4 (ATF4) | Endoplasmic reticulum (ER) stress, Amino acid deprivation | [31,32,33] |
| CCAAT enhancer binding protein homologous protein (CHOP) | ER stress | [31] |
| X-box-binding protein 1 (XBP1) | ER stress, Fasting | [30,34] |
| Carbohydrate-responsive element-binding protein (ChREBP) | Carbohydrate | [35,36] |
| Down-Regulation | ||
| Liver X receptor (LXR) | Cholesterol | [37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, Y.; Shimano, H. CREBH Regulates Systemic Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 1396. https://doi.org/10.3390/ijms19051396
Nakagawa Y, Shimano H. CREBH Regulates Systemic Glucose and Lipid Metabolism. International Journal of Molecular Sciences. 2018; 19(5):1396. https://doi.org/10.3390/ijms19051396
Chicago/Turabian StyleNakagawa, Yoshimi, and Hitoshi Shimano. 2018. "CREBH Regulates Systemic Glucose and Lipid Metabolism" International Journal of Molecular Sciences 19, no. 5: 1396. https://doi.org/10.3390/ijms19051396
APA StyleNakagawa, Y., & Shimano, H. (2018). CREBH Regulates Systemic Glucose and Lipid Metabolism. International Journal of Molecular Sciences, 19(5), 1396. https://doi.org/10.3390/ijms19051396