Calcium and Nuclear Signaling in Prostate Cancer


Abstract
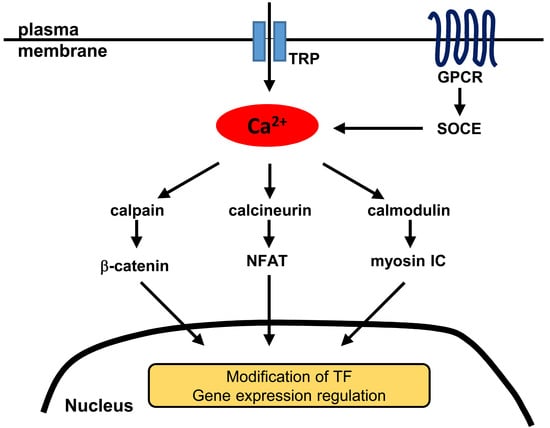
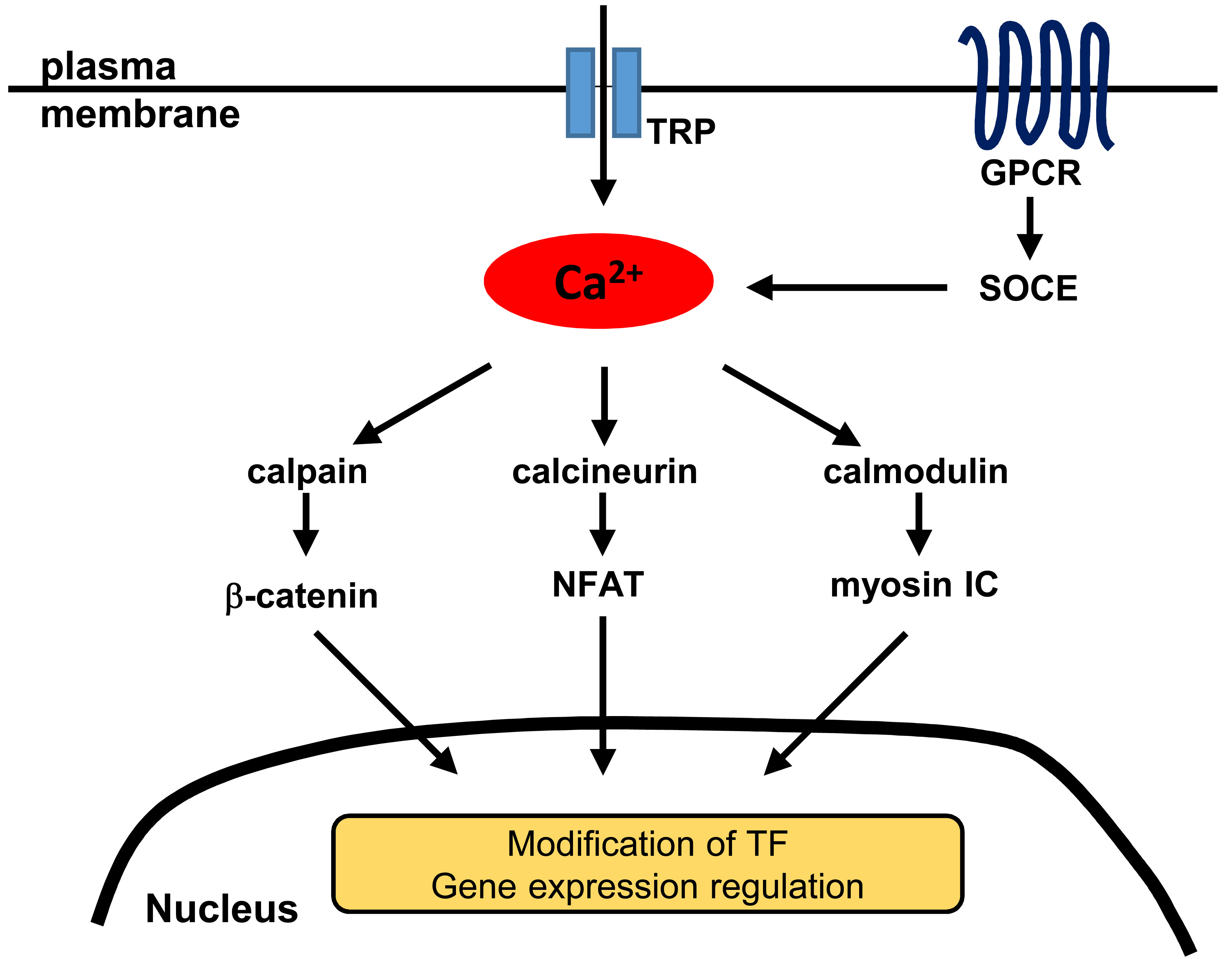
1. Introduction
2. Genomic Background
2.1. NKX3.1 Insufficiency
2.2. Amplification and Susceptibility in MYC
2.3. TMPRSS2-ERG Fusion
2.4. PTEN Loss
3. Developmental Pathways
4. Kinase Cascades
5. Anti-Senescence Signaling
6. Channels and Transporters
6.1. Transient Receptor Potential (TRP) Channels
6.2. Other Channels and Transporters
7. Additional Topics
7.1. Calpain Proteolysis
7.2. 14-3-3 Mediated Nucleocytoplasmic Redistribution
7.3. Autonomic-System Regulation
8. New Molecular Classes in Calcium Signaling to the Nucleus
8.1. GPCRs
8.2. Myosins
9. Summary and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| BKCa | Big potassium calcium-sensitive (channel) |
| BPH | Benign prostate hyperplasia |
| CAMK | Calcium/calmodulin-dependent kinase |
| CAMKK | Calcium/calmodulin-dependent kinase kinase |
| DcR2 | Decoy receptor 2 |
| DHT | Dihydrotestosterone |
| EGF | Epithelial growth factor |
| EMT | Epithelial-mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FAK | Focal adhesion kinase |
| FGF | Fibroblast growth factor |
| GPCR | G protein-coupled receptor |
| IκB | Inhibitor of κB |
| IKK | Inhibitor of κB kinase |
| InsP3R | Inositol 1,4,5-trisphosphate receptor |
| LNCaP | Lymph node cancer of prostate (cell line) |
| MAPK | Mitogen-activated protein kinase |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| NFAT | Nuclear factor of activated T-cells |
| NF-κB | Nuclear factor κB |
| NLS | Nuclear localization signal (sequence) |
| PI3K | Phosphoinositide 3-kinase |
| PIN | Prostate intraepithelial neoplasia |
| PKB | Protein kinase B |
| PKC | Protein kinase C |
| PSA | Prostate-specific antigen |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| SDF-1 | Stromal cell-derived factor 1 |
| SOCE | Store-operated calcium entry |
| TLR | Toll-like receptor |
| TRAMP | Transgenic adenocarcinoma of the mouse prostate |
| TRP | Transient receptor potential (channel) |
| YB-1 | Y-box binding protein 1 |
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.; Vickers, A. Spotlight on prostate cancer: The latest evidence and current controversies. BMC Med. 2015, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D. Calcium signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011171. [Google Scholar] [CrossRef] [PubMed]
- Cautain, B.; Hill, R.; de Pedro, N.; Link, W. Components and regulation of nuclear transport processes. FEBS J. 2015, 282, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Godfraind, T. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Ouderkirk, J.L.; Krendel, M. Non-muscle myosins in tumor progression, cancer cell invasion, and metastasis. Cytoskeleton 2014, 71, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Yang, W.X. Myosins as fundamental components during tumorigenesis: Diverse and indispensable. Oncotarget 2016, 7, 46785–46812. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Hofmann, W.A. Calcium-regulated import of myosin IC into the nucleus. Cytoskeleton 2016, 73, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Don-Salu-Hewage, A.S.; Chan, S.Y.; McAndrews, K.M.; Chetram, M.A.; Dawson, M.R.; Bethea, D.A.; Hinton, C.V. Cysteine (C)-X-C receptor 4 undergoes transportin 1-dependent nuclear localization and remains functional at the nucleus of metastatic prostate cancer cells. PLoS ONE 2013, 8, e57194. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Buck, M.R.; Vocke, C.D.; Pozzatti, R.O.; Duray, P.H.; Jennings, S.B.; Florence, C.D.; Zhuang, Z.; Bostwick, D.G.; Liotta, L.A.; Linehan, W.M. Allelic loss on chromosome 8p12-21 in microdissected prostatic intraepithelial neoplasia. Cancer Res. 1995, 55, 2959–2962. [Google Scholar] [PubMed]
- Bethel, C.R.; Faith, D.; Li, X.; Guan, B.; Hicks, J.L.; Lan, F.; Jenkins, R.B.; Bieberich, C.J.; De Marzo, A.M. Decreased NKX3.1 protein expression in focal prostatic atrophy, prostatic intraepithelial neoplasia, and adenocarcinoma: Association with gleason score and chromosome 8p deletion. Cancer Res. 2006, 66, 10683–10690. [Google Scholar] [CrossRef] [PubMed]
- Asatiani, E.; Huang, W.X.; Wang, A.; Rodriguez Ortner, E.; Cavalli, L.R.; Haddad, B.R.; Gelmann, E.P. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005, 65, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Bhatia-Gaur, R.; Donjacour, A.A.; Sciavolino, P.J.; Kim, M.; Desai, N.; Young, P.; Norton, C.R.; Gridley, T.; Cardiff, R.D.; Cunha, G.R.; et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999, 13, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Brand, T.; Zweigerdt, R.; Arnold, H. Targeted disruption of the Nkx3.1 gene in mice results in morphogenetic defects of minor salivary glands: Parallels to glandular duct morphogenesis in prostate. Mech. Dev. 2000, 95, 163–174. [Google Scholar] [CrossRef]
- Tanaka, M.; Komuro, I.; Inagaki, H.; Jenkins, N.A.; Copeland, N.G.; Izumo, S. Nkx3.1, a murine homolog of Ddrosophila bagpipe, regulates epithelial ductal branching and proliferation of the prostate and palatine glands. Dev. Dyn. 2000, 219, 248–260. [Google Scholar] [CrossRef]
- Abdulkadir, S.A.; Magee, J.A.; Peters, T.J.; Kaleem, Z.; Naughton, C.K.; Humphrey, P.A.; Milbrandt, J. Conditional loss of Nkx3.1 in adult mice induces prostatic intraepithelial neoplasia. Mol. Cell. Biol. 2002, 22, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bhatia-Gaur, R.; Banach-Petrosky, W.A.; Desai, N.; Wang, Y.; Hayward, S.W.; Cunha, G.R.; Cardiff, R.D.; Shen, M.M.; Abate-Shen, C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002, 62, 2999–3004. [Google Scholar] [PubMed]
- Lei, Q.; Jiao, J.; Xin, L.; Chang, C.J.; Wang, S.; Gao, J.; Gleave, M.E.; Witte, O.N.; Liu, X.; Wu, H. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell 2006, 9, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; DeWeese, T.L.; Nelson, W.G.; Abate-Shen, C. Loss-of-function of Nkx3.1 promotes increased oxidative damage in prostate carcinogenesis. Cancer Res. 2005, 65, 6773–6779. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.; Gelmann, E.P. NKX3.1 activates cellular response to DNA damage. Cancer Res. 2010, 70, 3089–3097. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; Bowen, C.; Gelmann, E.P. Inflammatory cytokines induce phosphorylation and ubiquitination of prostate suppressor protein NKX3.1. Cancer Res. 2008, 68, 6896–6901. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.; Jain, G.; Kießling, T.; Philip, S.; Rid, M.; Barth, T.T.; Möller, P.; Cronauer, M.V.; Marienfeld, R.B. Loss of the Tumor Suppressor NKX3.1 in Prostate Cancer Cells is Induced by Prostatitis Related Mitogens. J. Clin. Exp. Oncol. 2016, 5. [Google Scholar] [CrossRef]
- Sato, K.; Qian, J.; Slezak, J.M.; Lieber, M.M.; Bostwick, D.G.; Bergstralh, E.J.; Jenkins, R.B. Clinical significance of alterations of chromosome 8 in high-grade, advanced, nonmetastatic prostate carcinoma. J. Natl. Cancer Inst. 1999, 91, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.B.; Qian, J.; Lieber, M.M.; Bostwick, D.G. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 1997, 57, 524–531. [Google Scholar] [PubMed]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Esposito, D.; Duhagon, M.A.; Banfield, K.; Mehalko, J.; Liao, H.; Stephens, R.M.; Harris, T.J.; Munroe, D.J.; Wu, X. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 3001–3005. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Landan, G.; Pomerantz, M.; Jaschek, R.; Herman, P.; Reich, D.; Yan, C.; Khalid, O.; Kantoff, P.; Oh, W.; et al. Functional enhancers at the gene-poor 8q24 cancer-linked locus. PLoS Genet. 2009, 5, e1000597. [Google Scholar] [CrossRef] [PubMed]
- Al Olama, A.A.; Kote-Jarai, Z.; Giles, G.G.; Guy, M.; Morrison, J.; Severi, G.; Leongamornlert, D.A.; Tymrakiewicz, M.; Jhavar, S.; Saunders, E.; et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat. Genet. 2009, 41, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sulem, P.; Gudbjartsson, D.F.; Blondal, T.; Gylfason, A.; Agnarsson, B.A.; Benediktsdottir, K.R.; Magnusdottir, D.N.; Orlygsdottir, G.; Jakobsdottir, M.; et al. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat. Genet. 2009, 41, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef]
- Wang, J.; Kim, J.; Roh, M.; Franco, O.E.; Hayward, S.W.; Wills, M.L.; Abdulkadir, S.A. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 2010, 29, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Mamaeva, O.A.; Kim, J.; Feng, G.; McDonald, J.M. Calcium/calmodulin-dependent kinase II regulates notch-1 signaling in prostate cancer cells. J. Cell. Biochem. 2009, 106, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Tomlins, S.A.; Shen, R.; Nadeem, O.; Wang, L.; Wei, J.T.; Pienta, K.J.; Ghosh, D.; Rubin, M.A.; Chinnaiyan, A.M.; et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod. Pathol. 2007, 20, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Dhanasekaran, S.M.; Helgeson, B.E.; Cao, X.; Morris, D.S.; Menon, A.; Jing, X.; Cao, Q.; Han, B.; et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007, 448, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Iljin, K.; Wolf, M.; Edgren, H.; Gupta, S.; Kilpinen, S.; Skotheim, R.I.; Peltola, M.; Smit, F.; Verhaegh, G.; Schalken, J.; et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006, 66, 10242–10246. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, C.; Haudebourg, J.; Carpentier, X.; Valerio, L.; Amiel, J.; Michiels, J.F.; Pedeutour, F. Detection of the TMPRSS2-ETS fusion gene in prostate carcinomas: Retrospective analysis of 55 formalin-fixed and paraffin-embedded samples with clinical data. Cancer Genet. Cytogenet. 2008, 183, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.M.; Perner, S.; Genega, E.M.; Sanda, M.; Hofer, M.D.; Mertz, K.D.; Paris, P.L.; Simko, J.; Bismar, T.A.; Ayala, G.; et al. Characterization of TMPRSS2-ERG fusion high-grade prostatic intraepithelial neoplasia and potential clinical implications. Clin. Cancer Res. 2008, 14, 3380–3385. [Google Scholar] [CrossRef] [PubMed]
- Albadine, R.; Latour, M.; Toubaji, A.; Haffner, M.; Isaacs, W.B.; E, A.P.; Meeker, A.K.; Demarzo, A.M.; Epstein, J.I.; Netto, G.J. TMPRSS2-ERG gene fusion status in minute (minimal) prostatic adenocarcinoma. Mod. Pathol. 2009, 22, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced chromosomal proximity and gene fusions in prostate cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Klezovitch, O.; Risk, M.; Coleman, I.; Lucas, J.M.; Null, M.; True, L.D.; Nelson, P.S.; Vasioukhin, V. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc. Natl. Acad. Sci. USA 2008, 105, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2009, 41, 619–624. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Geybels, M.S.; Alumkal, J.J.; Luedeke, M.; Rinckleb, A.; Zhao, S.; Shui, I.M.; Bibikova, M.; Klotzle, B.; van den Brandt, P.A.; Ostrander, E.A.; et al. Epigenomic profiling of prostate cancer identifies differentially methylated genes in TMPRSS2:ERG fusion-positive versus fusion-negative tumors. Clin. Epigenet. 2015, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Geybels, M.S.; McCloskey, K.D.; Mills, I.G.; Stanford, J.L. Calcium Channel Blocker Use and Risk of Prostate Cancer by TMPRSS2:ERG Gene Fusion Status. Prostate 2017, 77, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiao, Y.; Asangani, I.A.; Ateeq, B.; Poliakov, A.; Cieslik, M.; Pitchiaya, S.; Chakravarthi, B.; Cao, X.; Jing, X.; et al. Development of Peptidomimetic Inhibitors of the ERG Gene Fusion Product in Prostate Cancer. Cancer Cell 2017, 31, 532–548. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, A.; Tan, S.H.; Mohamed, A.A.; Chen, Y.; Hu, Y.; Petrovics, G.; Sreenath, T.; Kagan, J.; Srivastava, S.; McLeod, D.G.; et al. Functional antagonism of TMPRSS2-ERG splice variants in prostate cancer. Genes Cancer 2014, 5, 273–284. [Google Scholar] [PubMed]
- Gimenez-Bonafe, P.; Fedoruk, M.N.; Whitmore, T.G.; Akbari, M.; Ralph, J.L.; Ettinger, S.; Gleave, M.E.; Nelson, C.C. YB-1 is upregulated during prostate cancer tumor progression and increases P-glycoprotein activity. Prostate 2004, 59, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Takeuchi, A.; Song, Y.; Yokomizo, A.; Kashiwagi, E.; Uchiumi, T.; Kuroiwa, K.; Tatsugami, K.; Fujimoto, N.; Oda, Y.; et al. Y-box binding protein-1 promotes castration-resistant prostate cancer growth via androgen receptor expression. Endocr. Relat. Cancer 2011, 18, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Heumann, A.; Kaya, O.; Burdelski, C.; Hube-Magg, C.; Kluth, M.; Lang, D.S.; Simon, R.; Beyer, B.; Thederan, I.; Sauter, G.; et al. Up regulation and nuclear translocation of Y-box binding protein 1 (YB-1) is linked to poor prognosis in ERG-negative prostate cancer. Sci. Rep. 2017, 7, 2056. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cai, Y.; Shao, L.J.; Siddiqui, J.; Palanisamy, N.; Li, R.; Ren, C.; Ayala, G.; Ittmann, M. Activation of NF-{kappa}B by TMPRSS2/ERG Fusion Isoforms through Toll-Like Receptor-4. Cancer Res. 2011, 71, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Lou, W.; Sun, M.; Chen, J.; Yue, J.; Kung, H.J.; Evans, C.P.; Zhou, Q.; Gao, A.C. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 2010, 70, 3309–3319. [Google Scholar] [CrossRef] [PubMed]
- Singareddy, R.; Semaan, L.; Conley-Lacomb, M.K.; St John, J.; Powell, K.; Iyer, M.; Smith, D.; Heilbrun, L.K.; Shi, D.; Sakr, W.; et al. Transcriptional regulation of CXCR4 in prostate cancer: Significance of TMPRSS2-ERG fusions. Mol. Cancer Res. 2013, 11, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.I.; Parsons, R.; Ittmann, M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin. Cancer Res. 1998, 4, 811–815. [Google Scholar] [PubMed]
- Schmitz, M.; Grignard, G.; Margue, C.; Dippel, W.; Capesius, C.; Mossong, J.; Nathan, M.; Giacchi, S.; Scheiden, R.; Kieffer, N. Complete loss of PTEN expression as a possible early prognostic marker for prostate cancer metastasis. Int. J. Cancer 2007, 120, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Sircar, K.; Yoshimoto, M.; Monzon, F.A.; Koumakpayi, I.H.; Katz, R.L.; Khanna, A.; Alvarez, K.; Chen, G.; Darnel, A.D.; Aprikian, A.G.; et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 2009, 218, 505–513. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, M.E.; Soung, P.; Perera, S.; Kaplan, I.; Loda, M.; Sellers, W.R. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999, 59, 4291–4296. [Google Scholar] [PubMed]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Kim, M.J.; Cardiff, R.D.; Desai, N.; Banach-Petrosky, W.A.; Parsons, R.; Shen, M.M.; Abate-Shen, C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Eltoum, I.E.; Roh, M.; Wang, J.; Abdulkadir, S.A. Interactions between cells with distinct mutations in c-MYC and Pten in prostate cancer. PLoS Genet. 2009, 5, e1000542. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Ouyang, X.; Banach-Petrosky, W.A.; Shen, M.M.; Abate-Shen, C. Emergence of androgen independence at early stages of prostate cancer progression in Nkx3.1; Pten mice. Cancer Res. 2006, 66, 7929–7933. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Hu, Y.C.; Lee, D.K.; Chang, C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol. Endocrinol. 2004, 18, 2409–2423. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Mulholland, D.J.; Valamehr, B.; Mosessian, S.; Sellers, W.R.; Wu, H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol. Cell. Biol. 2008, 28, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Nowak, D.G.; Narula, N.; Robinson, B.; Watrud, K.; Ambrico, A.; Herzka, T.M.; Zeeman, M.E.; Minderer, M.; Zheng, W.; et al. The nuclear transport receptor Importin-11 is a tumor suppressor that maintains PTEN protein. J. Cell Biol. 2017, 216, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.M.; Marchionni, L.; Huang, Z.; Simons, B.; Blackman, A.; Yu, W.; Parmigiani, G.; Berman, D.M. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene 2008, 27, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.; Mecham, B.; Dumpit, R.; Coleman, I.; Bhattacharjee, M.; Chen, Q.; Sikes, R.A.; Nelson, P.S. Conserved gene expression programs integrate mammalian prostate development and tumorigenesis. Cancer Res. 2009, 69, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Bruxvoort, K.J.; Charbonneau, H.M.; Giambernardi, T.A.; Goolsby, J.C.; Qian, C.N.; Zylstra, C.R.; Robinson, D.R.; Roy-Burman, P.; Shaw, A.K.; Buckner-Berghuis, B.D.; et al. Inactivation of Apc in the mouse prostate causes prostate carcinoma. Cancer Res. 2007, 67, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.B.; Phesse, T.J.; Clarke, A.R. K-ras and Wnt signaling synergize to accelerate prostate tumorigenesis in the mouse. Cancer Res. 2009, 69, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y.; Jiang, M.; Bierie, B.; Roy-Burman, P.; Shen, M.M.; Taketo, M.M.; Wills, M.; Matusik, R.J. Activation of β-Catenin in mouse prostate causes HGPIN and continuous prostate growth after castration. Prostate 2009, 69, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and β-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar] [CrossRef] [PubMed]
- Horvath, L.G.; Henshall, S.M.; Lee, C.S.; Kench, J.G.; Golovsky, D.; Brenner, P.C.; O’Neill, G.F.; Kooner, R.; Stricker, P.D.; Grygiel, J.J.; et al. Lower levels of nuclear β-catenin predict for a poorer prognosis in localized prostate cancer. Int. J. Cancer 2005, 113, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, H.C.; Girling, J.; Warren, A.Y.; Leung, H.; Mills, I.G.; Neal, D.E. Alterations in β-catenin expression and localization in prostate cancer. Prostate 2008, 68, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Symes, A.J.; Kane, C.A.; Freeman, A.; Nariculam, J.; Munson, P.; Thrasivoulou, C.; Masters, J.R.; Ahmed, A. A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS ONE 2010, 5, e10456. [Google Scholar] [CrossRef] [PubMed]
- Thrasivoulou, C.; Millar, M.; Ahmed, A. Activation of intracellular calcium by multiple Wnt ligands and translocation of β-catenin into the nucleus: A convergent model of Wnt/Ca2+ and Wnt/β-catenin pathways. J. Biol. Chem. 2013, 288, 35651–35659. [Google Scholar] [CrossRef] [PubMed]
- Nicotera, P.; Zhivotovsky, B.; Orrenius, S. Nuclear calcium transport and the role of calcium in apoptosis. Cell Calcium 1994, 16, 279–288. [Google Scholar] [CrossRef]
- Hsu, J.L.; Pan, S.L.; Ho, Y.F.; Hwang, T.L.; Kung, F.L.; Guh, J.H. Costunolide induces apoptosis through nuclear Ca2+ overload and DNA damage response in human prostate cancer. J. Urol. 2011, 185, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Gipp, J.; Bushman, W. The Sonic Hedgehog pathway stimulates prostate tumor growth by paracrine signaling and recapitulates embryonic gene expression in tumor myofibroblasts. Oncogene 2009, 28, 4480–4490. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Hedgehog signaling in prostate cancer and its therapeutic implication. Int. J. Mol. Sci. 2013, 14, 13979–14007. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Truong, S.; Nouri, M.; Moore, J.; Al Nakouzi, N.; Lubik, A.A.; Buttyan, R. Non-canonical activation of hedgehog in prostate cancer cells mediated by the interaction of transcriptionally active androgen receptor proteins with Gli3. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, V.D.; Gangula, R.D.; Freeman, K.W.; Li, R.; Zhang, Y.; Wang, F.; Ayala, G.E.; Peterson, L.E.; Ittmann, M.; Spencer, D.M. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 2007, 12, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Memarzadeh, S.; Xin, L.; Mulholland, D.J.; Mansukhani, A.; Wu, H.; Teitell, M.A.; Witte, O.N. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 2007, 12, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Xu, P.Z.; Peng, X.D.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.V.; Horvath, S.; Smith, B.L.; Crosby, K.; Lebel, L.A.; Schrage, M.; Said, J.; De Kernion, J.; Reiter, R.E.; Sawyers, C.L. Antibody-based profiling of the phosphoinositide 3-kinase pathway in clinical prostate cancer. Clin. Cancer Res. 2004, 10, 8351–8356. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Res. 2007, 67, 6535–6538. [Google Scholar] [CrossRef] [PubMed]
- Boormans, J.L.; Hermans, K.G.; van Leenders, G.J.; Trapman, J.; Verhagen, P.C. An activating mutation in AKT1 in human prostate cancer. Int. J. Cancer 2008, 123, 2725–2726. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Poulogiannis, G.; Pyne, S.; Jia, S.; Zou, L.; Signoretti, S.; Loda, M.; Cantley, L.C.; Roberts, T.M. A constitutively activated form of the p110beta isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 11002–11007. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Ouyang, X.; Banach-Petrosky, W.A.; Gerald, W.L.; Shen, M.M.; Abate-Shen, C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 14477–14482. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Alimonti, A.; Scaglioni, P.P.; Koutcher, J.A.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006, 441, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Yassine, M.; Slomianny, C.; Kondratska, K.; Gordienko, D.; Dewailly, E.; Lehen’kyi, V.; Skryma, R.; Prevarskaya, N. Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis. 2014, 5, e1193. [Google Scholar] [CrossRef] [PubMed]
- Uzgare, A.R.; Isaacs, J.T. Enhanced redundancy in Akt and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004, 64, 6190–6199. [Google Scholar] [CrossRef] [PubMed]
- Kinkade, C.W.; Castillo-Martin, M.; Puzio-Kuter, A.; Yan, J.; Foster, T.H.; Gao, H.; Sun, Y.; Ouyang, X.; Gerald, W.L.; Cordon-Cardo, C.; et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J. Clin. Investig. 2008, 118, 3051–3064. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Fradet, V.; Lessard, L.; Begin, L.R.; Karakiewicz, P.; Masson, A.M.; Saad, F. Nuclear factor-kappaB nuclear localization is predictive of biochemical recurrence in patients with positive margin prostate cancer. Clin. Cancer Res. 2004, 10, 8460–8464. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.A.; Lessard, L.; Mes-Masson, A.M.; Saad, F. Expression of NF-kappaB in prostate cancer lymph node metastases. Prostate 2004, 58, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Karakiewicz, P.I.; Bellon-Gagnon, P.; Alam-Fahmy, M.; Ismail, H.A.; Mes-Masson, A.M.; Saad, F. Nuclear localization of nuclear factor-kappaB p65 in primary prostate tumors is highly predictive of pelvic lymph node metastases. Clin. Cancer Res. 2006, 12, 5741–5745. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Altuwaijri, S.; Deng, F.; Chen, L.; Lal, P.; Bhanot, U.K.; Korets, R.; Wenske, S.; Lilja, H.G.; Chang, C.; et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 2009, 175, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Wang, Z.; Guimaraes, A.S.; Ouyang, X.; Figueiredo, J.L.; Ding, Z.; Jiang, S.; Guney, I.; Kang, G.H.; Shin, E.; et al. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PLoS ONE 2008, 3, e3949. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Gioeli, D.; Mandell, J.W.; Petroni, G.R.; Frierson, H.F., Jr.; Weber, M.J. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999, 59, 279–284. [Google Scholar] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Niu, H.; Ye, Y.; Hu, F.; Chen, S.; Li, X.; Luo, X.; Jiang, S.; Liu, Y.; et al. STIM1 accelerates cell senescence in a remodeled microenvironment but enhances the epithelial-to-mesenchymal transition in prostate cancer. Sci. Rep. 2015, 5, 11754. [Google Scholar] [CrossRef] [PubMed]
- van Noesel, M.M.; van Bezouw, S.; Salomons, G.S.; Voute, P.A.; Pieters, R.; Baylin, S.B.; Herman, J.G.; Versteeg, R. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Res. 2002, 62, 2157–2161. [Google Scholar] [PubMed]
- Sheikh, M.S.; Fornace, A.J., Jr. Death and decoy receptors and p53-mediated apoptosis. Leukemia 2000, 14, 1509–1513. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, K.; Yang, X.; Jung, C.; Gardner, T.; Kim, H.S.; Jeng, M.H.; Kao, C. NFATc1 with AP-3 site binding specificity mediates gene expression of prostate-specific-membrane-antigen. J. Mol. Biol. 2003, 330, 749–760. [Google Scholar] [CrossRef]
- Kavitha, C.V.; Deep, G.; Gangar, S.C.; Jain, A.K.; Agarwal, C.; Agarwal, R. Silibinin inhibits prostate cancer cells- and RANKL-induced osteoclastogenesis by targeting NFATc1, NF-kappaB, and AP-1 activation in RAW264.7 cells. Mol. Carcinog. 2014, 53, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; Carneiro, F.R.; Robbs, B.K.; Faget, D.V.; Viola, J.P. Cell cycle and apoptosis regulation by NFAT transcription factors: New roles for an old player. Cell Death Dis. 2016, 7, e2199. [Google Scholar] [CrossRef] [PubMed]
- Manda, K.R.; Tripathi, P.; Hsi, A.C.; Ning, J.; Ruzinova, M.B.; Liapis, H.; Bailey, M.; Zhang, H.; Maher, C.A.; Humphrey, P.A.; et al. NFATc1 promotes prostate tumorigenesis and overcomes PTEN loss-induced senescence. Oncogene 2016, 35, 3282–3292. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Jiang, X.; Qiao, H.; Zhai, B.; Zhang, L.; Zhang, Q.; Wu, Y.; Jiang, H.; Sun, X. STAT3 interacts with Skp2/p27/p21 pathway to regulate the motility and invasion of gastric cancer cells. Cell. Signal. 2013, 25, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhao, W.; Yang, D. Stat3 induces oncogenic Skp2 expression in human cervical carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 418, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Wissenbach, U.; Niemeyer, B.A.; Fixemer, T.; Schneidewind, A.; Trost, C.; Cavalie, A.; Reus, K.; Meese, E.; Bonkhoff, H.; Flockerzi, V. Expression of CaT-like, a novel calcium-selective channel, correlates with the malignancy of prostate cancer. J. Biol. Chem. 2001, 276, 19461–19468. [Google Scholar] [CrossRef] [PubMed]
- Fixemer, T.; Wissenbach, U.; Flockerzi, V.; Bonkhoff, H. Expression of the Ca2+-selective cation channel TRPV6 in human prostate cancer: A novel prognostic marker for tumor progression. Oncogene 2003, 22, 7858–7861. [Google Scholar] [CrossRef] [PubMed]
- Thebault, S.; Flourakis, M.; Vanoverberghe, K.; Vandermoere, F.; Roudbaraki, M.; Lehen’kyi, V.; Slomianny, C.; Beck, B.; Mariot, P.; Bonnal, J.L.; et al. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006, 66, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Flourakis, M.; Skryma, R.; Prevarskaya, N. TRPV6 channel controls prostate cancer cell proliferation via Ca2+/NFAT-dependent pathways. Oncogene 2007, 26, 7380–7385. [Google Scholar] [CrossRef] [PubMed]
- Raphael, M.; Lehen’kyi, V.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Vanden Abeele, F.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Monet, M.; Lehen’kyi, V.; Gackiere, F.; Firlej, V.; Vandenberghe, M.; Roudbaraki, M.; Gkika, D.; Pourtier, A.; Bidaux, G.; Slomianny, C.; et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010, 70, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Domaradzki, T.M.; Gosy, V.A.; Hofmann, W.A. Myosin isoform expressed in metastatic prostate cancer stimulates cell invasion. Sci. Rep. 2017, 7, 8476. [Google Scholar] [CrossRef] [PubMed]
- Tsavaler, L.; Shapero, M.H.; Morkowski, S.; Laus, R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001, 61, 3760–3769. [Google Scholar] [PubMed]
- Zhang, L.; Barritt, G.J. Evidence that TRPM8 is an androgen-dependent Ca2+ channel required for the survival of prostate cancer cells. Cancer Res. 2004, 64, 8365–8373. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed]
- Henshall, S.M.; Afar, D.E.; Hiller, J.; Horvath, L.G.; Quinn, D.I.; Rasiah, K.K.; Gish, K.; Willhite, D.; Kench, J.G.; Gardiner-Garden, M.; et al. Survival analysis of genome-wide gene expression profiles of prostate cancers identifies new prognostic targets of disease relapse. Cancer Res. 2003, 63, 4196–4203. [Google Scholar] [PubMed]
- Gkika, D.; Flourakis, M.; Lemonnier, L.; Prevarskaya, N. PSA reduces prostate cancer cell motility by stimulating TRPM8 activity and plasma membrane expression. Oncogene 2010, 29, 4611–4616. [Google Scholar] [CrossRef] [PubMed]
- Bidaux, G.; Flourakis, M.; Thebault, S.; Zholos, A.; Beck, B.; Gkika, D.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; et al. Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J. Clin. Investig. 2007, 117, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Vanoverberghe, K.; Vanden Abeele, F.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004, 11, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Vanden Abeele, F.; Skryma, R.; Shuba, Y.; Van Coppenolle, F.; Slomianny, C.; Roudbaraki, M.; Mauroy, B.; Wuytack, F.; Prevarskaya, N. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells. Cancer Cell 2002, 1, 169–179. [Google Scholar] [CrossRef]
- Chen, L.; Cao, R.; Wang, G.; Yuan, L.; Qian, G.; Guo, Z.; Wu, C.L.; Wang, X.; Xiao, Y. Downregulation of TRPM7 suppressed migration and invasion by regulating epithelial-mesenchymal transition in prostate cancer cells. Med. Oncol. 2017, 34, 127. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, A.I.; Sagredo, E.A.; Cappelli, C.; Baez, P.; Andaur, R.E.; Blanco, C.; Tapia, J.C.; Echeverria, C.; Cerda, O.; Stutzin, A.; et al. TRPM4 regulates Akt/GSK3-beta activity and enhances β-catenin signaling and cell proliferation in prostate cancer cells. Mol. Oncol. 2018, 12, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Sikka, S.C.; Huang, L.; Sun, C.; Xu, C.; Jia, D.; Abdel-Mageed, A.B.; Pottle, J.E.; Taylor, J.T.; Li, M. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2010, 13, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Haverstick, D.M.; Heady, T.N.; Macdonald, T.L.; Gray, L.S. Inhibition of human prostate cancer proliferation in vitro and in a mouse model by a compound synthesized to block Ca2+ entry. Cancer Res. 2000, 60, 1002–1008. [Google Scholar] [PubMed]
- Gray, L.S.; Perez-Reyes, E.; Gomora, J.C.; Haverstick, D.M.; Shattock, M.; McLatchie, L.; Harper, J.; Brooks, G.; Heady, T.; Macdonald, T.L. The role of voltage gated T-type Ca2+ channel isoforms in mediating “capacitative” Ca2+ entry in cancer cells. Cell Calcium 2004, 36, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar] [PubMed]
- Mariot, P.; Vanoverberghe, K.; Lalevee, N.; Rossier, M.F.; Prevarskaya, N. Overexpression of an alpha 1H (Cav3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J. Biol. Chem. 2002, 277, 10824–10833. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F.; Lesouhaitier, O.; Perrier, E.; Bockhorn, L.; Chiappe, A.; Lalevee, N. Aldosterone regulation of T-type calcium channels. J. Steroid Biochem. Mol. Biol. 2003, 85, 383–388. [Google Scholar] [CrossRef]
- Contreras, G.F.; Castillo, K.; Enrique, N.; Carrasquel-Ursulaez, W.; Castillo, J.P.; Milesi, V.; Neely, A.; Alvarez, O.; Ferreira, G.; Gonzalez, C.; et al. A BK (Slo1) channel journey from molecule to physiology. Channels 2013, 7, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Hoa, N.T.; Wilson, Z.; Arismendi-Morillo, G.; Kong, X.T.; Tajhya, R.B.; Beeton, C.; Jadus, M.R. Big Potassium (BK) ion channels in biology, disease and possible targets for cancer immunotherapy. Int. Immunopharmacol. 2014, 22, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2007, 26, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zheng, Z.; Li, D.; Chen, L.; Li, N.; Yi, X.; Yang, Y.; Guo, F.; Liu, W.; Xie, X.; et al. BKCa promotes growth and metastasis of prostate cancer through facilitating the coupling between alphavbeta3 integrin and FAK. Oncotarget 2016, 7, 40174–40188. [Google Scholar] [CrossRef] [PubMed]
- Legrand, G.; Humez, S.; Slomianny, C.; Dewailly, E.; Vanden Abeele, F.; Mariot, P.; Wuytack, F.; Prevarskaya, N. Ca2+ pools and cell growth. Evidence for sarcoendoplasmic Ca2+-ATPases 2B involvement in human prostate cancer cell growth control. J. Biol. Chem. 2001, 276, 47608–47614. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra186. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corra, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Vlietstra, R.J.; van Alewijk, D.C.; Hermans, K.G.; van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar] [PubMed]
- Carson, J.P.; Kulik, G.; Weber, M.J. Antiapoptotic signaling in LNCaP prostate cancer cells: A survival signaling pathway independent of phosphatidylinositol 3′-kinase and Akt/protein kinase B. Cancer Res. 1999, 59, 1449–1453. [Google Scholar] [PubMed]
- Khan, M.T.; Wagner, L., 2nd; Yule, D.I.; Bhanumathy, C.; Joseph, S.K. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2006, 281, 3731–3737. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Kobylewski, S.E.; Henderson, K.A.; Eckhert, C.D. Identification of ryanodine receptor isoforms in prostate DU-145, LNCaP, and PWR-1E cells. Biochem. Biophys. Res. Commun. 2012, 425, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Mariot, P.; Prevarskaya, N.; Roudbaraki, M.M.; Le Bourhis, X.; Van Coppenolle, F.; Vanoverberghe, K.; Skryma, R. Evidence of functional ryanodine receptor involved in apoptosis of prostate cancer (LNCaP) cells. Prostate 2000, 43, 205–214. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Kuefer, R.; Ethier, S.P.; Day, M.L. Cleavage of β-catenin by calpain in prostate and mammary tumor cells. Cancer Res. 2004, 64, 7237–7240. [Google Scholar] [CrossRef] [PubMed]
- Rios-Doria, J.; Day, K.C.; Kuefer, R.; Rashid, M.G.; Chinnaiyan, A.M.; Rubin, M.A.; Day, M.L. The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J. Biol. Chem. 2003, 278, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Rubio, M.V.; Zhang, Z.; Um, T.; Xie, Y.; Knoepp, S.M.; Snider, A.J.; Gibbs, T.C.; Meier, K.E. Effects of lysophosphatidic acid on calpain-mediated proteolysis of focal adhesion kinase in human prostate cancer cells. Prostate 2012, 72, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shukla, M.; Maclennan, G.T.; Fu, P.; Gupta, S. Deregulation of FOXO3A during prostate cancer progression. Int. J. Oncol. 2009, 34, 1613–1620. [Google Scholar] [PubMed]
- Shukla, S.; Bhaskaran, N.; Maclennan, G.T.; Gupta, S. Deregulation of FoxO3a accelerates prostate cancer progression in TRAMP mice. Prostate 2013, 73, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Kanai, F.; Stehn, J.; Xu, J.; Sarbassova, D.; Frangioni, J.V.; Dalal, S.N.; DeCaprio, J.A.; Greenberg, M.E.; Yaffe, M.B. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J. Cell Biol. 2002, 156, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yao, M.; Xu, J.; Quan, Y.; Zhang, K.; Yang, R.; Gao, W.Q. Autocrine Activation of CHRM3 Promotes Prostate Cancer Growth and Castration Resistance via CaM/CaMKK-Mediated Phosphorylation of Akt. Clin. Cancer Res. 2015, 21, 4676–4685. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Dong, B.J.; Quan, Y.; Chen, Q.; Chu, M.; Xu, J.; Xue, W.; Huang, Y.R.; Yang, R.; Gao, W.Q. Regulation of Prostate Development and Benign Prostatic Hyperplasia by Autocrine Cholinergic Signaling via Maintaining the Epithelial Progenitor Cells in Proliferating Status. Stem Cell Rep. 2016, 6, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.M.; Smith, S.; Hart, B.; Fletcher, L. CaM kinase control of AKT and LNCaP cell survival. J. Cell. Biochem. 2012, 113, 1514–1526. [Google Scholar] [CrossRef] [PubMed]
- Hekman, M.; Albert, S.; Galmiche, A.; Rennefahrt, U.E.; Fueller, J.; Fischer, A.; Puehringer, D.; Wiese, S.; Rapp, U.R. Reversible membrane interaction of BAD requires two C-terminal lipid binding domains in conjunction with 14-3-3 protein binding. J. Biol. Chem. 2006, 281, 17321–17336. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Chetram, M.A.; Don-Salu-Hewage, A.S.; Hinton, C.V. ROS enhances CXCR4-mediated functions through inactivation of PTEN in prostate cancer cells. Biochem. Biophys. Res. Commun. 2011, 410, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Makowska, K.A.; Hughes, R.E.; White, K.J.; Wells, C.M.; Peckham, M. Specific Myosins Control Actin Organization, Cell Morphology, and Migration in Prostate Cancer Cells. Cell Rep. 2015, 13, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Peckham, M. How myosin organization of the actin cytoskeleton contributes to the cancer phenotype. Biochem. Soc. Trans. 2016, 44, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Barylko, B.; Jung, G.; Albanesi, J.P. Structure, function, and regulation of myosin 1C. Acta Biochim. Pol. 2005, 52, 373–380. [Google Scholar] [PubMed]
- Ihnatovych, I.; Sielski, N.L.; Hofmann, W.A. Selective expression of myosin IC Isoform A in mouse and human cell lines and mouse prostate cancer tissues. PLoS ONE 2014, 9, e108609. [Google Scholar] [CrossRef] [PubMed]
- Ihnatovych, I.; Migocka-Patrzalek, M.; Dukh, M.; Hofmann, W.A. Identification and characterization of a novel myosin Ic isoform that localizes to the nucleus. Cytoskeleton 2012, 69, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Pestic-Dragovich, L.; Hozak, P.; Philimonenko, A.; Simerly, C.; Schatten, G.; de Lanerolle, P. Evidence for the presence of myosin I in the nucleus. J. Biol. Chem. 1997, 272, 17176–17181. [Google Scholar] [CrossRef] [PubMed]
- Zattelman, L.; Regev, R.; Usaj, M.; Reinke, P.Y.A.; Giese, S.; Samson, A.O.; Taft, M.H.; Manstein, D.J.; Henn, A. N-terminal splicing extensions of the human MYO1C gene fine-tune the kinetics of the three full-length myosin IC isoforms. J. Biol. Chem. 2017, 292, 17804–17818. [Google Scholar] [CrossRef] [PubMed]
- Dzijak, R.; Yildirim, S.; Kahle, M.; Novak, P.; Hnilicova, J.; Venit, T.; Hozak, P. Specific nuclear localizing sequence directs two myosin isoforms to the cell nucleus in calmodulin-sensitive manner. PLoS ONE 2012, 7, e30529. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, P.G.; Cyr, J.L. Calmodulin binding to recombinant myosin-1c and myosin-1c IQ peptides. BMC Biochem. 2002, 3, 31. [Google Scholar] [CrossRef]
- Nevzorov, I.; Sidorenko, E.; Wang, W.; Zhao, H.; Vartiainen, M.K. Myosin-1C uses a novel phosphoinositide-dependent pathway for nuclear localization. EMBO Rep. 2018, 19, 290–304. [Google Scholar] [CrossRef] [PubMed]
- De Lanerolle, P. Nuclear actin and myosins at a glance. J. Cell Sci. 2012, 125, 4945–4949. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhao, J.; Hoffmann-Rohrer, U.; Grummt, I. Nuclear myosin I acts in concert with polymeric actin to drive RNA polymerase I transcription. Genes Dev. 2008, 22, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Percipalle, P.; Fomproix, N.; Cavellan, E.; Voit, R.; Reimer, G.; Kruger, T.; Thyberg, J.; Scheer, U.; Grummt, I.; Farrants, A.K. The chromatin remodelling complex WSTF-SNF2h interacts with nuclear myosin 1 and has a role in RNA polymerase I transcription. EMBO Rep. 2006, 7, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.A.; Vargas, G.M.; Ramchandran, R.; Stojiljkovic, L.; Goodrich, J.A.; de Lanerolle, P. Nuclear myosin I is necessary for the formation of the first phosphodiester bond during transcription initiation by RNA polymerase II. J. Cell. Biochem. 2006, 99, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Kysela, K.; Philimonenko, A.A.; Philimonenko, V.V.; Janacek, J.; Kahle, M.; Hozak, P. Nuclear distribution of actin and myosin I depends on transcriptional activity of the cell. Histochem. Cell Biol. 2005, 124, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Philimonenko, V.V.; Zhao, J.; Iben, S.; Dingova, H.; Kysela, K.; Kahle, M.; Zentgraf, H.; Hofmann, W.A.; de Lanerolle, P.; Hozak, P.; et al. Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat. Cell Biol. 2004, 6, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Fomproix, N.; Percipalle, P. An actin-myosin complex on actively transcribing genes. Exp. Cell Res. 2004, 294, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Pestic-Dragovich, L.; Stojiljkovic, L.; Philimonenko, A.A.; Nowak, G.; Ke, Y.; Settlage, R.E.; Shabanowitz, J.; Hunt, D.F.; Hozak, P.; de Lanerolle, P. A myosin I isoform in the nucleus. Science 2000, 290, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Obrdlik, A.; Louvet, E.; Kukalev, A.; Naschekin, D.; Kiseleva, E.; Fahrenkrog, B.; Percipalle, P. Nuclear myosin 1 is in complex with mature rRNA transcripts and associates with the nuclear pore basket. FASEB J. 2010, 24, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.; Necchi, D.; Prosperi, E.; Biggiogera, M. Small ribosomal subunits associate with nuclear myosin and actin in transit to the nuclear pores. FASEB J. 2006, 20, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Mehta, I.S.; Elcock, L.S.; Amira, M.; Kill, I.R.; Bridger, J.M. Nuclear motors and nuclear structures containing A-type lamins and emerin: Is there a functional link? Biochem. Soc. Trans. 2008, 36, 1384–1388. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.H.; Carpenter, A.E.; Fuchsova, B.; Johnson, T.; de Lanerolle, P.; Belmont, A.S. Long-range directional movement of an interphase chromosome site. Curr. Biol. 2006, 16, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Venit, T.; Dzijak, R.; Kalendova, A.; Kahle, M.; Rohozkova, J.; Schmidt, V.; Rulicke, T.; Rathkolb, B.; Hans, W.; Bohla, A.; et al. Mouse nuclear myosin I knock-out shows interchangeability and redundancy of myosin isoforms in the cell nucleus. PLoS ONE 2013, 8, e61406. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, J.; Ye, F.; Zhang, M. Structure of myosin-1c tail bound to calmodulin provides insights into calcium-mediated conformational coupling. Nat. Struct. Mol. Biol. 2015, 22, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Pruschy, M.; Ju, Y.; Spitz, L.; Carafoli, E.; Goldfarb, D.S. Facilitated nuclear transport of calmodulin in tissue culture cells. J. Cell Biol. 1994, 127, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Visuttijai, K.; Pettersson, J.; Mehrbani Azar, Y.; van den Bout, I.; Orndal, C.; Marcickiewicz, J.; Nilsson, S.; Hornquist, M.; Olsson, B.; Ejeskar, K.; et al. Lowered Expression of Tumor Suppressor Candidate MYO1C Stimulates Cell Proliferation, Suppresses Cell Adhesion and Activates AKT. PLoS ONE 2016, 11, e0164063. [Google Scholar] [CrossRef] [PubMed]
- Letellier, E.; Schmitz, M.; Ginolhac, A.; Rodriguez, F.; Ullmann, P.; Qureshi-Baig, K.; Frasquilho, S.; Antunes, L.; Haan, S. Loss of Myosin Vb in colorectal cancer is a strong prognostic factor for disease recurrence. Br. J. Cancer 2017, 117, 1689–1701. [Google Scholar] [CrossRef] [PubMed]
- Buschman, M.D.; Field, S.J. MYO18A: An unusual myosin. Adv. Biol. Regul. 2018, 67, 84–92. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Category | Factor | Physiology | Partners | Calcium Regulation/Nuclear Signal |
|---|---|---|---|---|
| genomic | NKX3.1 insufficiency | development | p53, AKT, PTEN | Ca2+-regulated proteolysis |
| MYC amplification | cell cycle | NKX3.1, PIM1 | Ca2+-regulated Notch nuclear entry | |
| TMPRSS2-ERG fusion | EMT | PTEN, CACNA1D | NF-κB nuclear entry, Ca2+ channel expression | |
| PTEN loss | hormone independence | mTORC2, PI3K | AR nuclear localization | |
| developmental | WNT | cell motility | APC | Ca2+ wave, β-catenin nuclear entry |
| Hedgehog | signaling from stroma | AR | Gli1 nuclear entry | |
| FGF | signaling from stroma | ERK | nuclear entry of alternative translation isoforms | |
| kinases and phosphatases | AKT-mTOR | apoptosis | FOXO3A, Bim, p27 | pAKT accumulation in nuclear bodies, Ca2+-dependent inhibition |
| ERK, MAPK | hormone independence | RAS, RAF, AKT | pERK/pMAPK accumulation in nucleus | |
| calcineurin | anti-senescence | NFATc1, MYC, IL6, STAT3 | NFATc1 nuclear import |
| Category | Channel/Transporter | Related Cell Physiology | Partners | Calcium Regulation/Nuclear Signal |
|---|---|---|---|---|
| TRP channels | TRPM2 | proliferation | nuclear ADP-ribosylase | channel accumulation in nuclear clusters |
| TRPM4 | proliferation | β-catenin | β-catenin nuclear accumulation | |
| TRPM7 | EMT | E-cadherin, MMPs | PM Ca2+ channel | |
| TRPM8 | apoptosis | AR, PSA | lowered ER Ca2+ | |
| TRPV2 | invasion | MMPs | elevated basal Ca2+ | |
| TRPV6 | proliferation | NFAT, ORAI1, STIM1 | SOCE | |
| other channels, transporters | BKCa | invasion | integrin | Ca2+-activated phosphorylation of FAK |
| SERCA | proliferation | EGF, DHT | ER Ca2+ | |
| ORAI1 | apoptosis | ORAI3, arachidonic acid | capacitative entry via the gated heteromer | |
| mitochondrial uniporter | apoptosis | miR-25 | mitochondrial Ca2+ | |
| InsP3R | apoptosis | AKT, PTEN, FBXL2 | ER-to-mitochondria current |
| Category | Factor | Physiology | Partners | Calcium Regulation/Nuclear Signal |
|---|---|---|---|---|
| proteases | calpain | metastasis, EMT | AR, β-catenin, E-cadherin | nuclear entry of cleaved β-catenin |
| chaperones | 14-3-3 | apoptosis | AKT, FOXO3A, Bim | nuclear export of FOXO3A |
| autonomic regulation | CHRM3 | castration resistance | CAMKK, AKT, BAD | acetylcholine-induced Ca2+ influx |
| adrenergic receptors | proliferation | TPRC6, calcineurin, NFAT | store-independent entry | |
| GPCRs | CXCR4 | bone tropism | SDF-1, PTEN, ROS, importin-β1 | nuclear entry, intranuclear Ca2+ release |
| molecular motors | myosin IC | migration, invasion, metastasis | calmodulin, importin-β1, RNA polymerase I and II, nuclear actin | Ca2+-induced nuclear entry |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maly, I.V.; Hofmann, W.A. Calcium and Nuclear Signaling in Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 1237. https://doi.org/10.3390/ijms19041237
Maly IV, Hofmann WA. Calcium and Nuclear Signaling in Prostate Cancer. International Journal of Molecular Sciences. 2018; 19(4):1237. https://doi.org/10.3390/ijms19041237
Chicago/Turabian StyleMaly, Ivan V., and Wilma A. Hofmann. 2018. "Calcium and Nuclear Signaling in Prostate Cancer" International Journal of Molecular Sciences 19, no. 4: 1237. https://doi.org/10.3390/ijms19041237
APA StyleMaly, I. V., & Hofmann, W. A. (2018). Calcium and Nuclear Signaling in Prostate Cancer. International Journal of Molecular Sciences, 19(4), 1237. https://doi.org/10.3390/ijms19041237