Comparative Analysis of Repetitive DNA between the Main Vectors of Chagas Disease: Triatoma infestans and Rhodnius prolixus

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Characterization of Triatoma Infestans Satellite DNA Families in the Rhodnius Prolixus Genome
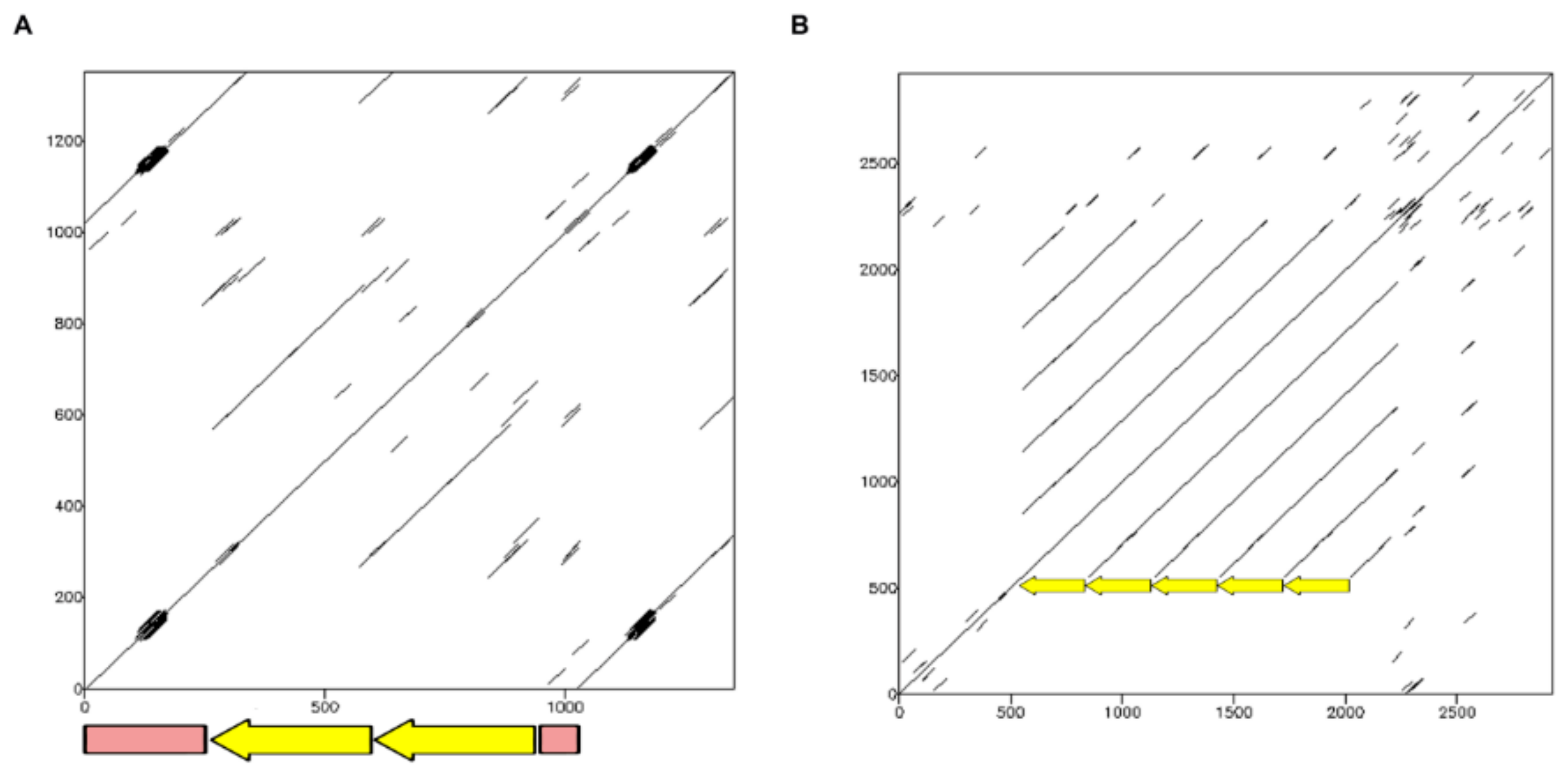
2.1.1. TinfSat04-1000
2.1.2. TinfSat12-84
2.1.3. TinfSat15-99
2.1.4. TinfSat33-372
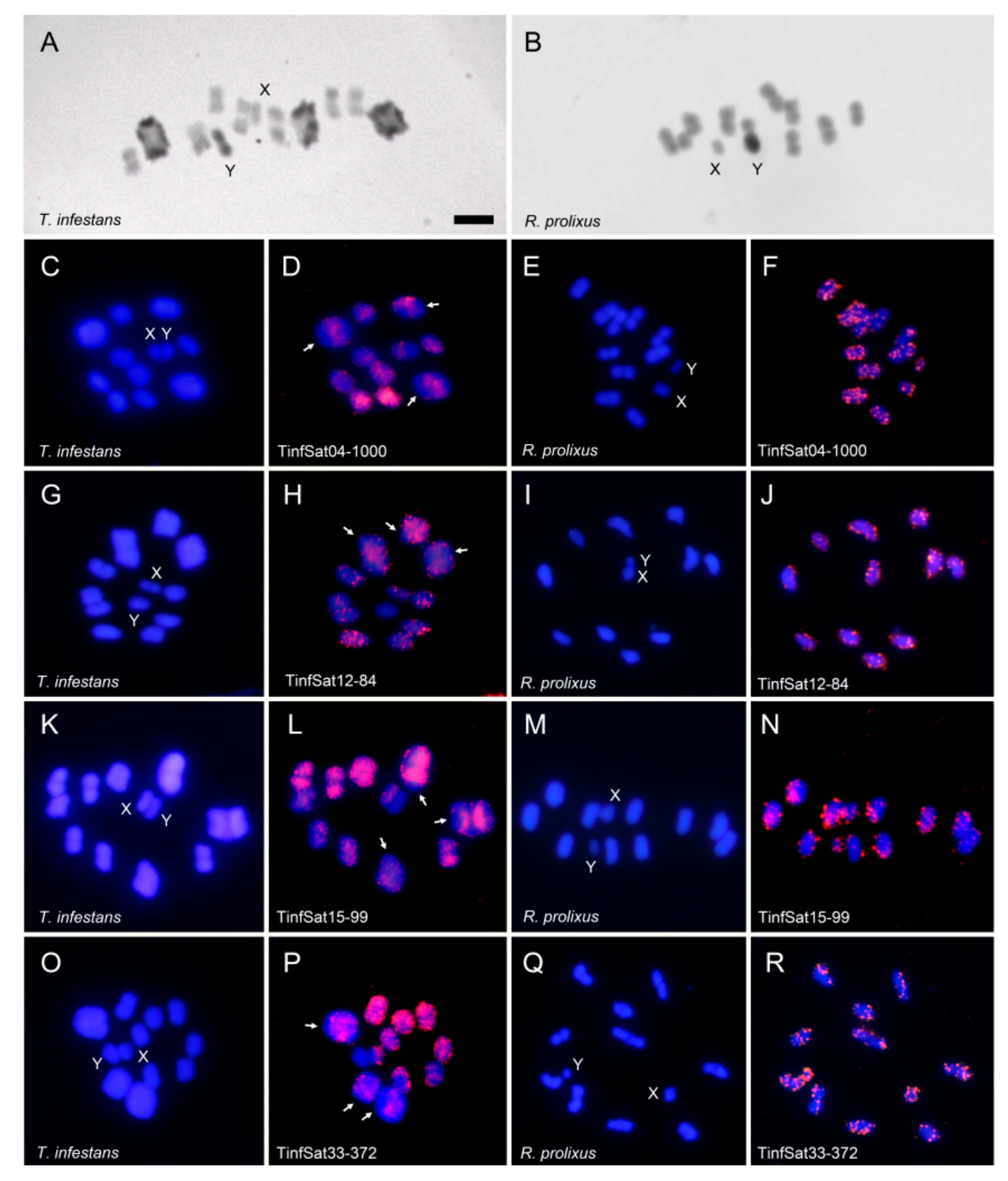
2.2. C-Banding and Chromosome Location of the Satellite DNA Families Shared between Triatoma infestans and Rhodnius prolixus
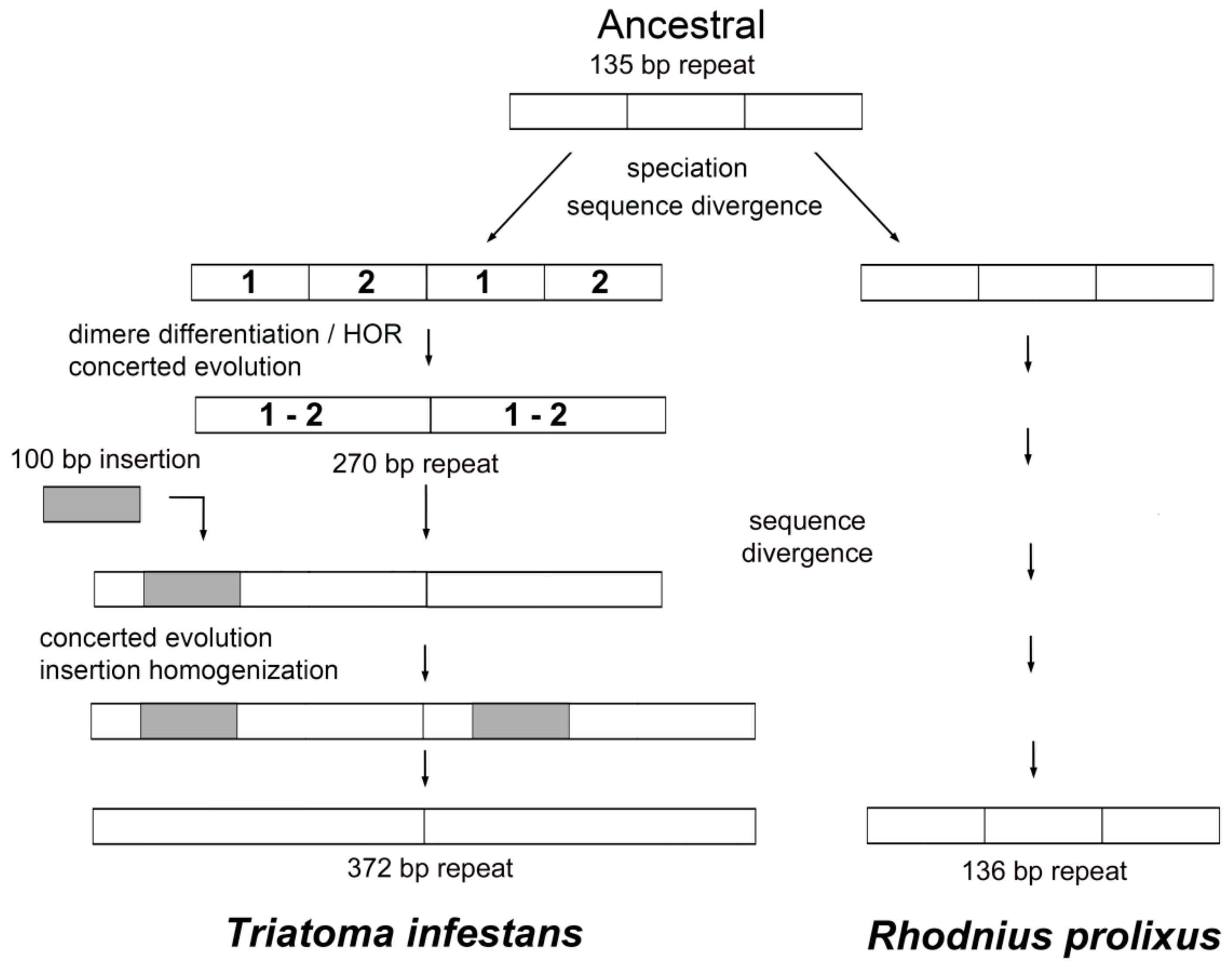
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Analyses
4.2. Insects and DNA Extraction
4.3. PCR Amplifications
4.4. Chromosome Preparations, C-Banding and FISH
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DAPI | 4′,6-Diamidine-2′-phenylindole |
| FISH | Fluorescence in situ hybridization |
| FITC | Fluorescein-5-isothiocyanate |
| GISH | Genomic in situ hybridization |
| HOR | Higher order repeat |
| satDNA | Satellite DNA |
| TE | Transposable element |
References
- Gascon, J.; Bern, C.; Pinazo, M.J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010, 115, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Schmunis, G.A.; Yadon, Z.E. Chagas disease: A Latin American health problem becoming a world health problem. Acta Trop. 2010, 115, 14–21. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Chagas Disease (American Trypanosomiasis). 2017. Available online: http://www.who.int/mediacentre/factsheets/fs340/en/index.html (accessed on 25 March 2018).
- Justi, S.A.; Galvão, C. The evolutionary origin of diversity in Chagas disease vectors. Trends Parasitol. 2017, 33, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Jannin, J.; Salvatella, R. The future of Chagas disease control. Trends Parasitol. 2006, 22, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Schofield, C.J. Elimination of Rhodnius prolixus in Central America. Parasit. Vectors 2012, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Chagas disease in Latin America: An epidemiological update based on 2010 estimates. Wkly. Epidemiol. Rec. 2015, 90, 33–44. [Google Scholar]
- Panzera, F.; Dujardin, J.P.; Nicolini, P.; Caraccio, M.N.; Rose, V.; Tellez, T.; Bermúdez, H.; Bargues, M.D.; Mas-Coma, S.; O’Connor, J.E.; et al. Genomic changes of Chagas disease vector, South America. Emerg. Infect. Dis. 2004, 10, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Bargues, M.D.; Klisiowicz, D.R.; Panzera, F.; Noireau, F.; Marcilla, A.; Pérez, R.; O’Connor, J.E.; Galvão, C.; Jurberg, J.; Carcavallo, R.U.; et al. Origin and phylogeography of the Chagas disease main vector Triatoma infestans based on nuclear rDNA sequences and genome size. Infect. Genet. Evol. 2006, 6, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Hernández, L.; Abrahan, L.; Moreno, M.; Gorla, D.; Catalá, S. Phenotypic variability associated to genomic changes in the main vector of Chagas disease in the southern cone of South America. Acta Trop. 2008, 106, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Fernández, G.M.; Girotti, J.R.; Juárez, M.P. Cuticular hydrocarbon pattern as a chemotaxonomy marker to assess intraspecific variability in Triatoma infestans, a major vector of Chagas’ disease. Med. Vet. Entomol. 2012, 26, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Panzera, F.; Ferreiro, M.J.; Pita, S.; Calleros, L.; Pérez, R.; Basmadjián, Y.; Guevara, Y.; Breniére, S.F.; Panzera, Y. Evolutionary and dispersal history of Triatoma infestans, main vector of Chagas disease, by chromosomal markers. Infect. Genet. Evol. 2014, 27, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lent, H.; Wygodzinsky, P. Revision of the Triatominae (Hemiptera, Reduviidae) and their significance as vector of Chagas disease. Bull. Am. Mus. Nat. Hist. 1979, 163, 123–520. [Google Scholar]
- Panzera, F.; Pérez, R.; Panzera, Y.; Ferrandis, I.; Ferreiro, M.J.; Calleros, L. Cytogenetics and genome evolution in the subfamily Triatominae (Hemiptera, Reduviidae). Cytogenet. Genome Res. 2010, 128, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Panzera, F.; Mora, P.; Vela, J.; Cuadrado, A.; Sánchez, A.; Palomeque, T.; Lorite, P. Comparative repeatome analysis on Triatoma infestans Andean and Non-Andean lineages, main vector of Chagas disease. PLoS ONE 2017, 12, e0181635. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, R.D.; Vionette-Amaral, R.J.; Lowenberger, C.; Rivera-Pomar, R.; Monteiro, F.A.; Minx, P.; Spieth, J.; Carvalho, A.B.; Panzera, F.; Lawson, D.; et al. Genome of Rhodnius prolixus, an insect vector of Chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc. Natl. Acad. Sci. USA 2015, 112, 14936–14941. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.S.; Weirauch, C. Evolutionary history of assassin bugs (Insecta: Hemiptera: Reduviidae): Insights from divergence dating and ancestral state reconstruction. PLoS ONE 2012, 7, e45523. [Google Scholar] [CrossRef] [PubMed]
- Justi, S.A.; Galvão, C.; Schrago, C.G. Geological changes of the Americas and their influence on the diversification of the Neotropical kissing bugs (Hemiptera: Reduviidae: Triatominae). PLoS Negl. Trop. Dis. 2016, 10, e0004527. [Google Scholar] [CrossRef] [PubMed]
- Mestrović, N.; Mravinac, B.; Pavlek, M.; Vojvoda-Zeljko, T.; Satović, E.; Plohl, M. Structural and functional liaisons between transposable elements and satellite DNAs. Chromosome Res. 2015, 23, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Bardella, V.B.; Aristeu, J.A.; Vanzela, A.L.L. Origin and distribution of AT-rich repetitive DNA families in Triatoma infestans (Heteroptera). Infect. Genet. Evol. 2014, 23, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Palomeque, T.; Carrillo, J.A.; Muñoz-López, M.; Lorite, P. Detection of a mariner-like element and a miniature inverted-repeat transposable element (MITE) associated with the heterochromatin from ants of the genus Messor and their possible involvement for satellite DNA evolution. Gene 2006, 371, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Palomeque, T.; Lorite, P. Satellite DNA in insects: A review. Heredity 2008, 100, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Meštrović, N.; Mravinac, B. Satellite DNA evolution. Genome Dyn. 2012, 7, 126–152. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Biscotti, M.A.; Canapa, A.; Forconi, M.; Olmo, E.; Barucca, M. Transcription of tandemly repetitive DNA: Functional roles. Chromosome Res. 2015, 23, 463–77. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Meštrović, N.; Mravinac, B. Centromere identity from the DNA point of view. Chromosoma 2014, 123, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Bardella, V.B.; Pita, S.; Vanzela, A.L.L.; Galvão, C.; Panzera, F. Heterochromatin base pair composition and diversification in holocentric chromosomes of kissing bugs (Hemiptera, Reduviidae). Mem. Inst. Oswaldo Cruz 2016, 111, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Panzera, F.; Sánchez, A.; Panzera, Y.; Palomeque, T.; Lorite, P. Distribution and evolution of repeated sequences in genomes of Triatominae (Hemiptera-Reduviidae) inferred from genomic in situ hybridization. PLoS ONE 2014, 9, e114298. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Lorite, P.; Vela, J.; Mora, P.; Palomeque, T.; Thi, K.P.; Panzera, F. Holocentric chromosome evolution in kissing bugs (Hemiptera-Reduviidae-Triatominae): Diversification of repeated sequences. Parasit. Vectors 2017, 10, 410. [Google Scholar] [CrossRef] [PubMed]
- Pita, S.; Panzera, F.; Sánchez, A.; Palomeque, T.; Lorite, P. Chromosome painting in triatomine insects reveals shared sequences between X chromosomes and autosomes. J. Med. Entomol. 2017, 54, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Ferree, P.M.; Prasad, S. How can satellite DNA divergence cause reproductive isolation? Let us count the chromosomal ways. Genet. Res. Int. 2012, 2012, 430136. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Palomeque, T.; Muñoz-López, M.; Carrillo, J.A.; Lorite, P. Characterization and evolutionary dynamics of a complex family of satellite DNA in the leaf beetle Chrysolina carnifex (Coleoptera, Chrysomelidae). Chromosome Res. 2005, 13, 795–807. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pita, S.; Mora, P.; Vela, J.; Palomeque, T.; Sánchez, A.; Panzera, F.; Lorite, P. Comparative Analysis of Repetitive DNA between the Main Vectors of Chagas Disease: Triatoma infestans and Rhodnius prolixus. Int. J. Mol. Sci. 2018, 19, 1277. https://doi.org/10.3390/ijms19051277
Pita S, Mora P, Vela J, Palomeque T, Sánchez A, Panzera F, Lorite P. Comparative Analysis of Repetitive DNA between the Main Vectors of Chagas Disease: Triatoma infestans and Rhodnius prolixus. International Journal of Molecular Sciences. 2018; 19(5):1277. https://doi.org/10.3390/ijms19051277
Chicago/Turabian StylePita, Sebastián, Pablo Mora, Jesús Vela, Teresa Palomeque, Antonio Sánchez, Francisco Panzera, and Pedro Lorite. 2018. "Comparative Analysis of Repetitive DNA between the Main Vectors of Chagas Disease: Triatoma infestans and Rhodnius prolixus" International Journal of Molecular Sciences 19, no. 5: 1277. https://doi.org/10.3390/ijms19051277
APA StylePita, S., Mora, P., Vela, J., Palomeque, T., Sánchez, A., Panzera, F., & Lorite, P. (2018). Comparative Analysis of Repetitive DNA between the Main Vectors of Chagas Disease: Triatoma infestans and Rhodnius prolixus. International Journal of Molecular Sciences, 19(5), 1277. https://doi.org/10.3390/ijms19051277