The Janus Face of VEGF in Stroke


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
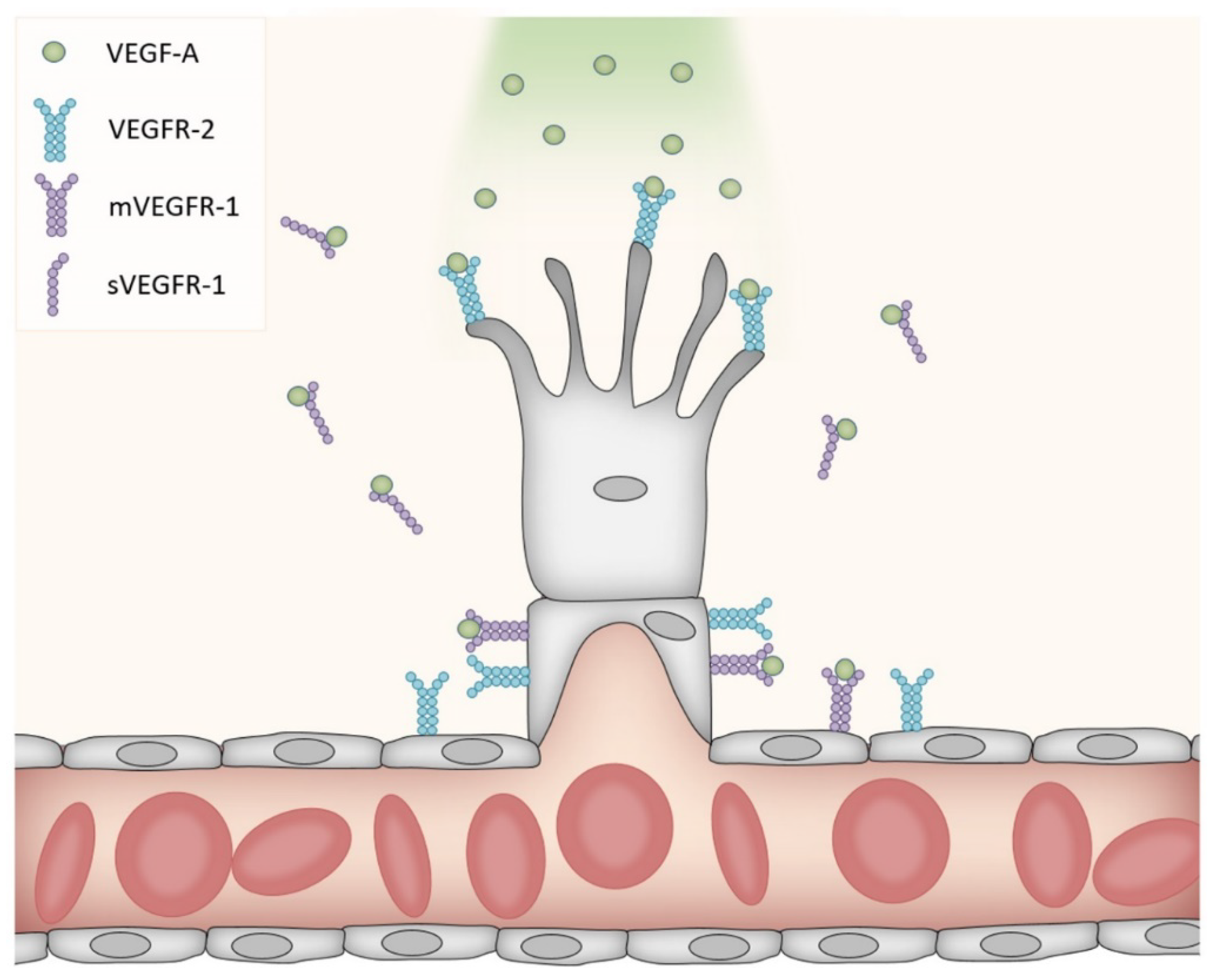
2. The VEGF Receptor Family
3. VEGF-A in the Prevention and Therapy of Stroke
3.1. VEGF-A Expression
3.2. VEGF-A in the Treatment of Stroke
3.2.1. Effects of VEGF-A in Angiogenesis
3.2.2. Effects of VEGF-A on Vasodilation
3.2.3. Acute Effects of VEGF-A on Vasopermeability
3.2.4. Effects of VEGF-A on Neuroprotection
3.2.5. Neurogenesis
3.3. VEGF-A in Stroke Prevention—Exercise and Preconditioning
3.3.1. Timing and Dosage
3.3.2. Hypoxic/Ischemic Preconditioning
3.3.3. Exercise
4. VEGF-A in Human Cerebral Stroke
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| VEGF | Vascular Endothelial Growth Factor |
| ERK | Extracellular Signal-Regulated Kinase |
| PI3K | Phosphoinositide 3-Kinase |
| HIF | Hypoxia Inducible Factor |
References
- Baron, J.-C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Wang, M.; Kumar, S. Prognostic value of blood vessel density in ischaemic stroke. Lancet 1993, 342, 742. [Google Scholar] [CrossRef]
- Marchal, G.; Serrati, C.; Rioux, P.; Petit-Taboue, M.C.; Viader, F.; de la Sayette, V.; Le Doze, F.; Lochon, P.; Derlon, J.M.; Orgogozo, J.M.; et al. Pet imaging of cerebral perfusion and oxygen consumption in acute ischaemic stroke: Relation to outcome. Lancet 1993, 341, 925–927. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Namiecinska, M.; Marciniak, K.; Nowak, J.Z. VEGF as an angiogenic, neurotrophic, and neuroprotective factor. Postepy Hig. Med. Dosw. 2005, 59, 573–583. [Google Scholar]
- Gora-Kupilas, K.; Josko, J. The neuroprotective function of vascular endothelial growth factor (VEGF). Folia Neuropathol. 2005, 43, 31–39. [Google Scholar] [PubMed]
- Cao, L.; Jiao, X.; Zuzga, D.S.; Liu, Y.; Fong, D.M.; Young, D.; During, M.J. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat. Genet. 2004, 36, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Dzietko, M.; Derugin, N.; Wendland, M.F.; Vexler, Z.S.; Ferriero, D.M. Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl. Stroke Res. 2013, 4, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, K.; Ng, Y.-S.; Zhong, L.; Bradley, J.; Schubert, W.; Jo, N.; Akita, J.; Samuelsson, S.J.; Robinson, G.S.; Adamis, A.P.; et al. Vascular endothelial growth factor-a is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 2007, 171, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zechariah, A.; Qu, Y.; Hermann, D.M. Effects of vascular endothelial growth factor in ischemic stroke. J. Neurosci. Res. 2012, 90, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.V.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Van Bruggen, N.; Thibodeaux, H.; Palmer, J.T.; Lee, W.P.; Fu, L.; Cairns, B.; Tumas, D.; Gerlai, R.; Williams, S.-P.; Campagne, M.V.L.; et al. Vegf antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J. Clin. Investig. 1999, 104, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Grassot, J.; Gouy, M.; Perriere, G.; Mouchiroud, G. Origin and molecular evolution of receptor tyrosine kinases with immunoglobulin-like domains. Mol. Biol. Evol. 2006, 23, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Almodovar, C.; Lambrechts, D.; Mazzone, M.; Carmeliet, P. Role and therapeutic potential of VEGF in the nervous system. Physiol. Rev. 2009, 89, 607–648. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to flt-1 but not to flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar] [PubMed]
- Meyer, R.D.; Mohammadi, M.; Rahimi, N. A single amino acid substitution in the activation loop defines the decoy characteristic of VEGFR-1/flt-1. J. Biol. Chem. 2006, 281, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.S.; Saint-Geniez, M.; Maldonado, A.E.; D’Amore, P.A. Vascular endothelial growth factor localization in the adult. Am. J. Pathol. 2006, 168, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.J.; Wei, Y.J.; Li, S.; Guo, W.; Chen, X.; Liu, S.Y.; He, J.J.; Yin, Q.; Yang, H.; Zhang, C.Q. Elevated expression of VEGF-c and its receptors, VEGFR-2 and VEGFR-3, in patients with mesial temporal lobe epilepsy. J. Mol. Neurosci. 2016, 59, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience 2002, 110, 589–604. [Google Scholar] [CrossRef]
- Marti, H.J.; Bernaudin, M.; Bellail, A.; Schoch, H.; Euler, M.; Petit, E.; Risau, W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am. J. Pathol. 2000, 156, 965–976. [Google Scholar] [CrossRef]
- Issa, R.; Krupinski, J.; Bujny, T.; Kumar, S.; Kaluza, J.; Kumar, P. Vascular endothelial growth factor and its receptor, KDR, in human brain tissue after ischemic stroke. Lab. Investig. 1999, 79, 417–425. [Google Scholar] [PubMed]
- Stowe, A.M.; Plautz, E.J.; Eisner-Janowicz, I.; Frost, S.B.; Barbay, S.; Zoubina, E.V.; Dancause, N.; Taylor, M.D.; Nudo, R.J. Vegf protein associates to neurons in remote regions following cortical infarct. J. Cereb. Blood Flow Metab. 2007, 27, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.M.; Plautz, E.J.; Nguyen, P.; Frost, S.B.; Eisner-Janowicz, I.; Barbay, S.; Dancause, N.; Sensarma, A.; Taylor, M.D.; Zoubina, E.V.; et al. Neuronal HIF-1α protein and VEGFR-2 immunoreactivity in functionally related motor areas following a focal m1 infarct. J. Cereb. Blood Flow Metab. 2008, 28, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Somanath, P.R.; Kozak, A.; Goc, A.; El-Remessy, A.B.; Ergul, A.; Johnson, M.H.; Alhusban, A.; Soliman, S.; Fagan, S.C. Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS ONE 2011, 6, e24551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Tsang, W.; Soltanian-Zadeh, H.; Morris, D.; Zhang, R.; Goussev, A.; Powers, C.; Yeich, T.; Chopp, M. Correlation of VEGF and angiopoietin expression with disruption of blood–brain barrier and angiogenesis after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Margaritescu, O.; Pirici, D.; Margaritescu, C. Vegf expression in human brain tissue after acute ischemic stroke. Rom. J. Morphol. Embryol. 2011, 52, 1283–1292. [Google Scholar] [PubMed]
- Khaibullina, A.A.; Rosenstein, J.M.; Krum, J.M. Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res. Dev. Brain Res. 2004, 148, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Krum, J.M.; Khaibullina, A. Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: Roles for VEGF in brain repair. Exp. Neurol. 2003, 181, 241–257. [Google Scholar] [CrossRef]
- Lee, M.Y.; Ju, W.K.; Cha, J.H.; Son, B.C.; Chun, M.H.; Kang, J.K.; Park, C.K. Expression of vascular endothelial growth factor MRNA following transient forebrain ischemia in rats. Neurosci. Lett. 1999, 265, 107–110. [Google Scholar] [CrossRef]
- Zan, L.; Zhang, X.; Xi, Y.; Wu, H.; Song, Y.; Teng, G.; Li, H.; Qi, J.; Wang, J. Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience 2014, 262, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Josko, J.; Mazurek, M. Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med. Sci. Monit. 2004, 10, Ra89–Ra98. [Google Scholar] [PubMed]
- Dery, M.A.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, M.; Nedelec, A.-S.; Divoux, D.; MacKenzie, E.T.; Petit, E.; Schumann-Bard, P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J. Cereb. Blood Flow Metab. 2002, 22, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Fandrey, J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R977–R988. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, Y.; Mahmood, A.; Meng, Y.; Qu, C.; Chopp, M. Erythropoietin mediates neurobehavioral recovery and neurovascular remodeling following traumatic brain injury in rats by increasing expression of vascular endothelial growth factor. Transl. Stroke Res. 2011, 2, 619–632. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Bi, Y.; Li, J.; Zhang, M.; Chen, X.; Zhang, K.; Li, J. IL-17 induces reactive astrocytes and up-regulation of vascular endothelial growth factor (VEGF) through JAK/STAT signaling. Sci. Rep. 2017, 7, 41779. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Kommineni, V.K.; William, A.; Detrick, B.; Hooks, J.J. Regulation of VEGF expression in human retinal cells by cytokines: Implications for the role of inflammation in age-related macular degeneration. J. Cell. Physiol. 2012, 227, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Andersson, K.A.; Haugen, O.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor hcar1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef] [PubMed]
- Thau-Zuchman, O.; Shohami, E.; Alexandrovich, A.G.; Leker, R.R. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J. Cereb. Blood Flow Metab. 2010, 30, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Drew, K.L.; Folkow, L.P.; Milton, S.L.; Park, T.J. No oxygen? No problem! Intrinsic brain tolerance to hypoxia in vertebrates. J. Exp. Biol. 2014, 217, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wang, S.; Yang, G.; Sun, X.; Zhao, S.; Lin, L.; Cheng, J.; Yang, W.; Cong, W.; Sun, W.; et al. HIF-1α contributes to hypoxia adaptation of the naked mole rat. Oncotarget 2017, 8, 109941–109951. [Google Scholar] [CrossRef] [PubMed]
- Manoonkitiwongsa, P.S. Critical questions for preclinical trials on safety and efficacy of vascular endothelial growth factor-based therapeutic angiogenesis for ischemic stroke. CNS Neurol. Disord. Drug Targets 2011, 10, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, C.; Jiang, H.; Li, Y.; Zhang, L.; Robin, A.; Katakowski, M.; Lu, M.; Chopp, M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J. Cereb. Blood Flow Metab. 2005, 25, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, K.S.; Park, I.H.; Kim, S.U. Human neural stem cells over-expressing VEGF provide neuroprotection, angiogenesis and functional recovery in mouse stroke model. PLoS ONE 2007, 2, e156. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.X.; Kazlauskas, A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012, 31, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-COA reductase inhibitor simvastatin activates the protein kinase AKT and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ruiz, M.; Fulton, D.; Sowa, G.; Languino, L.R.; Fujio, Y.; Walsh, K.; Sessa, W.C. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase AKT. Circ. Res. 2000, 86, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jiang, H.; Lu, D.; Qu, C.; Xiong, Y.; Zhou, D.; Chopp, M.; Mahmood, A. Induction of angiogenesis and modulation of vascular endothelial growth factor receptor-2 by simvastatin after traumatic brain injury. Neurosurgery 2011, 68, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, Z.; Avraham, H.; Avraham, S. Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J. Biol. Chem. 2000, 275, 20770–20774. [Google Scholar] [CrossRef] [PubMed]
- Kaya, D.; Gursoy-Ozdemir, Y.; Yemisci, M.; Tuncer, N.; Aktan, S.; Dalkara, T. Vegf protects brain against focal ischemia without increasing blood–brain permeability when administered intracerebroventricularly. J. Cereb. Blood Flow Metab. 2005, 25, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Wang, Y.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. The phosphatidylinositol-3 kinase/akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 2006, 20, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Dewil, M.; Lambrechts, D.; Sciot, R.; Shaw, P.J.; Ince, P.G.; Robberecht, W.; Van den Bosch, L. Vascular endothelial growth factor counteracts the loss of phospho-akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2007, 33, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Mir, M.; Olmos, G.; Llado, J. Vascular endothelial growth factor protects motoneurons from serum deprivation-induced cell death through phosphatidylinositol 3-kinase-mediated p38 mitogen-activated protein kinase inhibition. Neuroscience 2009, 158, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Duan, Y.; Ma, G.; Zhou, G.; Park-Windhol, C.; D’Amore, P.A.; Lei, H. Aav-crispr/cas9-mediated depletion of VEGFR2 blocks angiogenesis in vitro. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6082–6090. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Munoz, F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531. [Google Scholar] [CrossRef]
- Zhu, J.; Song, W.; Li, L.; Fan, X. Endothelial nitric oxide synthase: A potential therapeutic target for cerebrovascular diseases. Mol. Brain 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Kearney, J.B.; Johnson, J.H.; Rosenberg, M.P.; Kumar, R.; Bautch, V.L. The vascular endothelial growth factor (VEGF) receptor flt-1 (VEGFR-1) modulates flk-1 (VEGFR-2) signaling during blood vessel formation. Am. J. Pathol. 2004, 164, 1531–1535. [Google Scholar] [CrossRef]
- Chappell, J.C.; Taylor, S.M.; Ferrara, N.; Bautch, V.L. Local guidance of emerging vessel sprouts requires soluble flt-1. Dev. Cell 2009, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kearney, J.B.; Ambler, C.A.; Monaco, K.A.; Johnson, N.; Rapoport, R.G.; Bautch, V.L. Vascular endothelial growth factor receptor flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood 2002, 99, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Kappas, N.C.; Zeng, G.; Chappell, J.C.; Kearney, J.B.; Hazarika, S.; Kallianos, K.G.; Patterson, C.; Annex, B.H.; Bautch, V.L. The VEGF receptor flt-1 spatially modulates flk-1 signaling and blood vessel branching. J. Cell Biol. 2008, 181, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kilic, E.; Kilic, U.; Weber, B.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain 2005, 128, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Dieguez-Hurtado, R.; Schmidt, I.; Nakayama, M.; Jeong, H.W.; Arf, H.; Adams, S.; Ferrara, N.; Adams, R.H. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat. Commun. 2017, 8, 1574. [Google Scholar] [CrossRef] [PubMed]
- Bauters, C.; Asahara, T.; Zheng, L.P.; Takeshita, S.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Recovery of disturbed endothelium-dependent flow in the collateral-perfused rabbit ischemic hindlimb after administration of vascular endothelial growth factor. Circulation 1995, 91, 2802–2809. [Google Scholar] [CrossRef] [PubMed]
- Ku, D.D.; Zaleski, J.K.; Liu, S.; Brock, T.A. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am. J. Physiol. 1993, 265, H586–H592. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, J.P.; Almeida, A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim. Biophys. Acta 1999, 1411, 415–436. [Google Scholar] [CrossRef]
- Rees, D.D.; Palmer, R.M.; Moncada, S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Willmot, M.; Gray, L.; Gibson, C.; Murphy, S.; Bath, P.M. A systematic review of nitric oxide donors and l-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide 2005, 12, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, E.; Moskowitz, M.A.; Huang, Z.; Yoshida, T.; Irikura, K.; Dalkara, T. L-arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke 1994, 25, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Salom, J.B.; Orti, M.; Centeno, J.M.; Torregrosa, G.; Alborch, E. Reduction of infarct size by the no donors sodium nitroprusside and spermine/no after transient focal cerebral ischemia in rats. Brain Res. 2000, 865, 149–156. [Google Scholar] [CrossRef]
- Moisan, A.; Favre, I.M.; Rome, C.; Grillon, E.; Naegele, B.; Barbieux, M.; De Fraipont, F.; Richard, M.J.; Barbier, E.L.; Rémy, C.; et al. Microvascular plasticity after experimental stroke: A molecular and MRI study. Cerebrovasc. Dis. 2014, 38, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, M.R.; Ennis, S.R.; Sullivan, S.E.; Keep, R.F. Effects of intraventricular infusion of vascular endothelial growth factor on cerebral blood flow, edema, and infarct volume. Acta Neurochir. (Wien.) 2003, 145, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Tomita, H.; Taki, Y.; Kikuchi, Y.; Ono, C.; Yu, Z.; Sekiguchi, A.; Nouchi, R.; Kotozaki, Y.; Nakagawa, S.; et al. The VEGF gene polymorphism impacts brain volume and arterial blood volume. Hum. Brain Mapp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ames, A., 3rd; Wright, R.L.; Kowada, M.; Thurston, J.M.; Majno, G. Cerebral ischemia. II. The no-reflow phenomenon. Am. J. Pathol. 1968, 52, 437–453. [Google Scholar] [PubMed]
- Pedram, A.; Razandi, M.; Levin, E.R. Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J. Biol. Chem. 2002, 277, 44385–44398. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef] [PubMed]
- He, Y.X.; Liu, J.; Guo, B.; Wang, Y.X.; Pan, X.; Li, D.; Tang, T.; Chen, Y.; Peng, S.; Bian, Z.; et al. Src inhibitor reduces permeability without disturbing vascularization and prevents bone destruction in steroid-associated osteonecrotic lesions in rabbits. Sci. Rep. 2015, 5, 8856. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. Ssecks regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Bella, A.J.; Lin, G.; Tantiwongse, K.; Garcia, M.; Lin, C.S.; Brant, W.; Lue, T.F. Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: Part i. J. Sex. Med. 2006, 3, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, C.; Yuguchi, T.; Nishio, M.; Tomishima, T.; Fujinaka, T.; Taniguchi, M.; Nakajima, Y.; Kohmura, E.; Yoshimine, T. Src family kinase inhibitor pp1 reduces secondary damage after spinal cord compression in rats. J. Neurotrauma 2004, 21, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Lennmyr, F.; Ericsson, A.; Gerwins, P.; Akterin, S.; Ahlstrom, H.; Terent, A. Src family kinase-inhibitor pp2 reduces focal ischemic brain injury. Acta Neurol. Scand. 2004, 110, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P.; Paul, R.; Schwartzberg, P.L.; Hood, J.D.; Leng, J.; Cheresh, D.A. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 1999, 4, 915–924. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. Vegf controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of ve-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Shintani, S.; Weber, A.; Kirchmair, R.; Wood, M.; Cravens, A.; McSharry, H.; Iwakura, A.; Yoon, Y.-S.; Himes, N.; et al. Src blockade stabilizes a FLK/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Investig. 2004, 113, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Vilgrain, I.; Huber, P. Angiogenesis: The ve-cadherin switch. Trends Cardiovasc. Med. 2006, 16, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Tessema, M.; Wandinger-Ness, A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ. Res. 2006, 98, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (review). Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Maddahi, A.; Povlsen, G.K.; Edvinsson, L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J. Neuroinflamm. 2012, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.A.; Jin, K. From angiogenesis to neuropathology. Nature 2005, 438, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.F.; Krum, J.M.; Mani, N.; Rosenstein, J.M. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience 1999, 90, 1529–1541. [Google Scholar] [CrossRef]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef] [PubMed]
- Ogunshola, O.O.; Antic, A.; Donoghue, M.J.; Fan, S.Y.; Kim, H.; Stewart, W.B.; Madri, J.A.; Ment, L.R. Paracrine and autocrine functions of neuronal vascular endothelial growth factor (VEGF) in the central nervous system. J. Biol. Chem. 2002, 277, 11410–11415. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, J.M.; Mani, N.; Khaibullina, A.; Krum, J.M. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J. Neurosci. 2003, 23, 11036–11044. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor stimulates neurite outgrowth from cerebral cortical neurons via rho kinase signaling. J. Neurobiol. 2006, 66, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Tamatani, M.; Yamaguchi, A.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: Signal transduction cascades. FASEB J. 2001, 15, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Svensson, B.; Peters, M.; Konig, H.G.; Poppe, M.; Levkau, B.; Rothermundt, M.; Arolt, V.; Kogel, D.; Prehn, J.H. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J. Cereb. Blood Flow Metab. 2002, 22, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Irving, E.A.; Barone, F.C.; Reith, A.D.; Hadingham, S.J.; Parsons, A.A. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res. Mol. Brain Res. 2000, 77, 65–75. [Google Scholar] [CrossRef]
- Runden, E.; Seglen, P.O.; Haug, F.M.; Ottersen, O.P.; Wieloch, T.; Shamloo, M.; Laake, J.H. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: Evidence for a map kinase-dependent mechanism. J. Neurosci. 1998, 18, 7296–7305. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.; Alessandrini, A.; Cole, A.J.; Yee, A.G.; Furshpan, E.J. Inhibition of the p44/42 map kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc. Natl. Acad. Sci. USA 1998, 95, 11975–11980. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jin, K. Current perspectives on the link between neuroinflammation and neurogenesis. Metab. Brain Dis. 2015, 30, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, A.; Namura, S.; Moskowitz, M.A.; Bonventre, J.V. Mek1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 12866–12869. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Abe, K.; Itoyama, Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J. Cereb. Blood Flow Metab. 1998, 18, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.L.; Lu, S.D.; Wang, H.; Sun, F.Y. Intraventricular vascular endothelial growth factor antibody increases infarct volume following transient cerebral ischemia. Zhongguo Yao Li Xue Bao 1999, 20, 313–318. [Google Scholar] [PubMed]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.-L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Frisén, J. Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol. 2015, 13, e1002045. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Neurogenesis in the adult brain. J. Neurosci. 2002, 22, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Kempermann, G.; Palmer, T.D.; Peterson, D.A.; Ray, J. Multipotent progenitor cells in the adult dentate gyrus. J. Neurobiol. 1998, 36, 249–266. [Google Scholar] [CrossRef]
- Nilsson, M.; Perfilieva, E.; Johansson, U.; Orwar, O.; Eriksson, P.S. Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J. Neurobiol. 1999, 39, 569–578. [Google Scholar] [CrossRef]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Minami, M.; Lan, J.Q.; Mao, X.O.; Batteur, S.; Simon, R.P.; Greenberg, D.A. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. USA 2001, 98, 4710–4715. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. Vegf-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Investig. 2003, 111, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, K.; Mao, X.O.; Xie, L.; Banwait, S.; Marti, H.H.; Greenberg, D.A. Vegf-overexpressing transgenic mice show enhanced post-ischemic neurogenesis and neuromigration. J. Neurosci. Res. 2007, 85, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Cui, H.R.; Yang, S.Z.; Sun, H.P.; Qiu, M.H.; Feng, X.Y.; Sun, F.Y. Vegf enhance cortical newborn neurons and their neurite development in adult rat brain after cerebral ischemia. Neurochem. Int. 2009, 55, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar] [CrossRef] [PubMed]
- Segi-Nishida, E.; Warner-Schmidt, J.L.; Duman, R.S. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc. Natl. Acad. Sci. USA 2008, 105, 11352–11357. [Google Scholar] [CrossRef] [PubMed]
- Fournier, N.M.; Lee, B.; Banasr, M.; Elsayed, M.; Duman, R.S. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and pi3k/AKT-dependent signaling. Neuropharmacology 2012, 63, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Kirby, E.D.; Kuwahara, A.A.; Messer, R.L.; Wyss-Coray, T. Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc. Natl. Acad. Sci. USA 2015, 112, 4128–4133. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Fraser, J.L.; Yu, S.P.; Zhu, J.; Jiang, Y.-J.; Wei, L. The role of VEGF/VEGFR2 signaling in peripheral stimulation-induced cerebral neurovascular regeneration after ischemic stroke in mice. Exp. Brain Res. 2011, 214, 503. [Google Scholar] [CrossRef] [PubMed]
- Schanzer, A.; Wachs, F.P.; Wilhelm, D.; Acker, T.; Cooper-Kuhn, C.; Beck, H.; Winkler, J.; Aigner, L.; Plate, K.H.; Kuhn, H.G. Direct stimulation of adult neural stem cells in vitro and neurogenesis in vivo by vascular endothelial growth factor. Brain Pathol. 2004, 14, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Bao, W.L.; Qiu, M.H.; Zhang, L.M.; Lu, S.D.; Huang, Y.L.; Sun, F.Y. Role of vascular endothelial growth factor in neuronal DNA damage and repair in rat brain following a transient cerebral ischemia. J. Neurosci. Res. 2002, 70, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.-W.; Duan, C.-L.; Chen, X.-H.; Wang, Y.-Q.; Sun, X.; Zhang, Q.-W.; Cui, H.-R.; Sun, F.-Y. Neurogenic effect of VEGF is related to increase of astrocytes transdifferentiation into new mature neurons in rat brains after stroke. Neuropharmacology 2016, 108, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-P.; Liu, H.-J.; Liu, X.-F. VEGF promotes angiogenesis and functional recovery in stroke rats. J. Investig. Surg. 2010, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Manoonkitiwongsa, P.S.; Schultz, R.L.; McCreery, D.B.; Whitter, E.F.; Lyden, P.D. Neuroprotection of ischemic brain by vascular endothelial growth factor is critically dependent on proper dosage and may be compromised by angiogenesis. J. Cereb. Blood Flow Metab. 2004, 24, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Sacco, R.L.; Perez-Pinzon, M.A. Preconditioning the brain: Moving on to the next frontier of neurotherapeutics. Stroke 2012, 43, 1455–1457. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef]
- Koch, S. Preconditioning the human brain: Practical considerations for proving cerebral protection. Transl. Stroke Res. 2010, 1, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, X.; Peng, Y. Remote ischemic conditioning for acute ischemic stroke: Dawn in the darkness. Rev. Neurosci. 2016, 27, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Kirino, T.; Tsujita, Y.; Tamura, A. Induced tolerance to ischemia in gerbil hippocampal neurons. J. Cereb. Blood Flow Metab. 1991, 11, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Stagliano, N.E.; Perez-Pinzon, M.A.; Moskowitz, M.A.; Huang, P.L. Focal ischemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 1999, 19, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinzon, M.A.; Xu, G.P.; Dietrich, W.D.; Rosenthal, M.; Sick, T.J. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, C.; Du, X.; Liu, M.; Ji, X.; Du, H.; Zhao, H. Hypoxia inducible factor 1α plays a key role in remote ischemic preconditioning against stroke by modulating inflammatory responses in rats. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kawata, H.; Yoshida, K.; Kawamoto, A.; Kurioka, H.; Takase, E.; Sasaki, Y.; Hatanaka, K.; Kobayashi, M.; Ueyama, T.; Hashimoto, T.; et al. Ischemic preconditioning upregulates vascular endothelial growth factor mrna expression and neovascularization via nuclear translocation of protein kinase c epsilon in the rat ischemic myocardium. Circ. Res. 2001, 88, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Pacary, E.; Freret, T.; Divoux, D.; Petit, E.; Schumann-Bard, P.; Bernaudin, M. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: Identification of potential neuroprotective candidates for stroke. Neurobiol. Dis. 2006, 21, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Cho, J.H.; Kim, I.H.; Cho, G.S.; Cho, J.H.; Park, J.H.; Ahn, J.H.; Chen, B.H.; Shin, B.N.; Shin, M.C.; et al. Effects of ischemic preconditioning on VEGF and PFLK-1 immunoreactivities in the gerbil ischemic hippocampus after transient cerebral ischemia. J. Neurol. Sci. 2014, 347, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.; Wick, W.; Waltenberger, J.; Weller, M.; Dichgans, J.; Schulz, J.B. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J. Neurosci. 2002, 22, 6401–6407. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A.; Chalothorn, D.; Faber, J.E. Vascular endothelial growth factor-A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ. Res. 2008, 103, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Mackey, J.K.; Morrison, J.C.; Haigh, J.J.; Adams, R.H.; Faber, J.E. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ. Res. 2012, 111, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Zechariah, A.; ElAli, A.; Doeppner, T.R.; Jin, F.; Hasan, M.R.; Helfrich, I.; Mies, G.; Hermann, D.M. Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke 2013, 44, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Licht, T.; Rothe, G.; Kreisel, T.; Wolf, B.; Benny, O.; Rooney, A.G.; Ffrench-Constant, C.; Enikolopov, G.; Keshet, E. VEGF preconditioning leads to stem cell remodeling and attenuates age-related decay of adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E7828–E7836. [Google Scholar] [CrossRef] [PubMed]
- Limani, P.; Linecker, M.; Oberkofler, C.E.; Barmettler, G.; Kaech, A.; Graf, R.; Humar, B.; Clavien, P.A. Remote ischemic preconditioning: A novel strategy in rescuing older livers from ischemia-reperfusion injury in a rodent model. Ann. Surg. 2016, 264, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Blauenfeldt, R.A.; Andersen, G.; Hougaard, K.D.; Hoda, M.N.; Ding, Y.; Ji, X. Remote ischaemic conditioning-a new paradigm of self-protection in the brain. Nat. Rev. Neurol. 2015, 11, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Chen, C.; Li, X.R.; Ran, Y.Y.; Xu, T.; Zhang, Y.; Geng, X.K.; Zhang, Y.; Du, H.S.; Leak, R.K.; et al. Remote ischemic preconditioning-mediated neuroprotection against stroke is associated with significant alterations in peripheral immune responses. CNS Neurosci. Ther. 2016, 22, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Ren, C.; Chen, X.; Zhao, H. The chronic protective effects of limb remote preconditioning and the underlying mechanisms involved in inflammatory factors in rat stroke. PLoS ONE 2012, 7, e30892. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Asmaro, K.; Meng, L.; Liu, Y.; Ma, C.; Xi, C.; Li, G.; Ren, C.; Luo, Y.; Ling, F.; et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 2012, 79, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Mi, T.; Yu, F.; Ji, X.; Sun, Y.; Qu, D. The interventional effect of remote ischemic preconditioning on cerebral small vessel disease: A pilot randomized clinical trial. Eur. Neurol. 2016, 76, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yanamoto, H.; Hashimoto, N.; Nagata, I.; Kikuchi, H. Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res. 1998, 784, 239–249. [Google Scholar] [CrossRef]
- Tasaki, K.; Ruetzler, C.A.; Ohtsuki, T.; Martin, D.; Nawashiro, H.; Hallenbeck, J.M. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997, 748, 267–270. [Google Scholar] [CrossRef]
- Nawashiro, H.; Tasaki, K.; Ruetzler, C.A.; Hallenbeck, J.M. TNF-α pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1997, 17, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Ruetzler, C.A.; Tasaki, K.; Hallenbeck, J.M. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal ca1 neurons. J. Cereb. Blood Flow Metab. 1996, 16, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lotz, C.; Roewer, N.; Broscheit, J.-A. Comparison of volatile anesthetic-induced preconditioning in cardiac and cerebral system: Molecular mechanisms and clinical aspects. Eur. J. Med. Res. 2018, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Zitta, K.; Meybohm, P.; Bein, B.; Ohnesorge, H.; Steinfath, M.; Scholz, J.; Albrecht, M. Cytoprotective effects of the volatile anesthetic sevoflurane are highly dependent on timing and duration of sevoflurane conditioning: Findings from a human, in vitro hypoxia model. Eur. J. Pharmacol. 2010, 645, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Sheng, R.; Qin, Z.H. The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol. Sin. 2009, 30, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Gertz, K.; Lindauer, U.; Katchanov, J.; Schultze, J.; Schrock, H.; Nickenig, G.; Kuschinsky, W.; Dirnagl, U.; Laufs, U. Mechanisms of stroke protection by physical activity. Ann. Neurol. 2003, 54, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.H.; Luan, X.D.; Li, J.; Rafols, J.A.; Guthinkonda, M.; Diaz, F.G.; Ding, Y. Exercise-induced overexpression of angiogenic factors and reduction of ischemia/reperfusion injury in stroke. Curr. Neurovasc. Res. 2004, 1, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, X.; Luo, J.; Li, L.; Chen, X.; Huang, R.; Pei, Z. Physical exercise improves functional recovery through mitigation of autophagy, attenuation of apoptosis and enhancement of neurogenesis after MCAO in rats. BMC Neurosci. 2013, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.H.; Li, J.; Zhou, Y.; Rafols, J.A.; Clark, J.C.; Ding, Y. Cerebral angiogenesis and expression of angiogenic factors in aging rats after exercise. Curr. Neurovasc. Res. 2006, 3, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, M.; Allahtavakoli, M.; Abbasnejad, M.; Roohbakhsh, A.; Taghipour, Z.; Taghavi, M.; Khodadadi, H.; Shamsizadeh, A. Exercise preconditioning improves behavioral functions following transient cerebral ischemia induced by 4-vessel occlusion (4-vo) in rats. Arch. Iran. Med. 2013, 16, 697–704. [Google Scholar] [PubMed]
- Fukai, T.; Siegfried, M.R.; Ushio-Fukai, M.; Cheng, Y.; Kojda, G.; Harrison, D.G. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Investig. 2000, 105, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Hambrecht, R.; Wolf, A.; Gielen, S.; Linke, A.; Hofer, J.; Erbs, S.; Schoene, N.; Schuler, G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N. Engl. J. Med. 2000, 342, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Hornig, B.; Maier, V.; Drexler, H. Physical training improves endothelial function in patients with chronic heart failure. Circulation 1996, 93, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C.; Pritchard, K.; Seyedi, N.; Wang, J.; Hintze, T.H. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ. Res. 1994, 74, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Chang, Y.C.; Tu, Y.F.; Huang, C.C. VEGF-a/VEGFR-2 signaling leading to camp response element-binding protein phosphorylation is a shared pathway underlying the protective effect of preconditioning on neurons and endothelial cells. J. Neurosci. 2009, 29, 4356–4368. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.; Sprague, S.; Guo, M.; Davis, W.; Friedman, A.; Kumar, A.; Jimenez, D.F.; Ding, Y. Forced, not voluntary, exercise effectively induces neuroprotection in stroke. Acta Neuropathol. 2008, 115, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Lu, J.; Selfridge, J.E.; Burns, J.M.; Swerdlow, R.H. Lactate administration reproduces specific brain and liver exercise-related changes. J. Neurochem. 2013, 127, 91–100. [Google Scholar] [CrossRef]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Slowik, A.; Kumar, P.; Szczudlik, A.; Gaffney, J. Serial measurement of vascular endothelial growth factor and transforming growth factor-β1 in serum of patients with acute ischemic stroke. Stroke 2000, 31, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, R.; Ago, T.; Kamouchi, M.; Kuroda, J.; Kuwashiro, T.; Hata, J.; Sugimori, H.; Fukuda, K.; Gotoh, S.; Makihara, N.; et al. Clinical significance of plasma VEGF value in ischemic stroke—Research for biomarkers in ischemic stroke (rebios) study. BMC Neurol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Lee, K.Y.; Kim, Y.J.; Kim, S.H.; Koh, S.H.; Lee, Y.J. Serum VEGF levels in acute ischaemic strokes are correlated with long-term prognosis. Eur. J. Neurol. 2010, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. https://doi.org/10.3390/ijms19051362
Geiseler SJ, Morland C. The Janus Face of VEGF in Stroke. International Journal of Molecular Sciences. 2018; 19(5):1362. https://doi.org/10.3390/ijms19051362
Chicago/Turabian StyleGeiseler, Samuel J., and Cecilie Morland. 2018. "The Janus Face of VEGF in Stroke" International Journal of Molecular Sciences 19, no. 5: 1362. https://doi.org/10.3390/ijms19051362
APA StyleGeiseler, S. J., & Morland, C. (2018). The Janus Face of VEGF in Stroke. International Journal of Molecular Sciences, 19(5), 1362. https://doi.org/10.3390/ijms19051362