Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1

Abstract
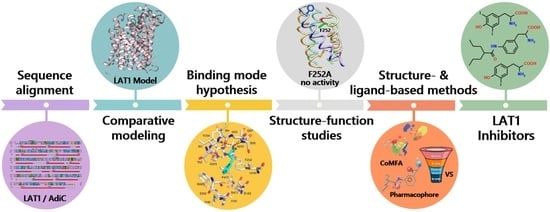
1. Introduction
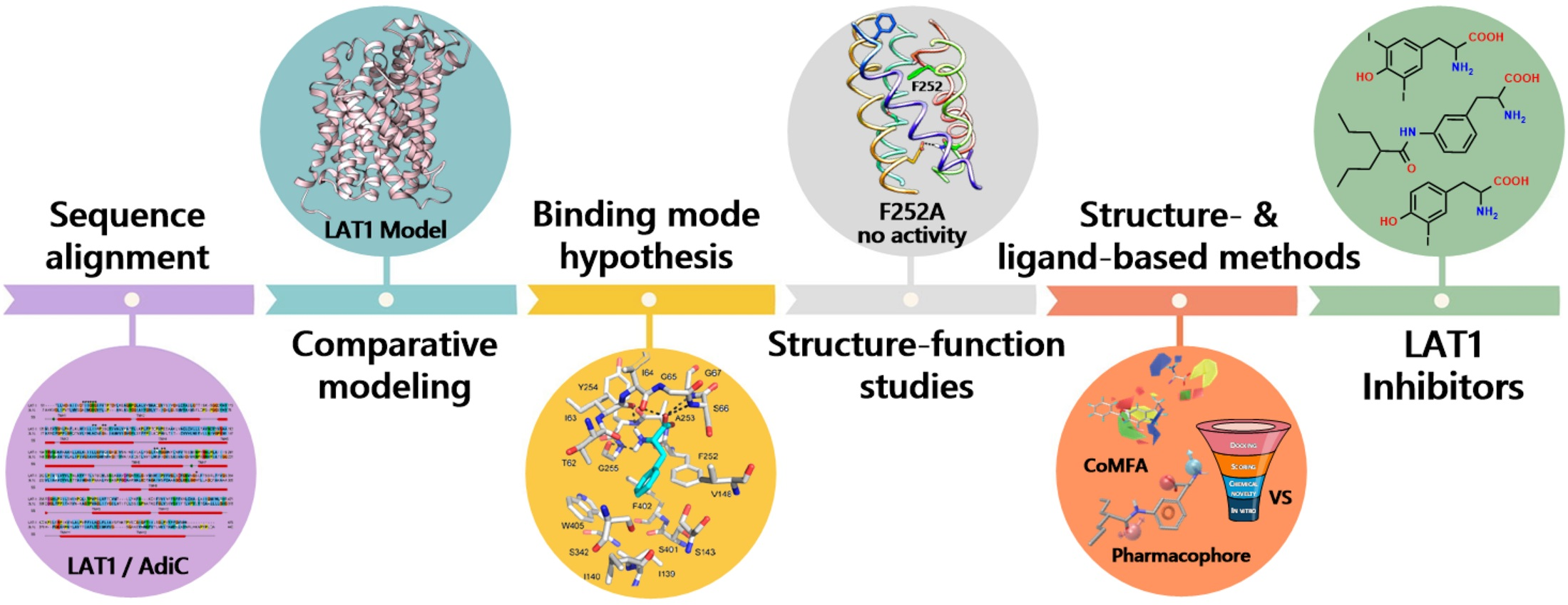
2. Structure of LAT1–4F2hc
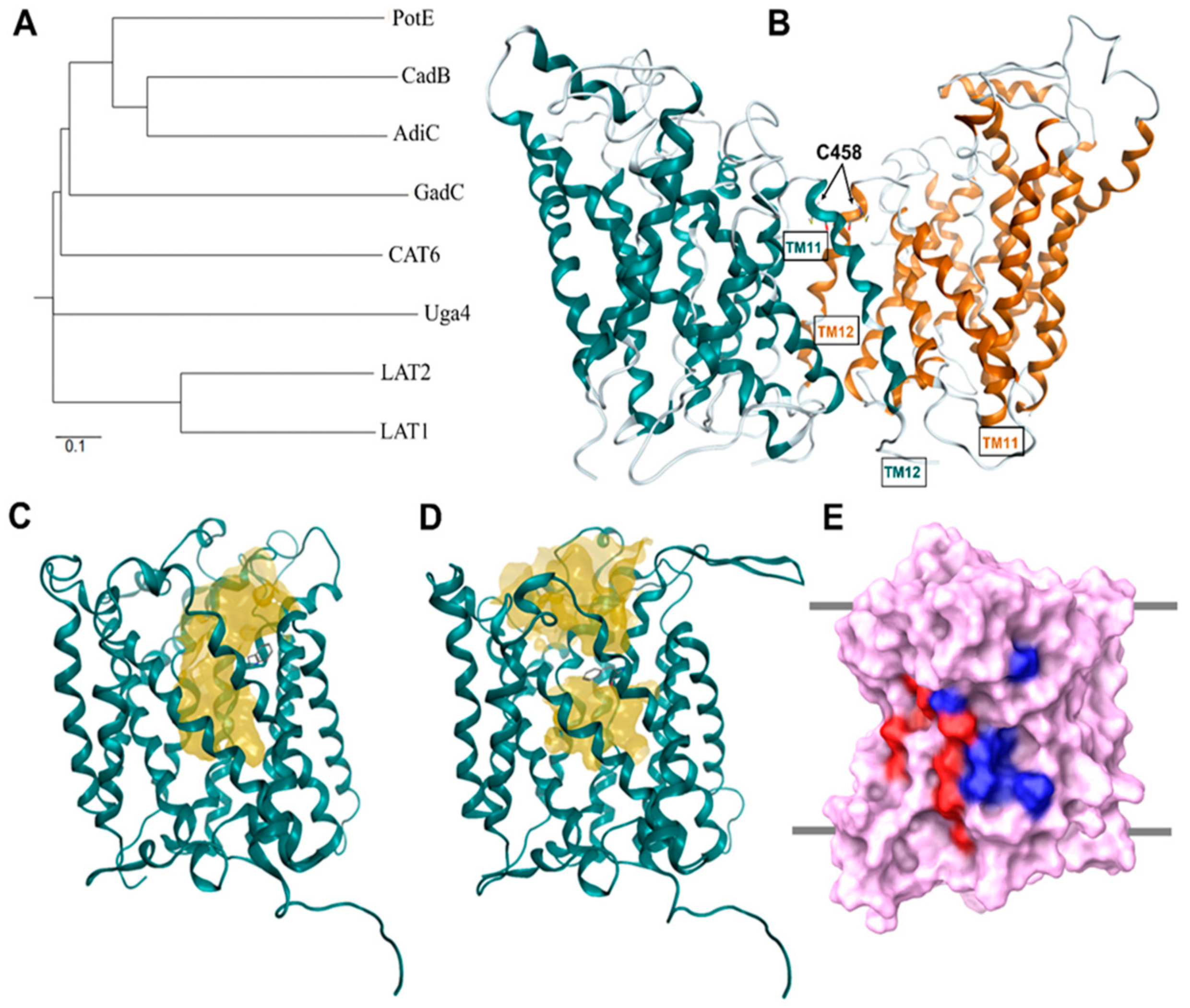
3. Structure of LAT1 Homolog AdiC
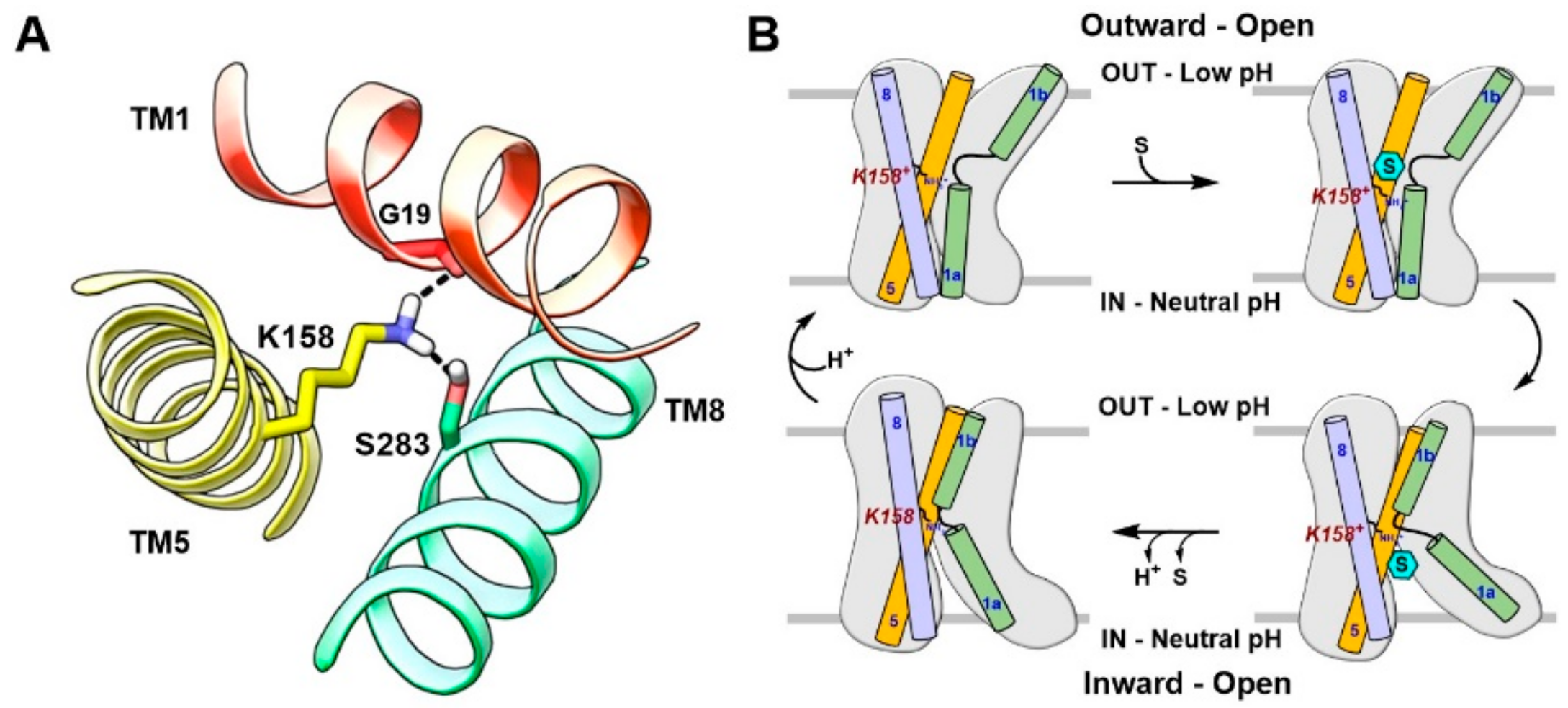
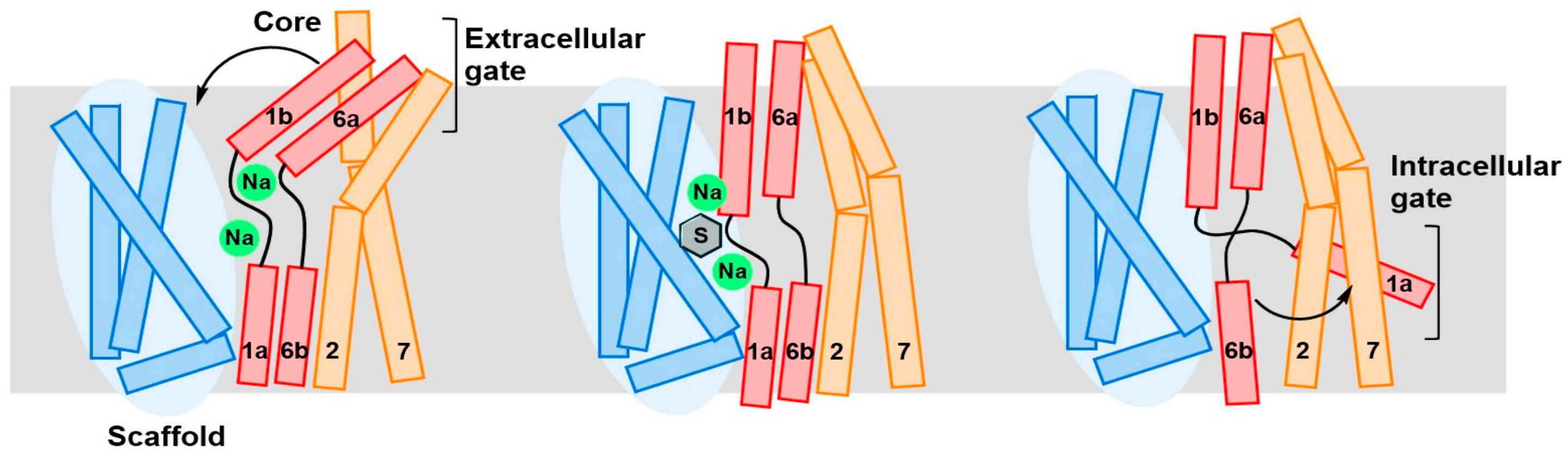
4. Substrate Translocation Mechanism
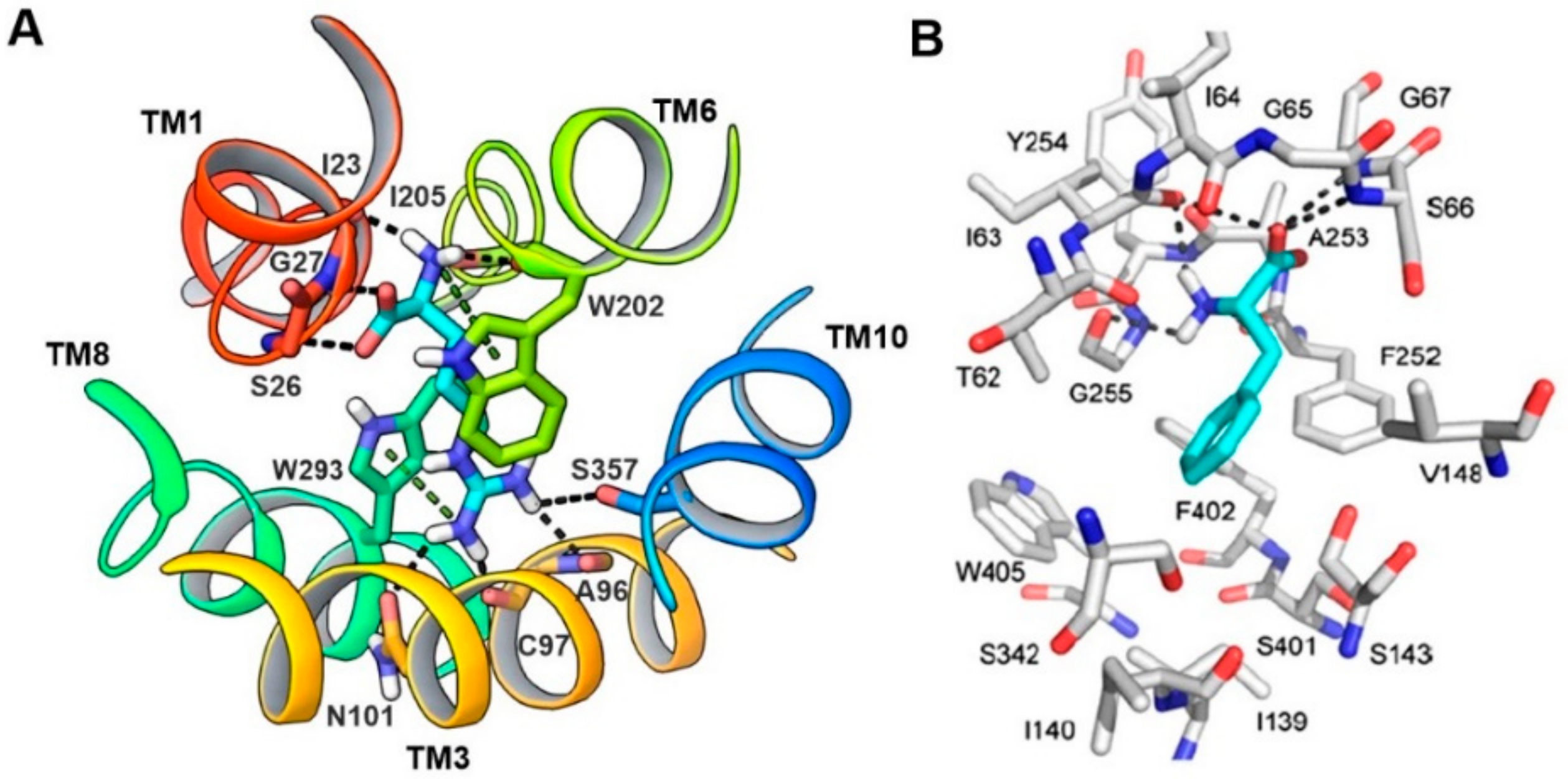
5. Homology Modeling of LAT1
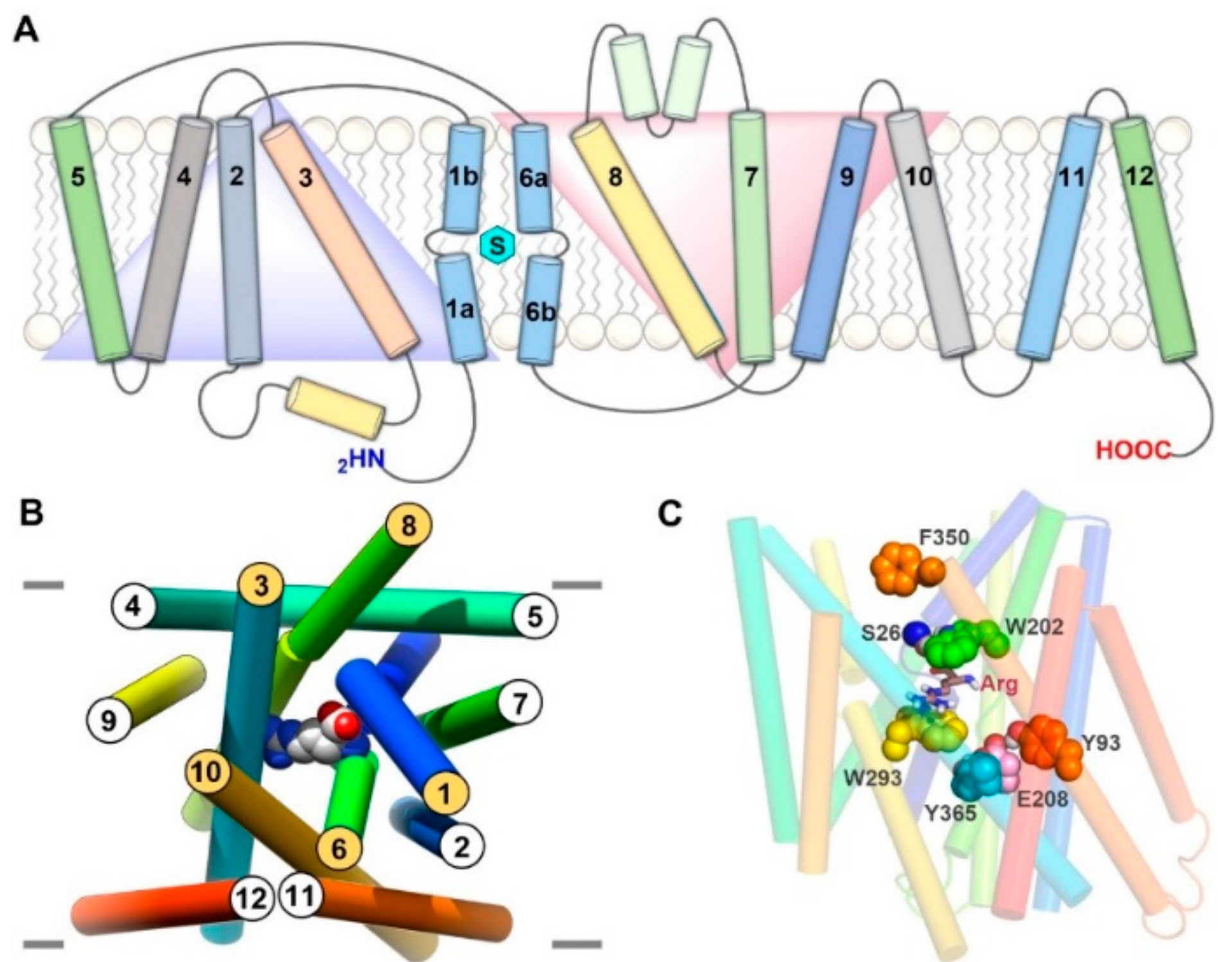
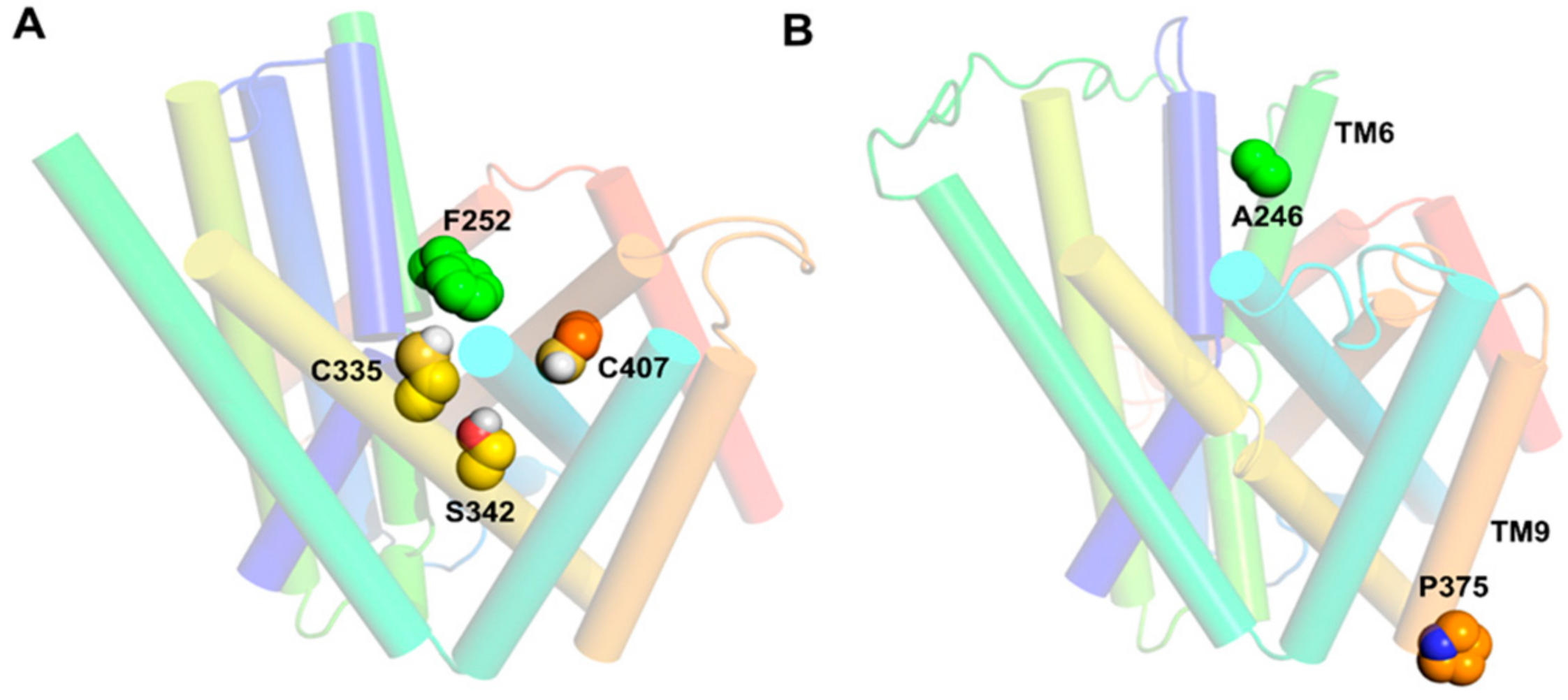
6. LAT1 Structure–Function Studies
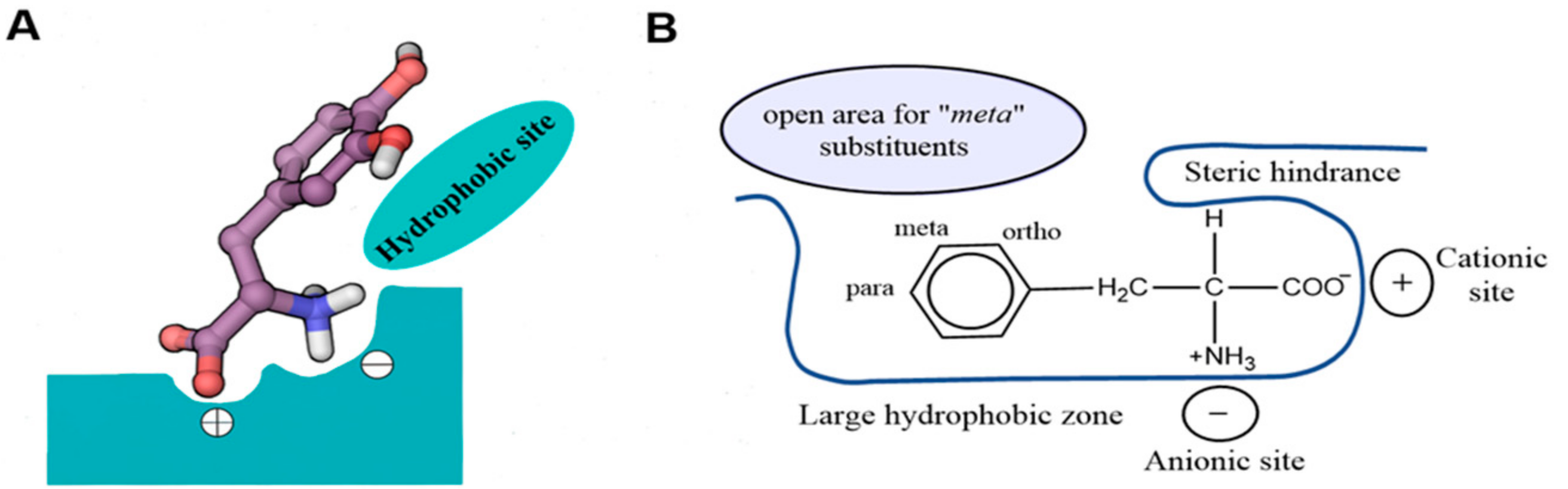
7. Binding Hypothesis of LAT1 Ligands
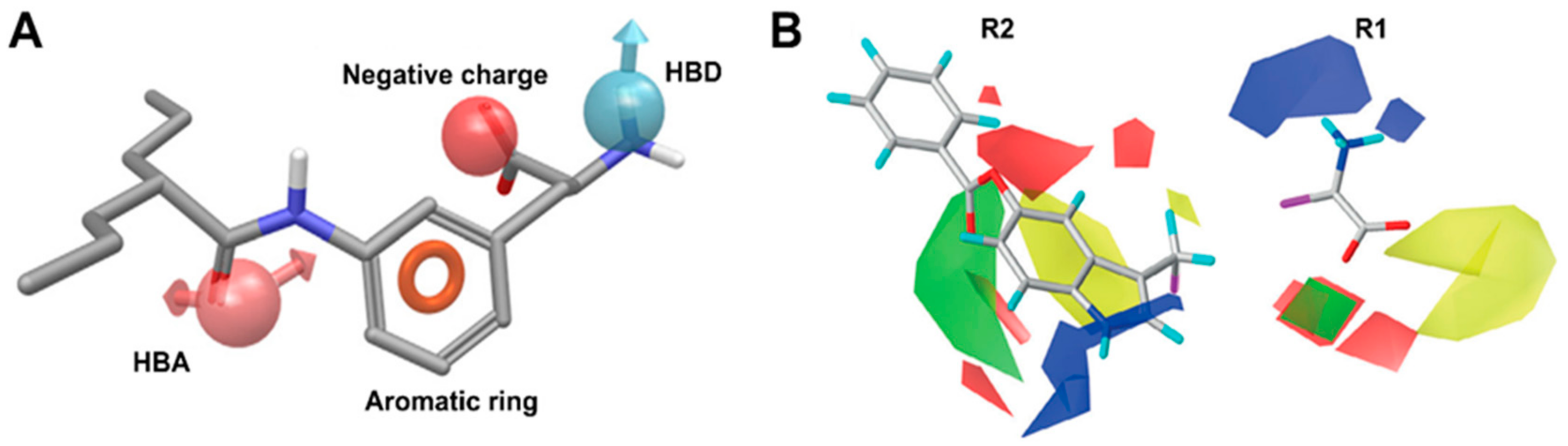
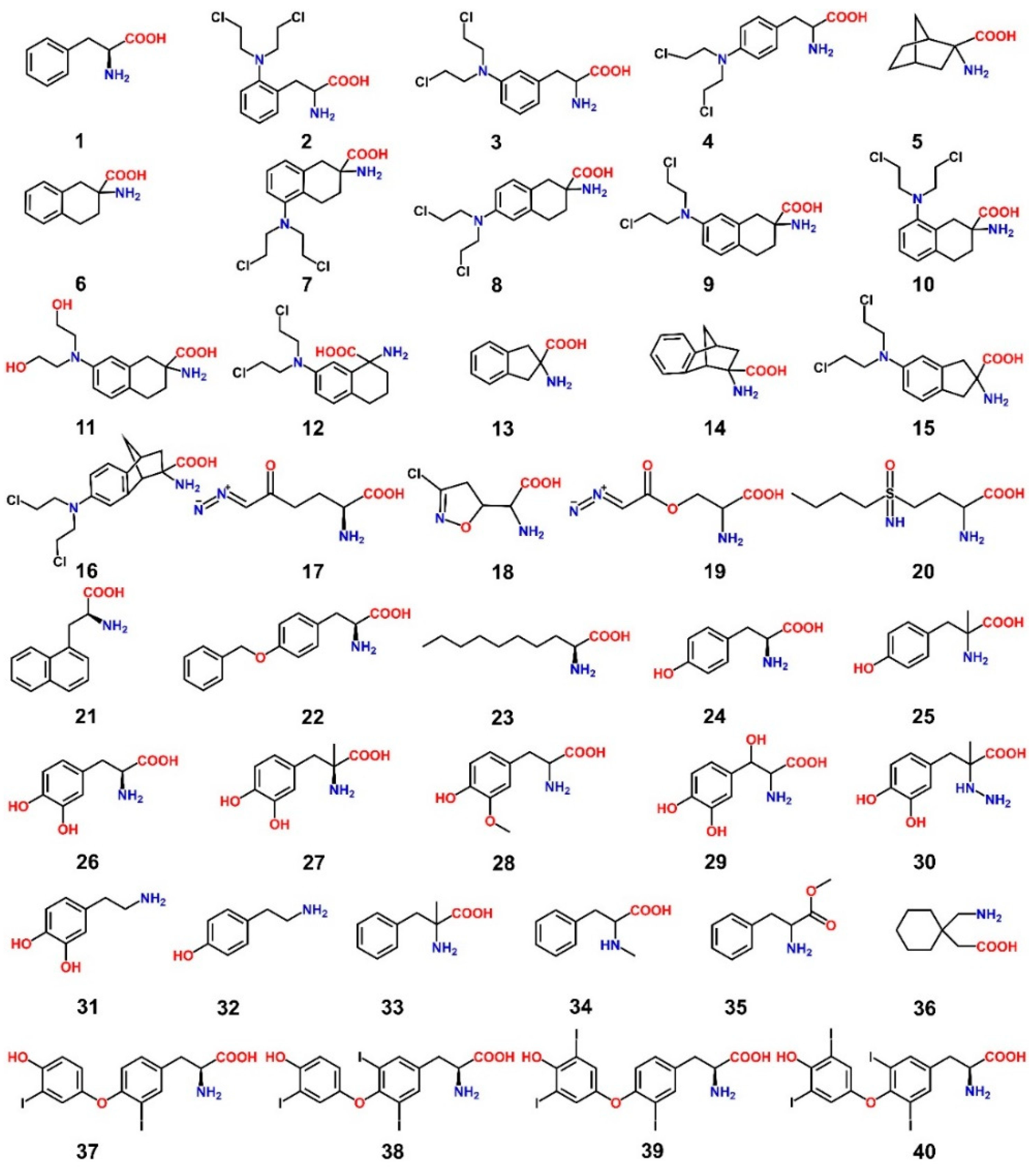
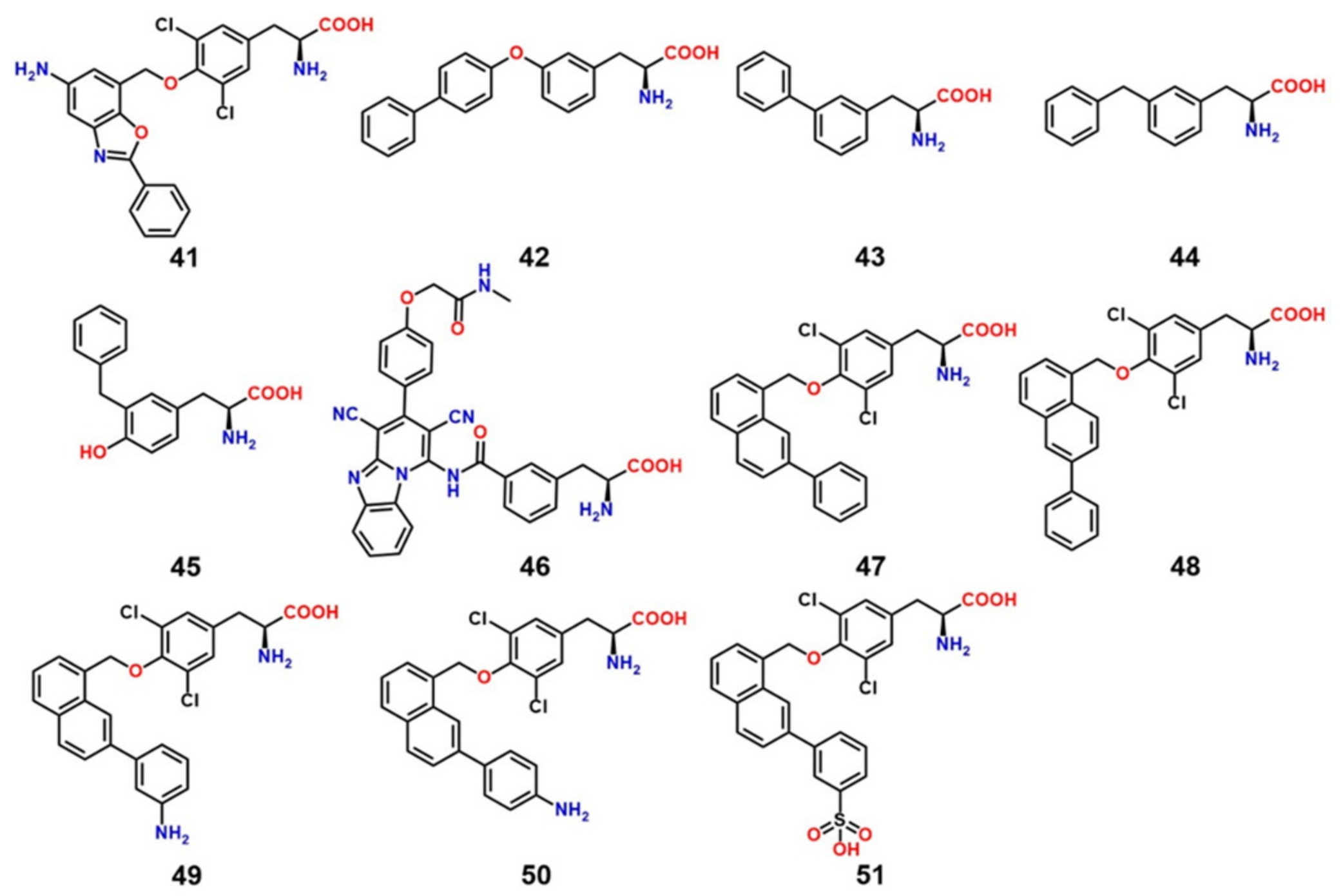
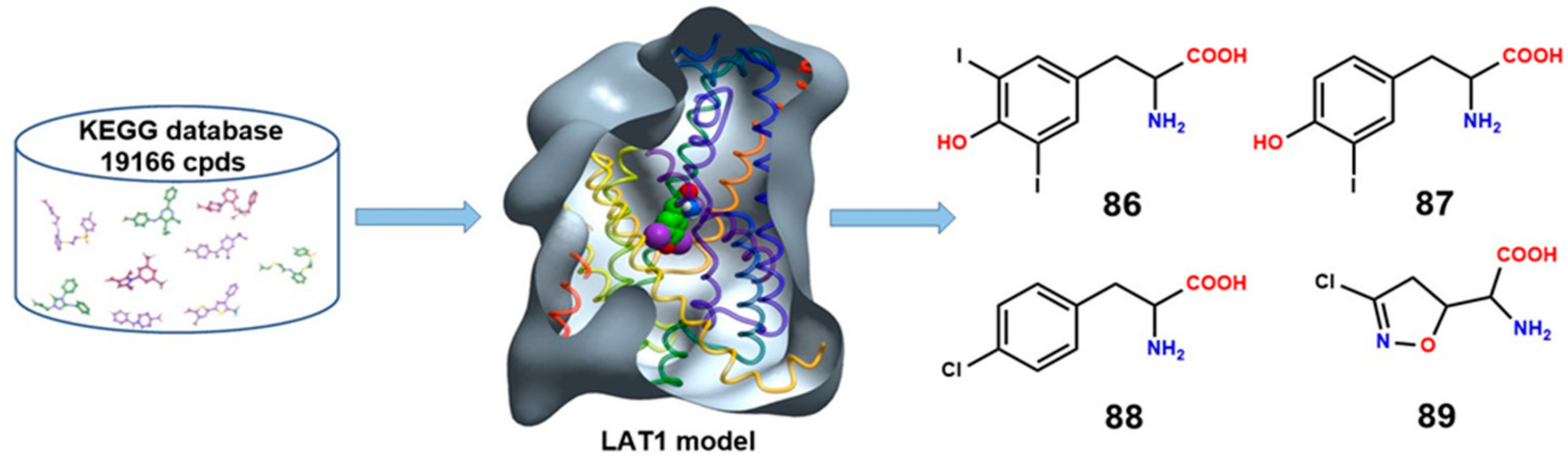
8. LAT1 Ligand Discovery
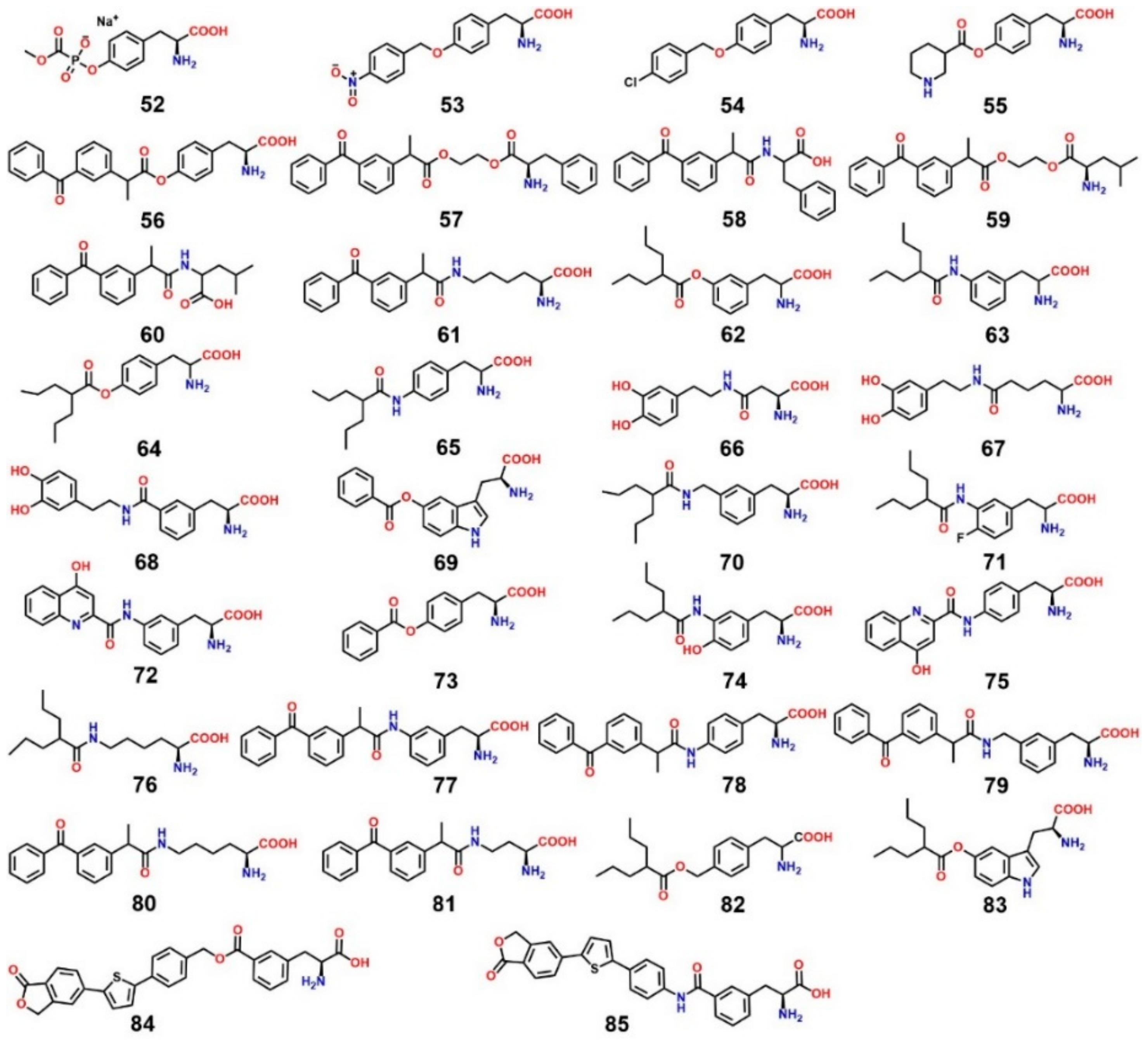
9. LAT1-Mediated Prodrug Delivery
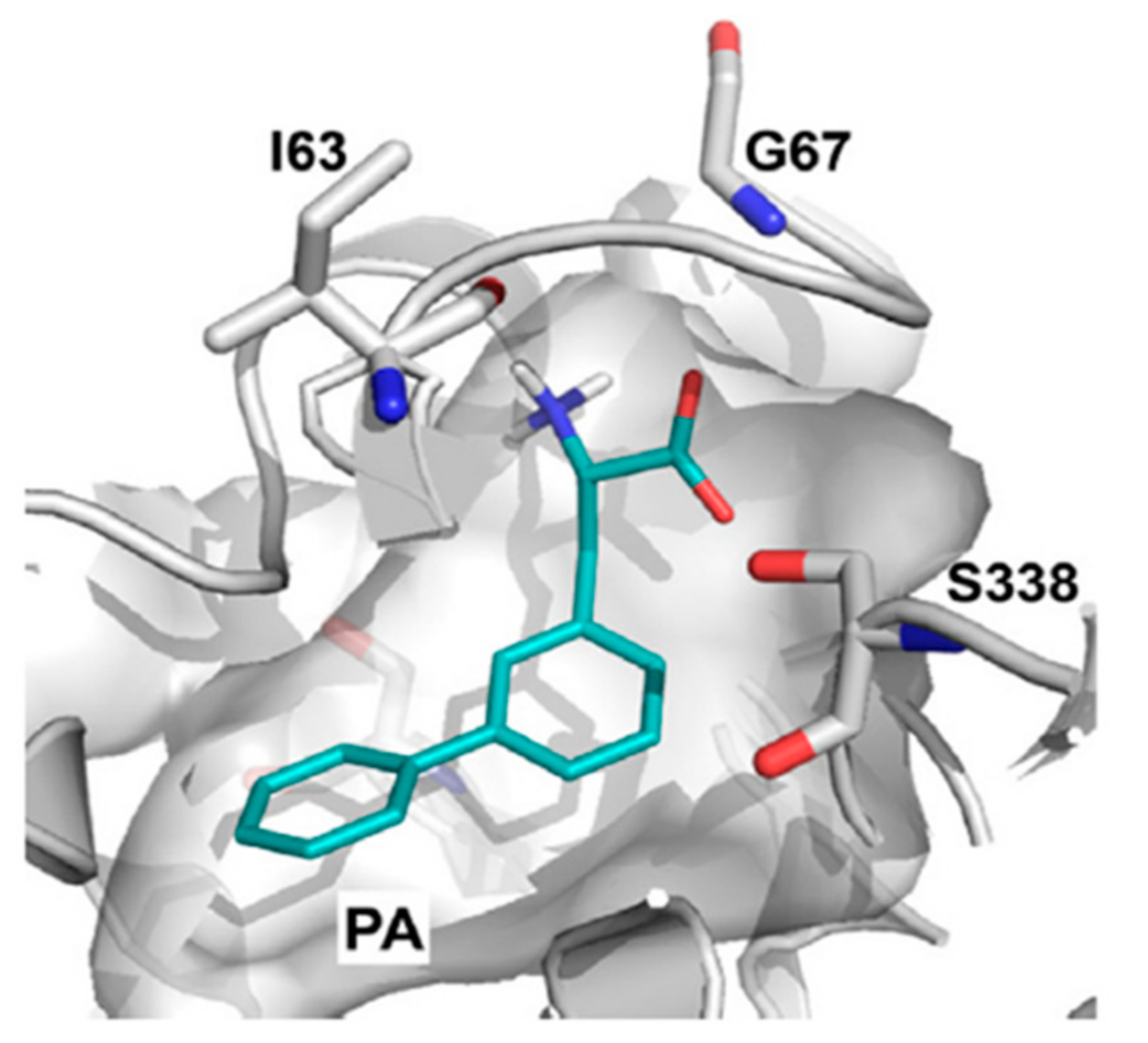
10. Structure-Based Ligand Discovery of LAT1
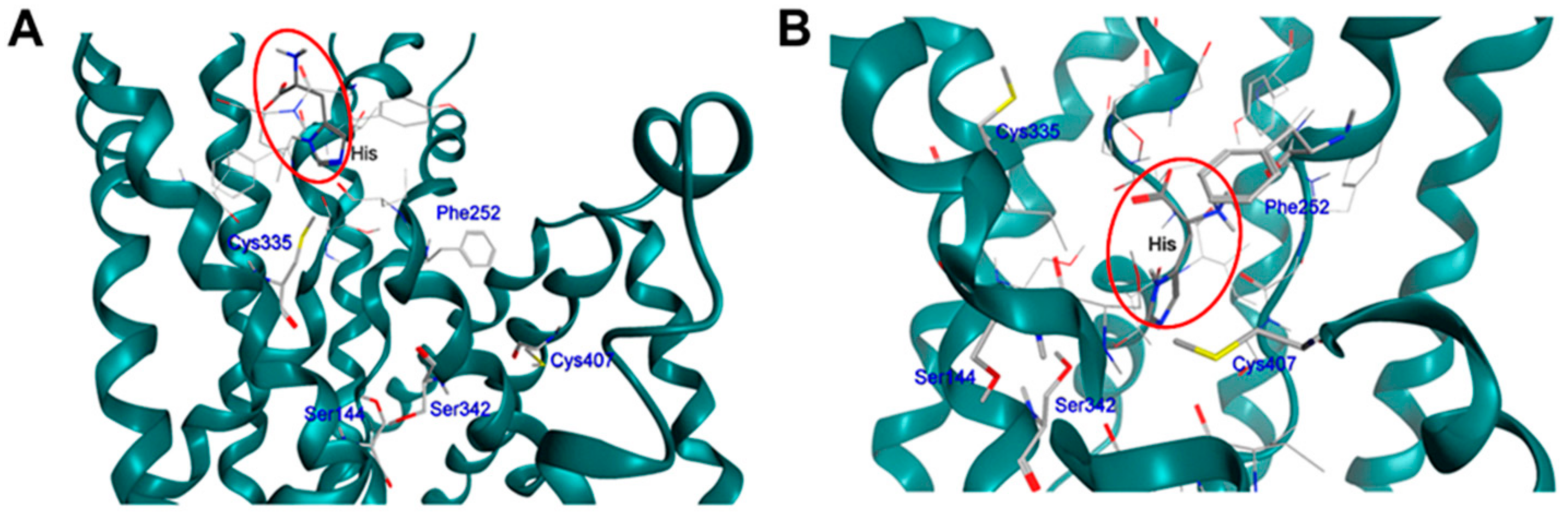
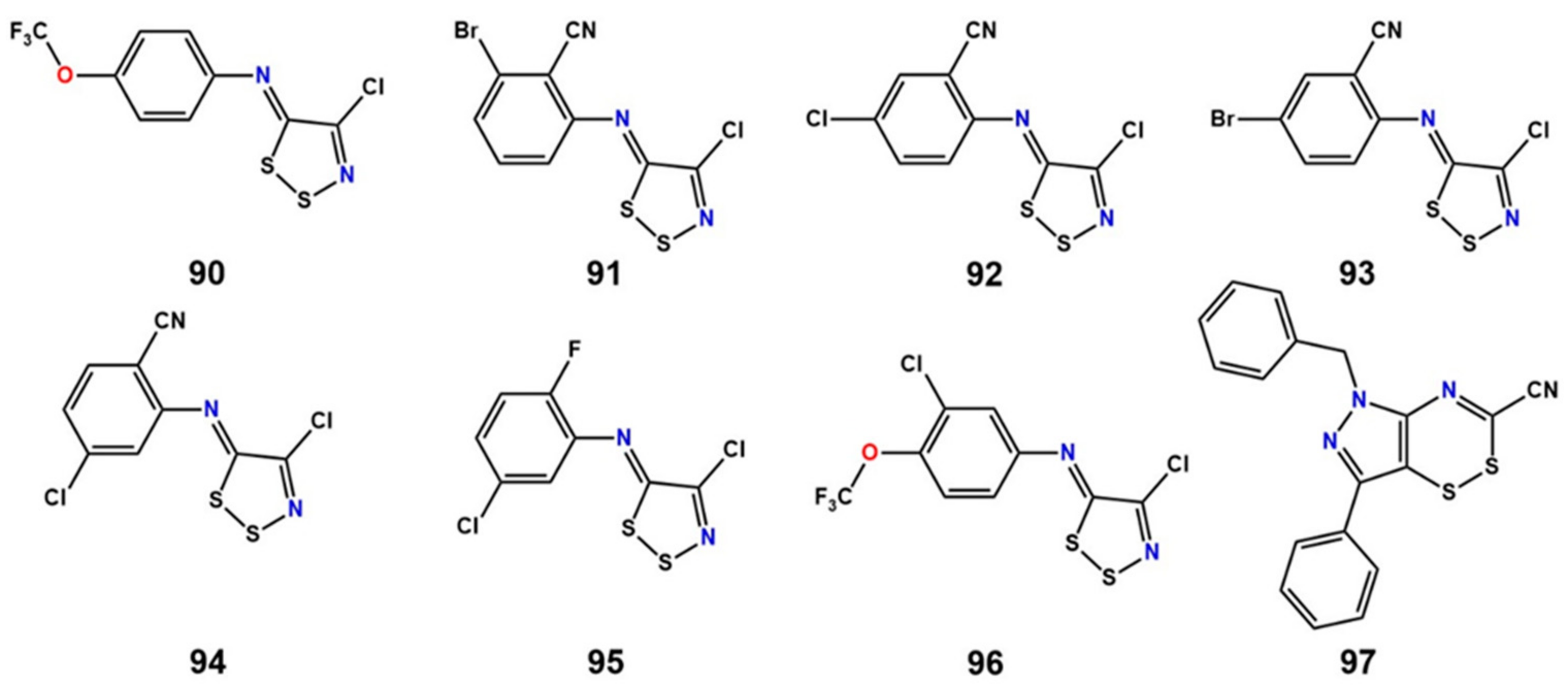
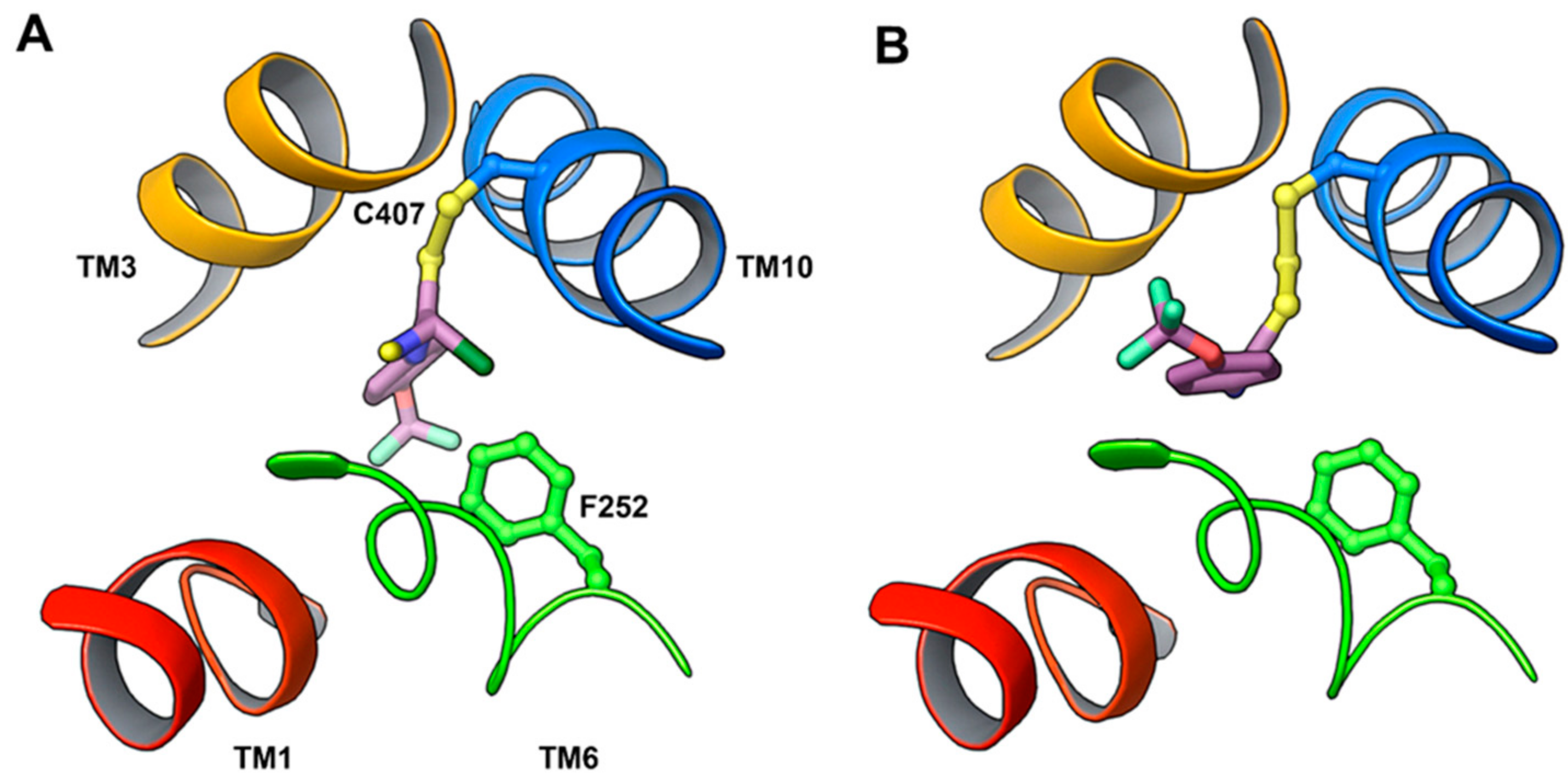
11. Covalent Inhibition of LAT1
12. Conclusions and Future Outlooks
Acknowledgments
Conflicts of Interest
References
- Verrey, F.; Closs, E.I.; Wagner, C.A.; Palacin, M.; Endou, H.; Kanai, Y. CATs and HATs: The SLC7 family of amino acid transporters. Pflüg. Arch. 2004, 447, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Fotiadis, D.; Kanai, Y.; Palacín, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Aspects Med. 2013, 34, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Segawa, H.; Miyamoto, K.I.; Uchino, H.; Takeda, E.; Endou, H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Kinne, A.; Bräuer, A.U.; Sapin, R.; Klein, M.O.; Köhrle, J.; Wirth, E.K.; Schweizer, U. Developmental and cell type-specific expression of thyroid hormone transporters in the mouse brain and in primary brain cells. Glia 2011, 59, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.-B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immnol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Smith, Q.R. Carrier-mediated transport to enhance drug delivery to brain. Int. Congr. Ser. 2005, 1277, 63–74. [Google Scholar] [CrossRef]
- Pardridge, W.M. Brain metabolism: A perspective from the blood-brain barrier. Physiol. Rev. 1983, 63, 1481–1535. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 2002, 21, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Scalise, M.; Galluccio, M.; Pochini, L.; Albanese, L.M.; Indiveri, C. LAT1 is the transport competent unit of the LAT1/CD98 heterodimeric amino acid transporter. Int. J. Biochem. Cell Biol. 2015, 67, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Sato, M.; Yang, H.; Miyagawa, F.; Harasaki, M.; Tomita, K.; Matsuoka, S.; Noma, A.; Iwai, K.; Minato, N. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J. Biol. Chem. 1999, 274, 3009–3016. [Google Scholar] [CrossRef] [PubMed]
- Poncet, N.; Mitchell, F.E.; Ibrahim, A.F.; McGuire, V.A.; English, G.; Arthur, J.S.; Shi, Y.B.; Taylor, P.M. The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS ONE 2014, 9, e89547. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Ohnishi, A.; Promsuk, J.; Shimizu, S.; Kanai, Y.; Shiokawa, Y.; Nagane, M. Enhanced tumor growth elicited by l-type amino acid transporter 1 in human malignant glioma cells. Neurosurgery 2008, 62, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Tanaka, S.; Ishizuka, T.; Kanai, Y.; et al. l-type amino acid transporter 1 and CD98 expression in primary and metastatic sites of human neoplasms. Cancer Sci. 2008, 99, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Betsunoh, H.; Fukuda, T.; Anzai, N.; Nishihara, D.; Mizuno, T.; Yuki, H.; Masuda, A.; Yamaguchi, Y.; Abe, H.; Yashi, M.; et al. Increased expression of system large amino acid transporter (LAT)-1 mRNA is associated with invasive potential and unfavorable prognosis of human clear cell renal cell carcinoma. BMC Cancer 2013, 13, 509. [Google Scholar] [CrossRef] [PubMed]
- Ebara, T.; Kaira, K.; Saito, J.; Shioya, M.; Asao, T.; Takahashi, T.; Sakurai, H.; Kanai, Y.; Kuwano, H.; Nakano, T. l-type amino-acid transporter 1 expression predicts the response to preoperative hyperthermo-chemoradiotherapy for advanced rectal cancer. Anticancer Res. 2010, 30, 4223–4227. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Horiguchi, J.; Nakajima, H.; Kanai, Y.; Oyama, T. Correlation of l-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci. 2012, 103, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Nawashiro, H.; Otani, N.; Shinomiya, N.; Fukui, S.; Ooigawa, H.; Shima, K.; Matsuo, H.; Kanai, Y.; Endou, H. l-type amino acid transporter 1 as a potential molecular target in human astrocytic tumors. Int. J. Cancer 2006, 119, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Sakata, T.; Ferdous, G.; Tsuruta, T.; Satoh, T.; Baba, S.; Muto, T.; Ueno, A.; Kanai, Y.; Endou, H.; Okayasu, I. l-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer. Pathol. Int. 2009, 59, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Ogata, S.; Nakanishi, K.; Ozeki, Y.; Hiroi, S.; Tominaga, S.; Aida, S.; Matsuo, H.; Sakata, T.; Kawai, T. LAT1 expression in non-small-cell lung carcinomas: Analyses by semiquantitative reverse transcription-PCR (237 cases) and immunohistochemistry (295 cases). Lung Cancer (Amsterdam, Netherlands) 2010, 68, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Park, K.J.; Park, J.R.; Kanai, Y.; Endou, H.; Park, J.C.; Kim, D.K. The RNA interference of amino acid transporter LAT1 inhibits the growth of KB human oral cancer cells. Anticancer Res. 2006, 26, 2943–2948. [Google Scholar] [PubMed]
- Marshall, A.D.; van Geldermalsen, M.; Otte, N.J.; Anderson, L.A.; Lum, T.; Vellozzi, M.A.; Zhang, B.K.; Thoeng, A.; Wang, Q.; Rasko, J.E.; et al. LAT1 is a putative therapeutic target in endometrioid endometrial carcinoma. Int. J. Cancer 2016, 139, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Jutabha, P.; Endou, H.; Anzai, N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol. Rep. 2012, 28, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Sakamoto, S.; Matsushima, J.; Kimura, T.; Ueda, T.; Mizokami, A.; Kanai, Y.; Ichikawa, T. Up-Regulation of LAT1 during Antiandrogen Therapy Contributes to Progression in Prostate Cancer Cells. J. Urol. 2016, 195, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Cho, H.T.; Williams, L.; Zhu, A.; Liang, K.; Huang, K.; Wu, H.; Jiang, C.; Hong, S.; Crowe, R.; et al. Potential Biomarker of l-type Amino Acid Transporter 1 in Breast Cancer Progression. Nucl. Med. Mol. Imaging 2011, 45, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutté, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic Disruption of the Multifunctional CD98/LAT1 Complex Demonstrates the Key Role of Essential Amino Acid Transport in the Control of mTORC1 and Tumor Growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Tamai, I. Solute carrier transporters as targets for drug delivery and pharmacological intervention for chemotherapy. J. Pharm. Sci. 2011, 100, 3731–3750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Holst, J. l-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015, 5, 1281–1294. [Google Scholar] [PubMed]
- Wang, Q.; Tiffen, J.; Bailey, C.G.; Lehman, M.L.; Ritchie, W.; Fazli, L.; Metierre, C.; Feng, Y.J.; Li, E.; Gleave, M.; et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: Effects on cell cycle, cell growth, and tumor development. J. Natl. Cancer Inst. 2013, 105, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K.; et al. Inhibition of l-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 2010, 30, 4819–4828. [Google Scholar] [PubMed]
- Kim, C.S.; Cho, S.H.; Chun, H.S.; Lee, S.Y.; Endou, H.; Kanai, Y.; Kim, D.K. BCH, an inhibitor of system L amino acid transporters, induces apoptosis in cancer cells. Biol. Pharm. Bull. 2008, 31, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Shennan, D.B.; Thomson, J. Inhibition of system L (LAT1/CD98hc) reduces the growth of cultured human breast cancer cells. Oncol. Rep. 2008, 20, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Rosell, A.; Álvarez-Marimon, E.; Zorzano, A.; Fotiadis, D.; Palacín, M. Expression of human heteromeric amino acid transporters in the yeast Pichia pastoris. Protein Expr. Purif. 2013, 87, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Meury, M.; Álvarez-Marimon, E.; Costa, M.; Pérez-Cano, L.; Zorzano, A.; Fernández-Recio, M.; Palacín, M.; Fotiadis, D. Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. Proc. Natl. Acad. Sci. USA 2014, 111, 2966–2971. [Google Scholar]
- Estévez, R.; Camps, M.; Rojas, A.M.; Testar, X.; Devés, R.; Hediger, M.A.; Zorzano, A.; Palacín, M. The amino acid transport system y+L/4F2hc is a heteromultimeric complex. FASEB J. 1998, 12, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Bippes, C.A.; Zeltina, A.; Casagrande, F.; Ratera, M.; Palacin, M.; Muller, D.J.; Fotiadis, D. Substrate binding tunes conformational flexibility and kinetic stability of an amino acid antiporter. J. Biol. Chem. 2009, 284, 18651–18663. [Google Scholar] [CrossRef] [PubMed]
- Saier, M.H.; Tran, C.V.; Barabote, R.D. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef] [PubMed]
- Fort, J.; de la Ballina, L.R.; Burghardt, H.E.; Ferrer-Costa, J.; Turnay, J.; Ferrer-Orta, C.; Usón, I.; Usón, I.; Zorzano, A.; Fernández-Recio, J.; Orozco, M.; et al. The structure of human 4F2hc ectodomain provides a model for homodimerization and electrostatic interaction with plasma membrane. J. Biol. Chem. 2007, 282, 31444–31452. [Google Scholar]
- Gao, X.; Zhou, L.; Jiao, X.; Lu, F.; Yan, C.; Zeng, X.; Wang, J.; Shi, Y. Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 2010, 463, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, P.L.; Goehring, A.; Shankaranarayanan, A.; Gouaux, E. Structure and mechanism of a Na+-independent amino acid transporter. Science 2009, 325, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lu, P.; Yan, C.; Fan, C.; Yin, P.; Wang, J.; Shi, Y. Structure and mechanism of a glutamate-GABA antiporter. Nature 2012, 483, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lu, F.; Zhou, L.; Dang, S.; Sun, L.; Li, X.; Wang, J.; Shi, Y. Structure and mechanism of an amino acid antiporter. Science 2009, 324, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Krammer, E.-M.; Ghaddar, K.; André, B.; Prévost, M. Unveiling the Mechanism of Arginine Transport through AdiC with Molecular Dynamics Simulations: The Guiding Role of Aromatic Residues. PLoS ONE 2016, 11, e0160219. [Google Scholar] [CrossRef] [PubMed]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.J.; Sali, A.; Giacomini, K.M. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Weinstein, H. Conformational Rearrangements to the Intracellular Open States of the LeuT and ApcT Transporters Are Modulated by Common Mechanisms. Biophys. J. 2010, 99, L103–L105. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Jayaram, H.; Shane, T.; Kolmakova-Partensky, L.; Wu, F.; Williams, C.; Xiong, Y.; Miller, C. Structure of a prokaryotic virtual proton pump at 3.2 A resolution. Nature 2009, 460, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Ilgü, H.; Jeckelmann, J.-M.; Gapsys, V.; Ucurum, Z.; de Groot, B.L.; Fotiadis, D. Insights into the molecular basis for substrate binding and specificity of the wild-type l-arginine/agmatine antiporter AdiC. Proc. Natl. Acad. Sci. USA 2016, 113, 10358–10363. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, L.; Ratera, M.; Paladino, A.; Bartoccioni, P.; Errasti-Murugarren, E.; Valencia, E.; Portella, G.; Bial, S.; Zorzano, A.; Fita, I.; et al. Molecular basis of substrate-induced permeation by an amino acid antiporter. Proc. Natl. Acad. Sci. USA 2011, 108, 3935–3940. [Google Scholar] [CrossRef] [PubMed]
- Forrest, L.R.; Rudnick, G. The rocking bundle: A mechanism for ion-coupled solute flux by symmetrical transporters. Physiology 2009, 24, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal structure of a bacterial homologue of Na+/Cl−dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [Google Scholar]
- Dickens, D.; Webb, S.D.; Antonyuk, S.; Giannoudis, A.; Owen, A.; Rädisch, S.; Hasnain, S.S.; Pirmohamed, M. Transport of gabapentin by LAT1 (SLC7A5). Biochem. Pharmacol. 2013, 85, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Galluccio, M.; Scalise, M.; Parravicini, C.; Palazzolo, L.; Eberini, I.; Indiveri, C. Novel insights into the transport mechanism of the human amino acid transporter LAT1 (SLC7A5). Probing critical residues for substrate translocation. Biochim. Biophys. Acta 2017, 1861, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Panni, S.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Transport mechanism and regulatory properties of the human amino acid transporter ASCT2 (SLC1A5). Amino Acids 2014, 46, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Capobianco, L.; Krämer, R.; Palmieri, F. Kinetics of the reconstituted dicarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta 1989, 977, 187–193. [Google Scholar] [CrossRef]
- Khafizov, K.; Staritzbichler, R.; Stamm, M.; Forrest, L.R. A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry 2010, 49, 10702–10713. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.; Boudker, O. Shared Molecular Mechanisms of Membrane Transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Kim, B.-H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Hersh, B.M.; Farooq, F.T.; Barstad, D.N.; Blankenhorn, D.L.; Slonczewski, J.L. A glutamate-dependent acid resistance gene in Escherichia coli. J. Bacteriol. 1996, 178, 3978–3981. [Google Scholar] [CrossRef] [PubMed]
- Castanie-Cornet, M.P.; Penfound, T.A.; Smith, D.; Elliott, J.F.; Foster, J.W. Control of acid resistance in Escherichia coli. J. Bacteriol. 1999, 181, 3525–3535. [Google Scholar] [PubMed]
- Soksawatmaekhin, W.; Kuraishi, A.; Sakata, K.; Kashiwagi, K.; Igarashi, K. Excretion and uptake of cadaverine by CadB and its physiological functions in Escherichia coli. Mol. Microbiol. 2004, 51, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Soksawatmaekhin, W.; Uemura, T.; Fukiwake, N.; Kashiwagi, K.; Igarashi, K. Identification of the Cadaverine Recognition Site on the Cadaverine-Lysine Antiporter CadB. J. Biol. Chem. 2006, 281, 29213–29220. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, K.; Miyamoto, S.; Suzuki, F.; Kobayashi, H.; Igarashi, K. Excretion of putrescine by the putrescine-ornithine antiporter encoded by the potE gene of Escherichia coli. Proc. Natl. Acad. Sci. USA 1992, 89, 4529–4533. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, K.; Kuraishi, A.; Tomitori, H.; Igarashi, A.; Nishimura, K.; Shirahata, A.; Igarashi, K. Identification of the Putrescine Recognition Site on Polyamine Transport Protein PotE. J. Biol. Chem. 2000, 275, 36007–36012. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, K.; Shibuya, S.; Tomitori, H.; Kuraishi, A.; Igarashi, K. Excretion and Uptake of Putrescine by the PotE Protein in Escherichia coli. J. Biol. Chem. 1997, 272, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Kolmakova-Partensky, L.; Miller, C. A Bacterial Arginine-Agmatine Exchange Transporter Involved in Extreme Acid Resistance. J. Biol. Chem. 2007, 282, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, F.; Ratera, M.; Schenk, A.D.; Chami, M.; Valencia, E.; Lopez, J.M.; Torrents, D.; Engel, A.; Palacin, M.; Fotiadis, D. Projection Structure of a Member of the Amino Acid/Polyamine/Organocation Transporter Superfamily. J. Biol. Chem. 2008, 283, 33240–33248. [Google Scholar] [CrossRef] [PubMed]
- Dickens, D.; Chiduza, G.N.; Wright, G.S.A.; Pirmohamed, M.; Antonyuk, S.V.; Hasnain, S.S. Modulation of LAT1 (SLC7A5) transporter activity and stability by membrane cholesterol. Sci. Rep. 2017, 7, 43580. [Google Scholar] [CrossRef] [PubMed]
- Penmatsa, K.; Wang, H.; Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2015-1: PrimeX; Schrödinger, LLC: New York, NY, USA, 2015.
- The PyMOL Molecular Graphics System, version 1.6; Schrödinger, LLC: New York, NY, USA, 2015.
- Schrödinger Release 2015-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2015.
- Oppedisano, F.; Indiveri, C. Reconstitution into liposomes of the B degrees-like glutamine-neutral amino acid transporter from renal cell plasma membrane. Biochim. Biophys. Acta 2008, 1778, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Tărlungeanu, D.C.; Deliu, E.; Dotter, C.P.; Kara, M.; Janiesch, P.C.; Scalise, M.; Galluccio, M.; Tesulov, M.; Morelli, E.; Sonmez, F.M.; et al. Impaired Amino Acid Transport at the Blood Brain Barrier Is a Cause of Autism Spectrum Disorder. Cell 2016, 167, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, H. Transport of amino acid-related compounds mediated by l-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Peura, L.; Malmioja, K.; Leppänen, J.; Laine, K.; Poso, A.; Lahtela-Kakkonen, M.; Rautio, J. Structure-activity relationship study of compounds binding to large amino acid transporter 1 (LAT1) based on pharmacophore modeling and in situ rat brain perfusion. Eur. J. Pharm. Sci. 2013, 48, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lahtela-Kakkonen, M.; Huttunen, K.M.; et al. Quantitative insight into the design of compounds recognized by the l-type amino acid transporter 1 (LAT1). Chem. Med. Chem. 2014, 9, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D. Topomer CoMFA: A Design Methodology for Rapid Lead Optimization. J. Med. Chem. 2003, 46, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Smith, Q.R. Drug delivery to brain and the role of carrier-mediated transport. In Frontiers in Cerebral Vascular Biology, Advances in Experimental Medicine and Biology; Drewes, L.R., Betz, A.L., Eds.; Springer: Boston, MA, USA, 1993. [Google Scholar]
- Killian, D.M.; Hermeling, S.; Chikhale, P.J. Targeting the cerebrovascular large neutral amino acid transporter (LAT1) isoform using a novel disulfide-based brain drug delivery system. Drug Deliv. 2007, 14, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Jalkanen, A.; Lehtonen, M.; Forsberg, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Knuuti, J.; Rautio, J. Brain uptake of ketoprofen-lysine prodrug in rats. Int. J. Pharm. 2010, 399, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bergel, F.; Stock, J.A. Cytotoxic alpha amino acids and peptides. Br. Emp. Cancer Campaign. A 1953, 31, 6–7. [Google Scholar]
- Bergel, F.; Stock, J.A. Cytoactive amino acids and derivatives. I. Substituted phenylalanines. J. Chem. Soc. 1954, 2409–2417. [Google Scholar] [CrossRef]
- Larionov, L.F.; Khokhlov, A.S.; Shkodinskaja, E.M.; Vasina, S.; Troosheikina, V.I.; Novikova, M.A. Studies on the anti-tumour activity of p-di-(2-chloroethyl) aminophenylalanine (sarcolysine). Lancet 1955, 269, 169–171. [Google Scholar] [CrossRef]
- White, F.R. Sarcolysin and related compounds. Cancer Chemother. Rep. 1960, 6, 61–93. [Google Scholar]
- Young, R.C.; Canellos, G.P.; Chabner, B.A.; Schein, P.S.; Hubbard, S.P.; DeVita, V.T. Chemotherapy of advanced ovarian carcinoma: A prospective randomized comparison of phenylalanine mustard and high dose cyclophosphamide. Gynecol. Oncol. 1974, 2, 489–497. [Google Scholar] [CrossRef]
- Schmidt, L.H.; Fradkin, R.; Sullivan, R.; Flowers, A. Comparative Pharmacology of Alkylating Agents. I. Cancer Chemother. Rep. 1965, 18 (Suppl. 2), 1–401. [Google Scholar]
- Vistica, D.T.; Fuller, R.; Dillon, N.; Petro, B.J. Rational Basis for Chemotherapy; Chabner, B.A., Ed.; Alan R. Liss: New York, NY, USA, 1983; p. 475. [Google Scholar]
- Haines, D.R.; Fuller, R.W.; Ahmad, S.; Vistica, D.T.; Marquez, V.E. Selective cytotoxicity of a system L specific amino acid nitrogen mustard. J. Med. Chem. 1987, 30, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Takasato, Y.; Rapoport, S.I.; Smith, Q.R. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am. J. Physiol. Heart Circ. Physiol. 1984, 247, H484–H493. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Greig, N.H.; Vistica, D.T.; Rapoport, S.I.; Smith, Q.R. Affinity of antineoplastic amino acid drugs for the large neutral amino acid transporter of the blood-brain barrier. Cancer Chemother. Pharmacol. 1991, 29, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Smith, Q.R.; Aoyagi, M.; Rapoport, S.I. Structural specificity of the brain capillary neutral amino acid transporter. Soc. Neurosci. Abstr. 1989, 15, 1025. [Google Scholar]
- Takada, Y.; Vistica, D.T.; Greig, N.H.; Purdon, D.; Rapoport, S.I.; Smith, Q.R. Rapid high-affinity transport of a chemotherapeutic amino acid across the blood-brain barrier. Cancer Res. 1992, 52, 2191–2196. [Google Scholar] [PubMed]
- Matharu, J.; Oki, J.; Worthen, D.R.; Smith, Q.R.; Crooks, P.A. Regiospecific and conformationally restrained analogs of melphalan and DL-2-NAM-7 and their affinities for the large neutral amino acid transporter (system LAT1) of the blood-brain barrier. Bioorg. Med. Chem. Lett. 2010, 20, 3688–3691. [Google Scholar] [CrossRef] [PubMed]
- Yunger, L.M.; Cramer, R.D. Measurement of correlation of partition coefficients of polar amino acids. Mol. Pharmacol. 1981, 20, 602–608. [Google Scholar] [PubMed]
- Chollet, J.F.; Delétage, C.; Faucher, M.; Miginiac, L.; Bonnemain, J.L. Synthesis and structure-activity relationships of some pesticides with an α-amino acid function. Biochim. Biophys. Acta 1997, 1336, 331–341. [Google Scholar] [CrossRef]
- Friesema, E.C.; Docter, R.; Moerings, E.P.; Verrey, F.; Krenning, E.P.; Hennemann, G.; Visser, T.J. Thyroid hormone transport by the heterodimeric human system L amino acid transporter. Endocrinology 2001, 142, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- Kinne, A.; Wittner, M.; Wirth, E.K.; Hinz, K.M.; Schülein, R.; Köhrle, J.; Krause, G. Involvement of the l-Type Amino Acid Transporter Lat2 in the Transport of 3,3′-Diiodothyronine across the Plasma Membrane. Eur. Thyroid J. 2015, 4, 42–50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bodoy, S.; Martín, L.; Zorzano, A.; Palacín, M.; Estévez, R.; Bertran, J. Identification of LAT4, a novel amino acid transporter with system L activity. J. Biol. Chem. 2005, 280, 12002–12011. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; et al. l-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Yun, D.W.; Lee, S.A.; Park, M.G.; Kim, J.S.; Yu, S.K.; Park, M.R.; Kim, S.G.; Oh, J.S.; Kim, C.S.; Kim, H.J.; et al. JPH203, an l-type amino acid transporter 1-selective compound, induces apoptosis of YD-38 human oral cancer cells. J. Pharmacol. Sci. 2014, 124, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. l-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Augustyn, E.; Finke, K.; Zur, A.A.; Hansen, L.; Heeren, N.; Chien, H.C.; Lin, L.; Giacomini, K.M.; Colas, C.; Schlessinger, A.; et al. LAT-1 activity of meta-substituted phenylalanine and tyrosine analogs. Bioorg. Med. Chem. Lett. 2016, 26, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Gynther, M.; Huttunen, J.; Puris, E.; Spicer, J.A.; Denny, W.A. A Selective and Slowly Reversible Inhibitor of l-Type Amino Acid Transporter 1 (LAT1) Potentiates Antiproliferative Drug Efficacy in Cancer Cells. J. Med. Chem. 2016, 59, 5740–5751. [Google Scholar] [CrossRef] [PubMed]
- Kongpracha, P.; Nagamori, S.; Wiriyasermkul, P.; Tanaka, Y.; Kaneda, K.; Okuda, S.; Ohgaki, R.; Kanai, Y. Structure-activity relationship of a novel series of inhibitors for cancer type transporter l-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci. 2017, 133, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Soares-da-Silva, P. l-DOPA transport properties in an immortalised cell line of rat capillary cerebral endothelial cells, RBE 4. Brain Res. 1999, 829, 143–150. [Google Scholar] [CrossRef]
- Van Bree, J.B.; Audus, K.L.; Borchardt, R.T. Carrier-mediated transport of baclofen across monolayers of bovine brain endothelial cells in primary culture. Pharm. Res. 1988, 5, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; et al. XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, G.J.; Lam, H.Y.; Begleiter, A. Active carrier-mediated transport of melphalan by two separate amino acid transport systems in LPC-1 plasmacytoma cells in vitro. J. Biol. Chem. 1979, 254, 1057–1064. [Google Scholar] [PubMed]
- Gynther, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Mannila, A.; Nevalainen, T.; Savolainen, J.; Järvinen, T.; Rautio, J. Large neutral amino acid transporter enables brain drug delivery via prodrugs. J. Med. Chem. 2008, 51, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.; Nicholls, D.; Irwin, W.J.; Freeman, S. Drug delivery via active transport at the blood-brain barrier: Affinity of a prodrug of phosphonoformate for the large amino acid transporter. Int. J. Pharm. 1994, 104, 157–167. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Jain-Vakkalagadda, B.; Yang, C.; Pal, D.; Mitra, A.K. Carrier mediated uptake of l-tyrosine and its competitive inhibition by model tyrosine linked compounds in a rabbit corneal cell line (SIRC)--strategy for the design of transporter/receptor targeted prodrugs. Int. J. Pharm. 2002, 247, 115–125. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Palagiano, F.; Saija, A.; Nava, F.; Trombetta, D.; de Caprariis, P. Synthesis, stability, and pharmacological evaluation of nipecotic acid prodrugs. J. Pharm. Sci. 1999, 88, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Peura, L.; Malmioja, K.; Laine, K.; Leppänen, J.; Gynther, M.; Isotalo, A.; Rautio, J. Large Amino Acid Transporter 1 (LAT1) Prodrugs of Valproic Acid: New Prodrug Design Ideas for Central Nervous System Delivery. Mol. Pharm. 2011, 8, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Peura, L.; Malmioja, K.; Huttunen, K.; Leppänen, J.; Hämäläinen, M.; Forsberg, M.M.; Gynther, M.; Rautio, J.; Laine, K. Design, synthesis and brain uptake of LAT1-targeted amino acid prodrugs of dopamine. Pharm. Res. 2013, 30, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Puris, E.; Gynther, M.; Huttunen, J.; Petsalo, A.; Huttunen, K.M. l-type amino acid transporter 1 utilizing prodrugs: How to achieve effective brain delivery and low systemic exposure of drugs. J. Control. Release 2017, 261, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Peura, L.; Vernerová, M.; Leppänen, J.; Kärkkäinen, J.; Lehtonen, M.; Rautio, J.; Huttunen, K.M. Amino Acid Promoieties Alter Valproic Acid Pharmacokinetics and Enable Extended Brain Exposure. Neurochem. Res. 2016, 41, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Pickering, D.S.; Spicer, J.A.; Denny, W.A.; Huttunen, K.M. Systemic and Brain Pharmacokinetics of Perforin Inhibitor Prodrugs. Mol. Pharm. 2016, 13, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Yamada, T.; Hamajima, M.; Itoh, M.; Katayama, T.; Bork, P.; Goto, S.; Kanehisa, M. KEGG Atlas mapping for global analysis of metabolic pathways. Nucleic Acids Res. 2008, 36, W423–W426. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Shoichet, B.K. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C. Biochemical mechanisms of drug action: What does it take for success? Nat. Rev. Drug Discov. 2004, 3, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Kaptein, A. Irreversible Protein Kinase Inhibitors: Balancing the Benefits and Risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Li, J.Y.; Chu, C.; Ogoshi, F.; Wise, P.; Pardridge, W.M. Site-directed mutagenesis of cysteine residues of large neutral amino acid transporter LAT1. Biochim. Biophys. Acta BBA Biomembr. 2005, 1715, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Scalise, M.; Koyioni, M.; Koutentis, P.; Catto, M.; Eberini, I.; Parravicini, C.; Palazzolo, L.; Pisani, L.; Galluccio, M.; et al. Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem. Pharmacol. 2017, 143, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Oppedisano, F.; Catto, M.; Koutentis, P.A.; Nicolotti, O.; Pochini, L.; Koyioni, M.; Introcaso, A.; Michaelidou, S.S.; Carotti, A.; Indiveri, C. Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: Proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol. Appl. Pharmacol. 2012, 265, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Zur, A.A.; Chien, H.C.; Augustyn, E.; Flint, A.; Heeren, N.; Finke, K.; Hernandez, C.; Hansen, L.; Miller, S.; Lin, L.; et al. LAT1 activity of carboxylic acid bioisosteres: Evaluation of hydroxamic acids as substrates. Bioorg. Med. Chem. Lett. 2016, 26, 5000–5006. [Google Scholar] [CrossRef] [PubMed]
- Scholfield, M.R.; Zanden, C.M.V.; Carter, M.; Ho, P.S. Halogen bonding (X-bonding): A biological perspective. Protein Sci. Publ. Protein Soc. 2013, 22, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Brammer, L.; Bruton, E.A.; Sherwood, P. Understanding the Behavior of Halogens as Hydrogen Bond Acceptors. Cryst. Growth Des. 2001, 1, 277–290. [Google Scholar] [CrossRef]
- Zhou, P.-P.; Qiu, W.-Y.; Liu, S.; Jin, N.-Z. Halogen as halogen-bonding donor and hydrogen-bonding acceptor simultaneously in ring-shaped H3N·X(Y)·HF (X = Cl, Br and Y = F, Cl, Br) complexes. Phys. Chem. Chem. Phys. 2011, 13, 7408–7418. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.; Meck, R.; Yunis, A. The inhibition of gamma-glutamyl transpeptidase from human pancreatic carcinoma cells by (alpha S,5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (AT-125; NSC-163501). Res. Commun. Chem. Pathol. Pharmacol. 1980, 27, 175–182. [Google Scholar] [PubMed]
- Kreuzer, J.; Bach, N.C.; Forler, D.; Sieber, S.A. Target discovery of acivicin in cancer cells elucidates its mechanism of growth inhibition. Chem. Sci. 2015, 6, 237–245. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref./Year | Template | Organism | PDB ID | Resolution (Å) | Conformation | Protomeric Composition | Substrate |
|---|---|---|---|---|---|---|---|
| [44]/2013 | AdiC | E. coli | 3L1L | 3.0 | Outward-occluded | Monomer | Arginine |
| [44]/2013 | ApcT | M. jannaschii | 3GI9 | 2.48 | Inward-open | Monomer | - |
| [51]/2013 | AdiC | E. coli | 3L1L | 3.0 | Outward-occluded | Monomer | Arginine |
| [51]/2013 | ApcT | E. coli | 3GIA | 2.32 | Inward-open | Monomer | - |
| [52]/2017 | AdiC | E. coli | 3L1L | 3.0 | Outward-occluded | Monomer | Arginine |
| [52]/2017 | AdiC | E. coli | 3OB6 | 3.0 | Outward-open | Homodimer | Arginine |
| [52]/2017 | AdiC | E. coli | 3LRB | 3.61 | Outward-open | Homodimer | - |
| Cpd | Ref. | Experimental Model | Assay Type | Biological Activity |
|---|---|---|---|---|
| 1 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | a 95 ± 8 mM |
| 2 | [91] | in vivo tumor models | cytotoxicity assay | N/A |
| 3 | [91] | in vivo tumor models | cytotoxicity assay | N/A |
| 4 | [91] | in vivo tumor models | cytotoxicity assay | N/A |
| 4 | [93] | Murine L1210 leukemic cells | uptake inhibition of [14C]-BCH | b 111.6 ± 7.7 μM |
| 4 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 55 ± 4 μM |
| 4 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 49.1 μM |
| 5 | [96] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | ≤b 100 μM |
| 6 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 7.7 ± 0.8 μM |
| 7 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 8.5 ± 0.6 μM |
| 8 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 68 ± 9 μM |
| 9 | [93] | Murine L1210 leukemic cells | uptake inhibition of [14C]-BCH | b 0.22 ± 0.02 μM |
| 9 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 0.079 ± 0.006 μM |
| 10 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 252 ± 44 μM |
| 11 | [97] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 1.3 ± 0.01 μM |
| 12 | [97] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 730 ± 57 μM |
| 13 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 12.5 ± 1.1 μM |
| 14 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 26 ± 1 μM |
| 15 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 5.0 ± 0.6 μM |
| 16 | [98] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 2.1 ± 0.2 μM |
| 17 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | a 2.7 ± 0.1 mM |
| 18 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | a 3.4 ± 0.2 mM |
| 19 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | a 6.2 ± 0.3 mM |
| 20 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | a 0.21 ± 0.01 mM |
| 21 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | <b 10 μM |
| 22 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | <b 10 μM |
| 23 | [95] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | <b 10 μM |
| 24 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 31.1 μM |
| 25 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 107 μM |
| 26 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 67.2 μM |
| 27 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 405 μM |
| 28 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 56.4 μM |
| 29 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | inactive |
| 30 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | inactive |
| 31 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | inactive |
| 32 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | inactive |
| 33 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | N/A |
| 34 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | inactive |
| 35 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | inactive |
| 36 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 340 μM |
| 37 | [101] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake assay | c 7.9 μM |
| 38 | [97] | in situ rat brain perfusion | uptake inhibition of l-[14C]-leucine | b 1.0 ± 0.1 μM |
| 38 | [78] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake inhibition of l-[14C]-phenylalanine | b 5.8 μM |
| 38 | [101] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake assay | c 0.8 μM |
| 39 | [101] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake assay | c 12.5 μM |
| 40 | [101] | Human LAT1−4F2hc expressing Xenopus laevis oocytes | uptake assay | c 7.9 μM |
| 41 | [104] | Human LAT1 expressing S2 cells | uptake inhibition of l-[14C]-leucine | d 0.14 μM |
| 41 | [104] | HT-29 cells | uptake inhibition of l-[14C]-leucine | d 0.06 μM |
| 42 | [104] | Human LAT1 expressing S2 cells | uptake inhibition of l-[14C]-leucine | d 2.0 μM |
| 43 | [107] | Human LAT1 expressing HEK cells | uptake inhibition of [14C]-gabapentin | d 7.3 μM |
| 44 | [107] | Human LAT1 expressing HEK cells | uptake inhibition of [14C]-gabapentin | d 6.6 μM |
| 45 | [107] | Human LAT1 expressing HEK cells | uptake inhibition of [14C]-gabapentin | d 9.1 μM |
| 46 | [108] | Human breast cancer cells (MCF-7) | uptake inhibition of l-[14C]-leucine | d 18.2 μM |
| 47 | [109] | HEK293-Human LAT1 cells | uptake inhibition of l-[14C]-leucine | d 1.98 ± 1.07 μM b 2.1 ± 0.12 μM |
| 48 | [109] | HEK293-Human LAT1 cells | uptake inhibition of l-[14C]-leucine | inactive |
| 49 | [109] | HEK293-Human LAT1 cells | uptake inhibition of l-[14C]-leucine | N/A |
| 50 | [109] | HEK293-Human LAT1 cells | uptake inhibition of l-[14C]-leucine | N/A |
| 51 | [109] | HEK293-Human LAT1 cells | uptake inhibition of l-[14C]-leucine | N/A |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Ecker, G.F. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. https://doi.org/10.3390/ijms19051278
Singh N, Ecker GF. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. International Journal of Molecular Sciences. 2018; 19(5):1278. https://doi.org/10.3390/ijms19051278
Chicago/Turabian StyleSingh, Natesh, and Gerhard F. Ecker. 2018. "Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1" International Journal of Molecular Sciences 19, no. 5: 1278. https://doi.org/10.3390/ijms19051278
APA StyleSingh, N., & Ecker, G. F. (2018). Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. International Journal of Molecular Sciences, 19(5), 1278. https://doi.org/10.3390/ijms19051278