Assembly and Functional Analysis of an S/MAR Based Episome with the Cystic Fibrosis Transmembrane Conductance Regulator Gene


 , ,
, , 
Abstract
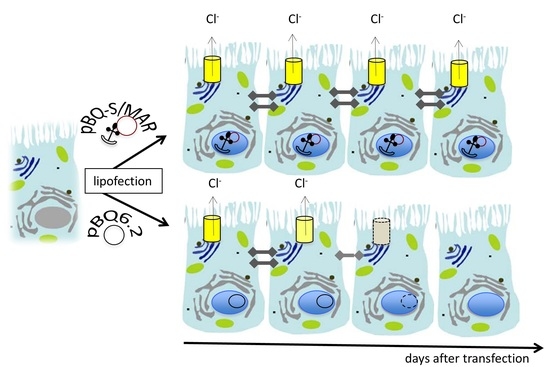
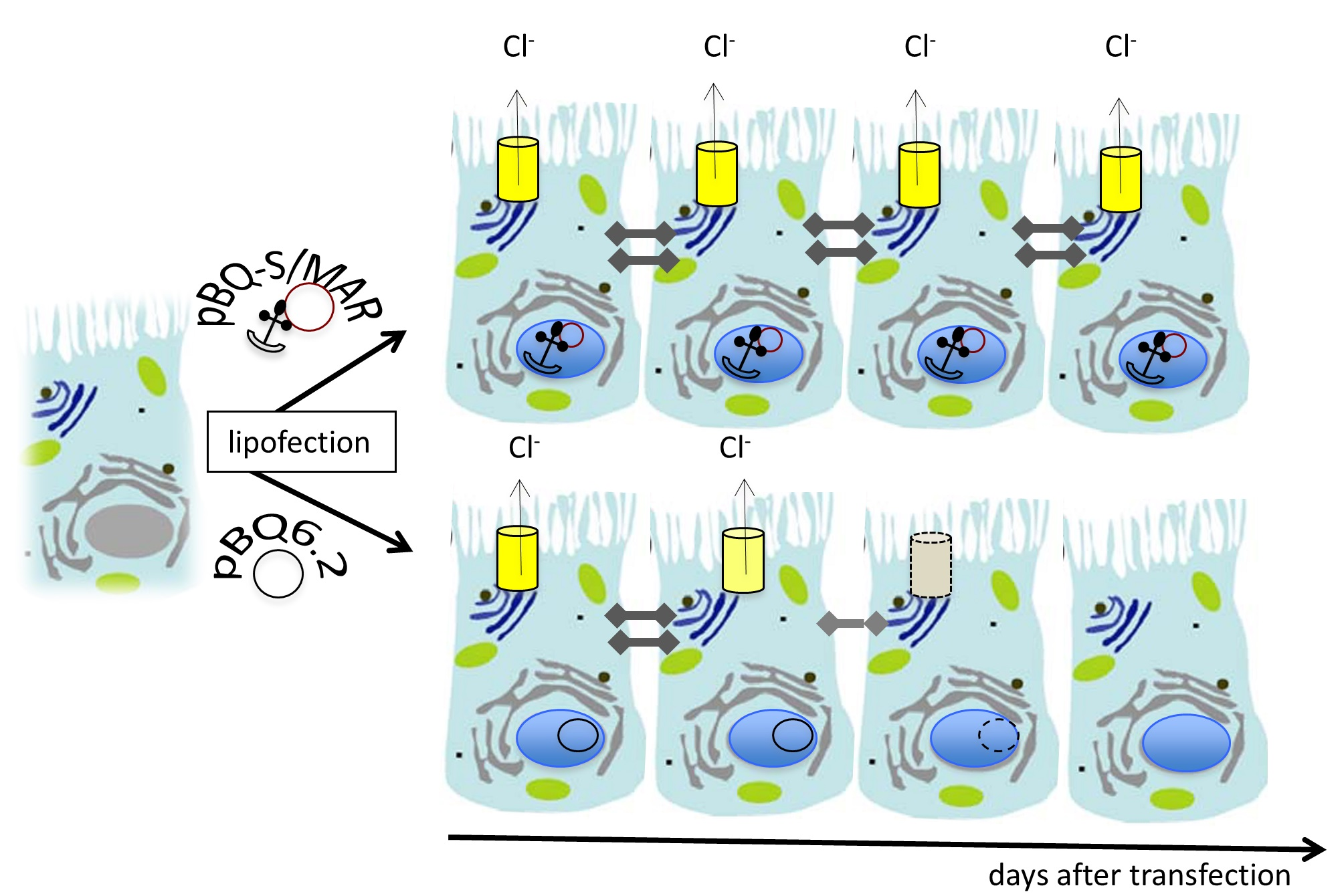
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
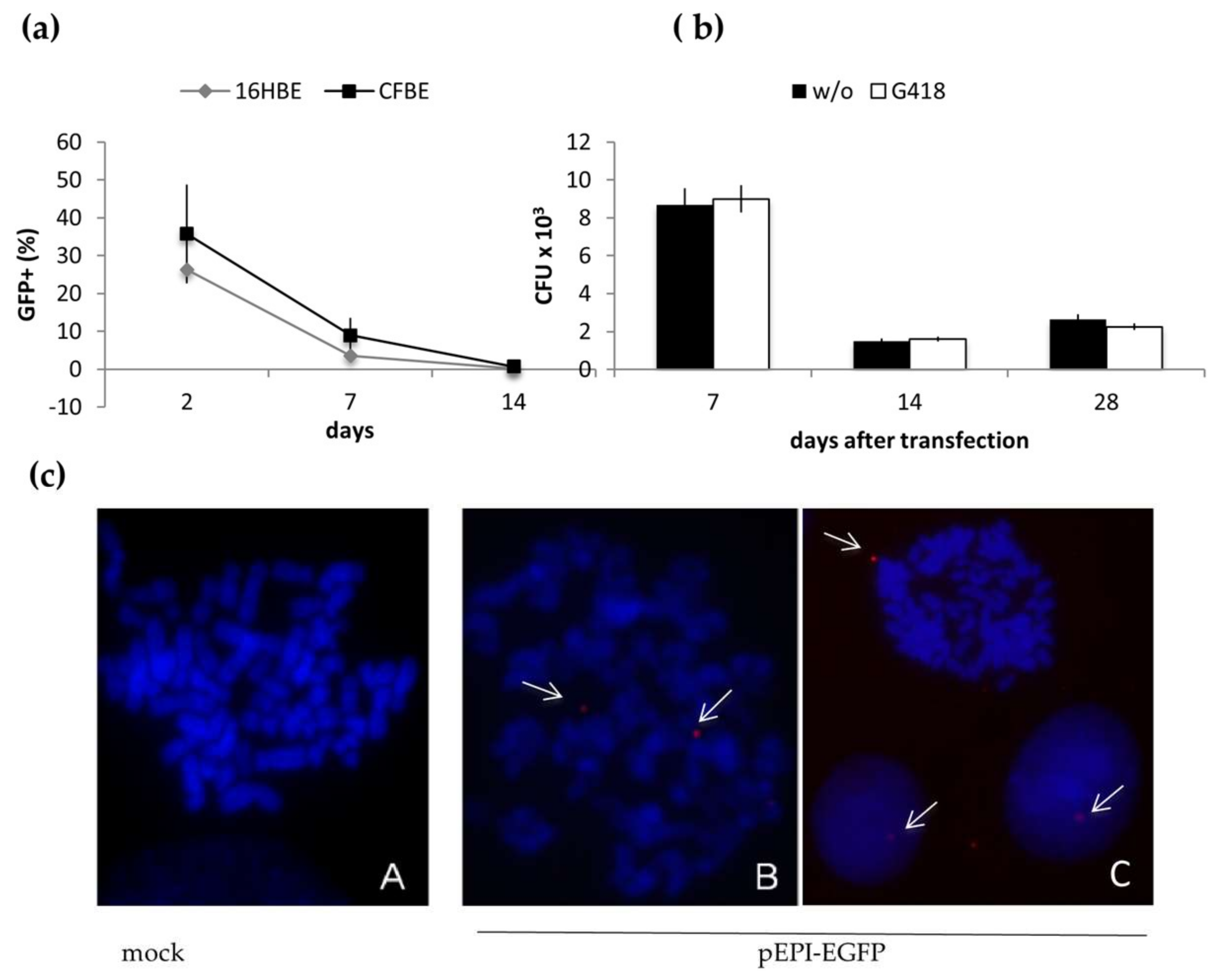
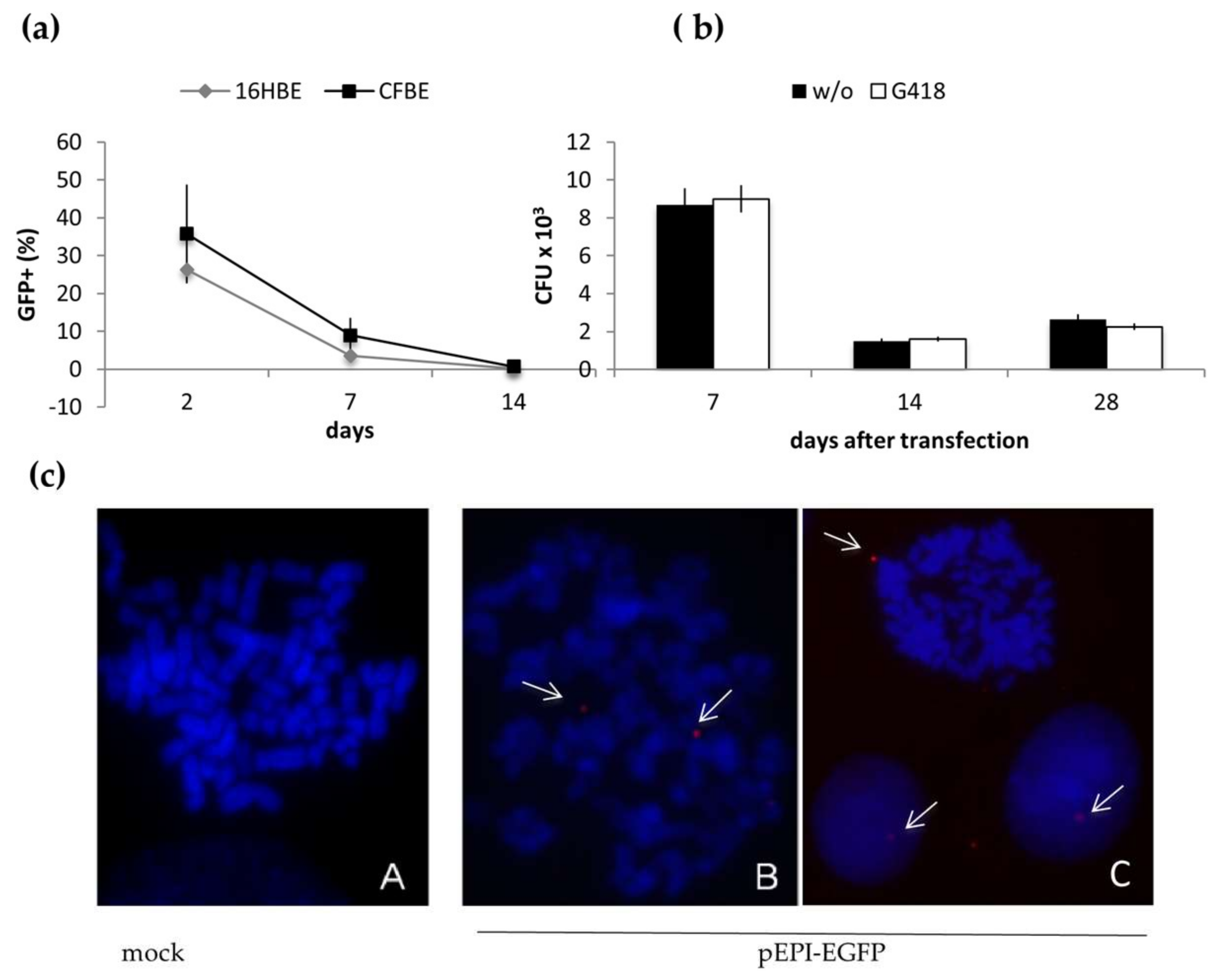
2.1. Long-Term Maintenance of pEPI-EGFP in Epithelial Cells
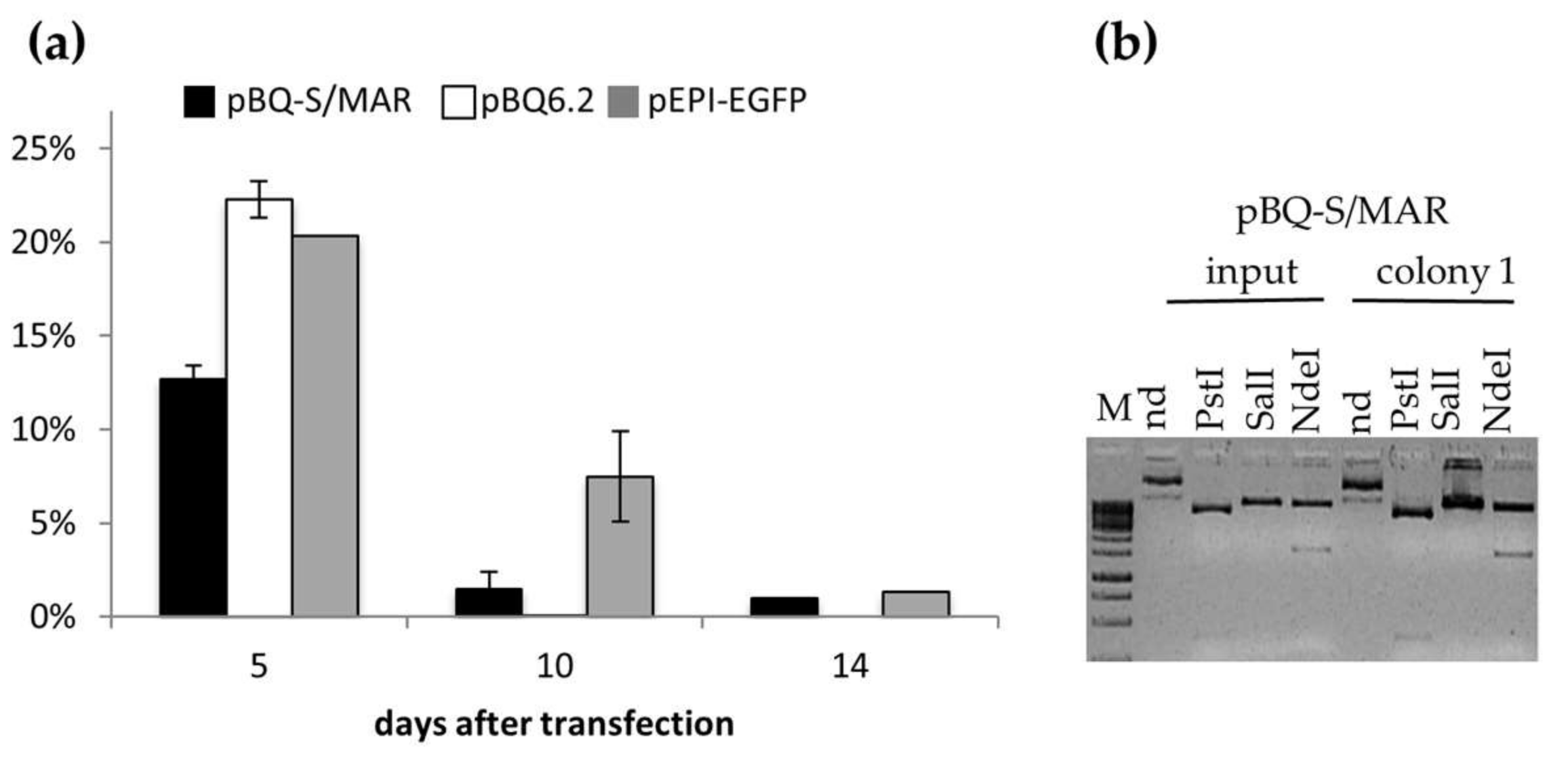
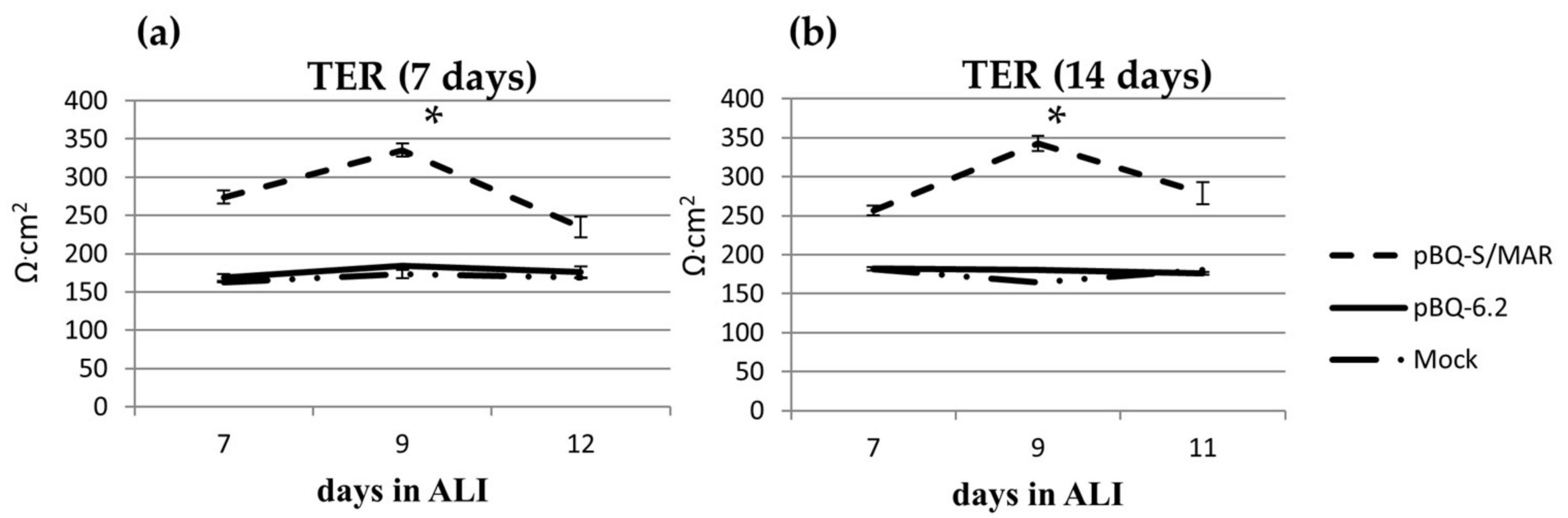
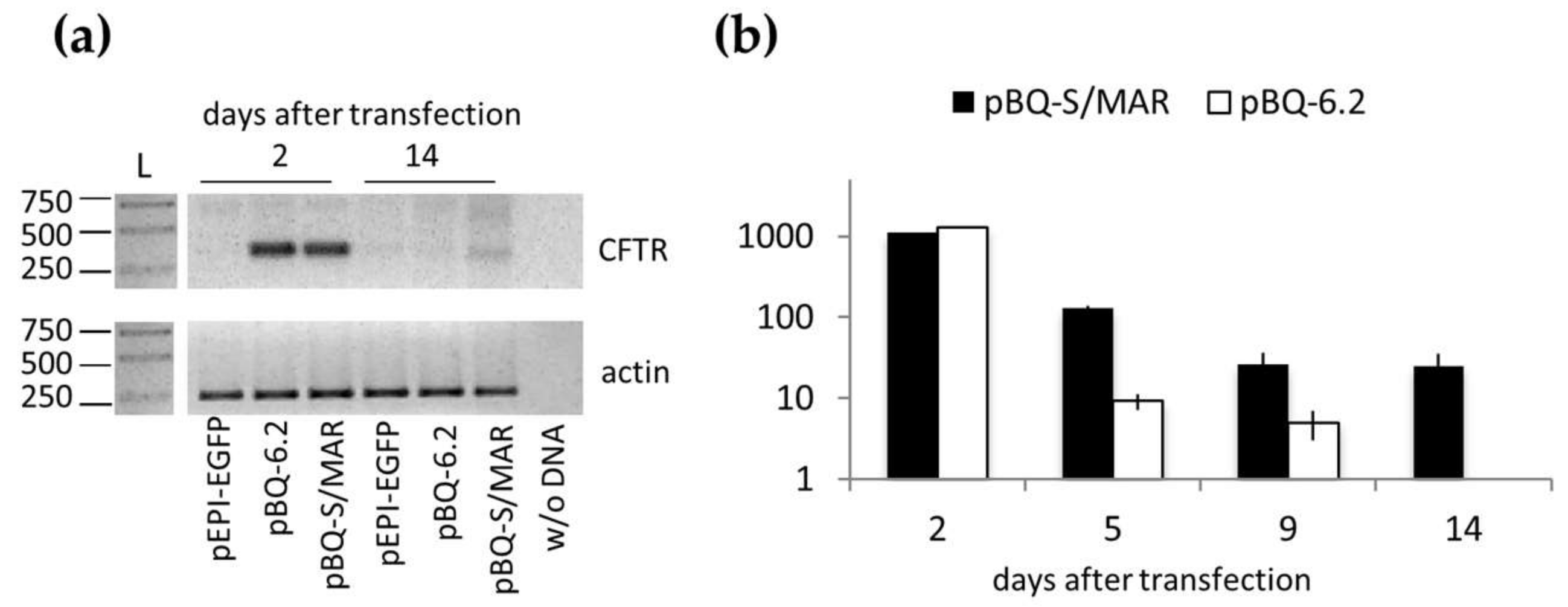
2.2. Assembly and Analysis of the S/MAR-Based CFTR Vector
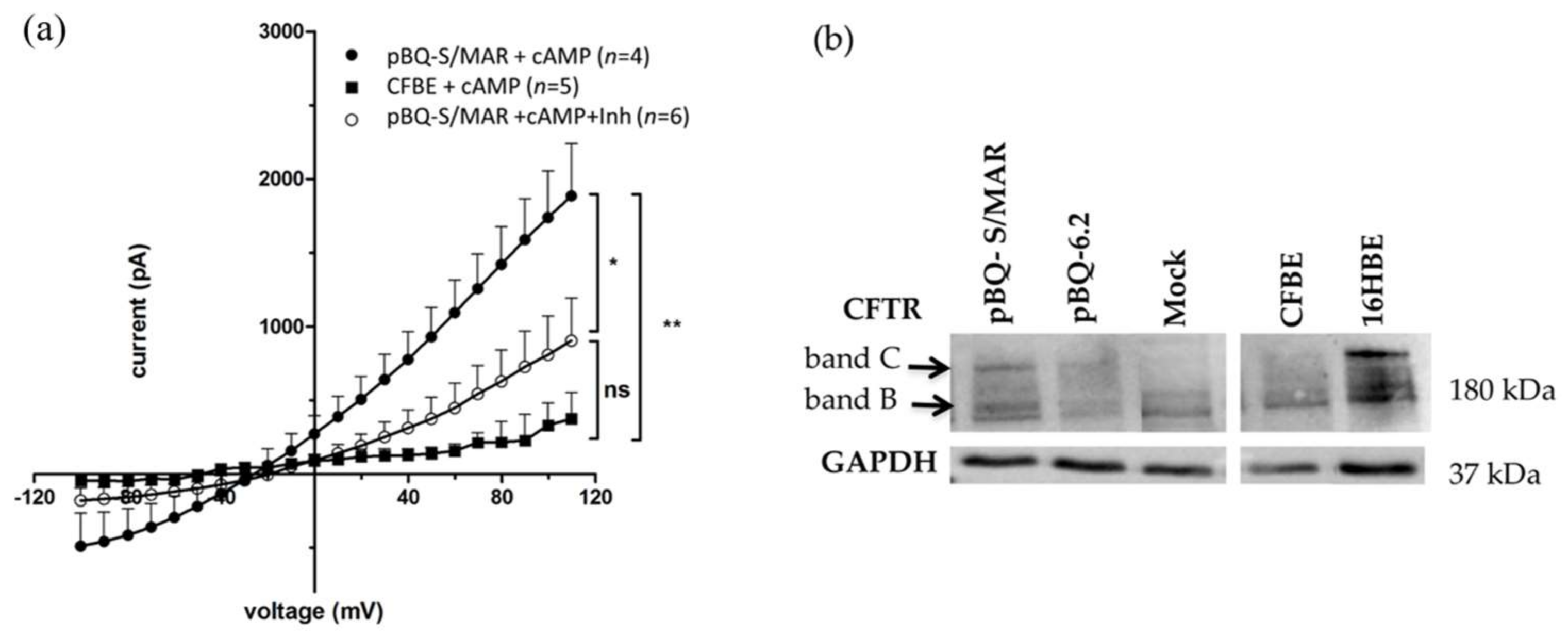
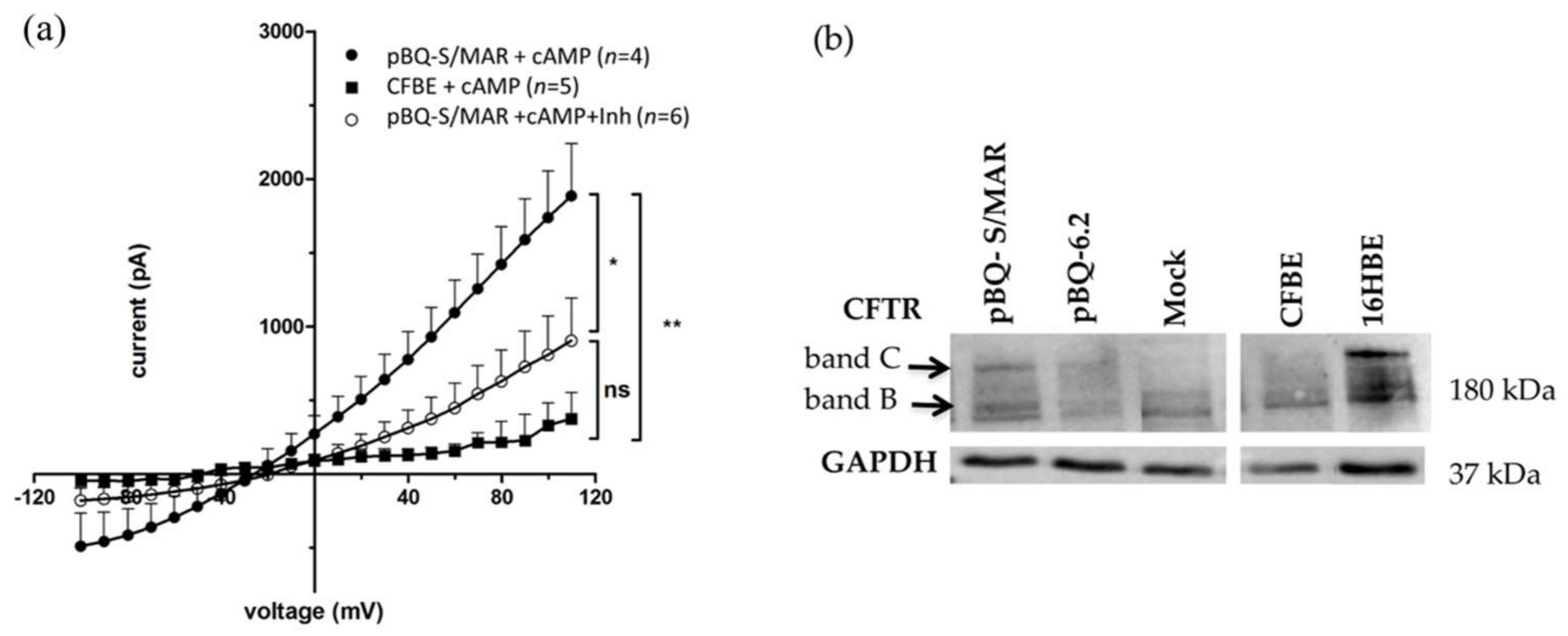
2.3. CFTR Expression and Function in CFBE Cells
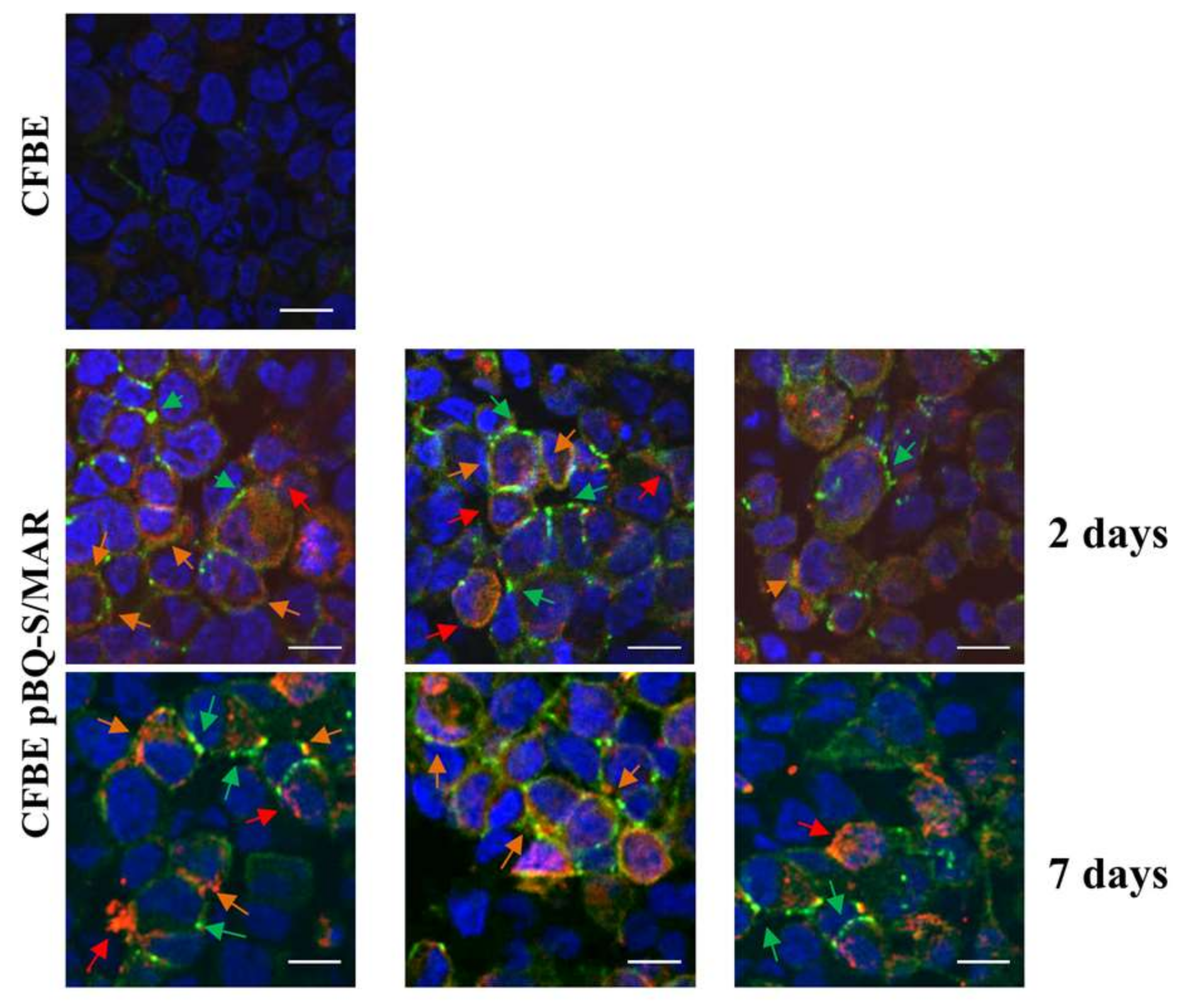
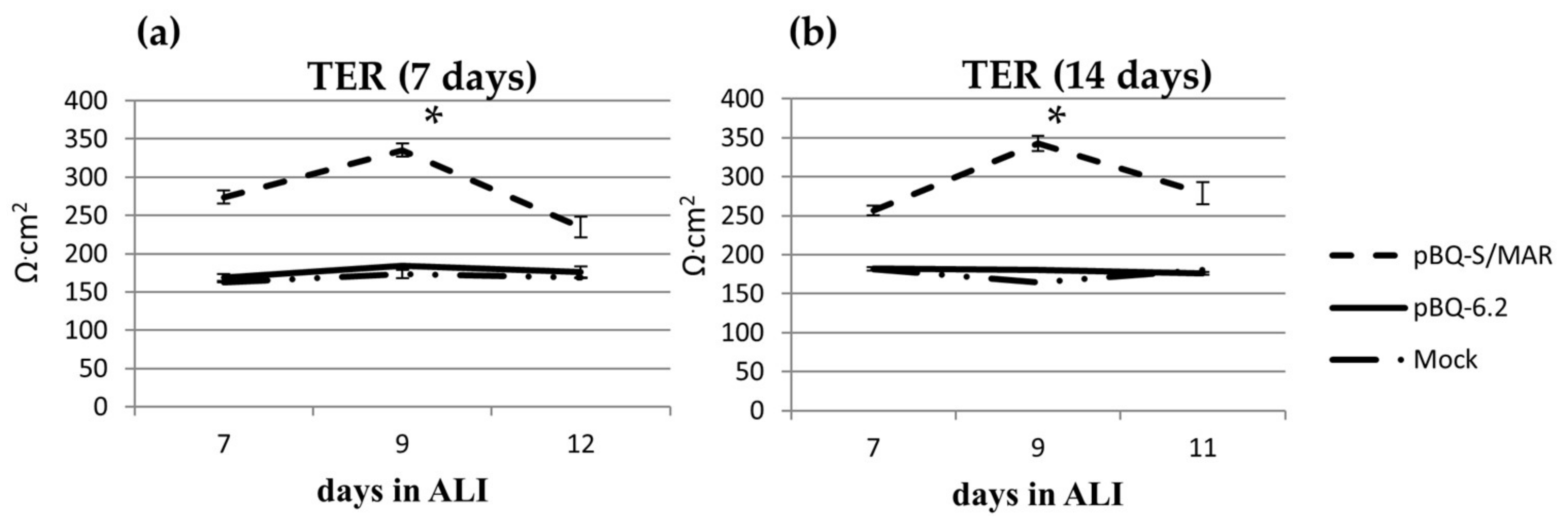
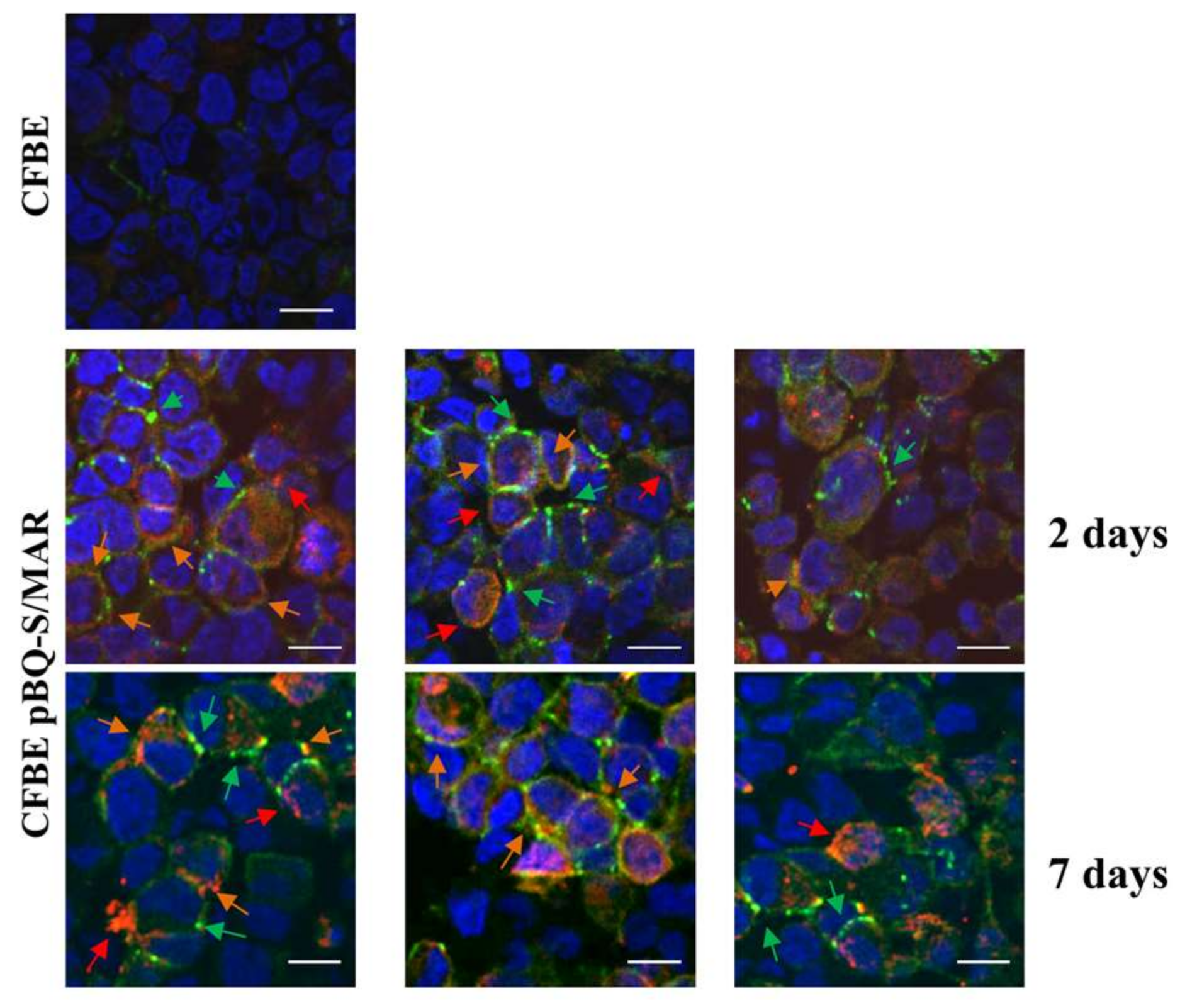
2.4. CFTR Expression in CFBE Cells Improves Tight Junction Organization and Function
3. Discussion
4. Materials and Methods
4.1. Plasmids and pBQ S/MAR Construction
4.2. Cells, Transfection and Culture Conditions
4.3. E. coli Rescue
4.4. RNA and Protein Analysis
4.5. Immunofluorescence and Confocal Microscopy
4.6. Electrophysiology
4.7. Data Analysis and Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Elborn, J.S. Cystic Fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [PubMed]
- Amaral, M.D. Novel personalized therapies for cystic fibrosis: Treating the basic defect in all patients. J. Intern. Med. 2015, 277, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoňez, C.L.; Gelle, D.E. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Campbell, P.W.; De Boek, K.; Donaldson, S.H.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Elborn, I.S.; Ramsey, B.W.; Boyle, M.P.; Konstan, M.W.; Huang, X.; Marigowda, G.; Waltz, D.; Wainwright, C.E. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: A pooled analysis. Lancet Respir. Med. 2016, 4, 617–626. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish ∆F508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246–297. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.R.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of ∆F508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246–296. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, M.; Bruno, S.M.; Pierandrei, S.; Ferraguti, G.; Stamato, A.; Narzi, F.; Amato, A.; Cimino, G.; Bertasi, S.; Quattrucci, S.; et al. A Genotypic-Oriented View of CFTR Genetics Highlights Specific Mutational Patterns Underlying Clinical Macrocategories of Cystic Fibrosis. Mol. Med. 2015, 21, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Quinton, P.M. Cystic fibrosis: Impaired bicarbonate secretion and mucoviscidosis. Lancet 2008, 372, 415–417. [Google Scholar] [CrossRef]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [PubMed]
- Castellani, S.; Guerra, L.; Favia, M.; Di Gioia, S.; Casavola, V.; Conese, M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: Role of ezrin and the RhoA/ROCK pathway. Lab. Investig. 2012, 92, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Favia, M.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellai, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol. Biol. Cell 2010, 21, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Lasalvia, M.; Castellani, S.; D’Antonio, P.; Perna, G.; Carbone, A.; Colia, A.L.; Maffione, A.B.; Capozzi, V.; et al. Human airway epithelial cells investigated by atomic force microscopy: A hint to cystic fibrosis epithelial pathology. Exp. Cell Res. 2016, 348, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.R.; Hyde, S.C. Delivery of genes into the CF airway. Thorax 2014, 69, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Boyd, A.C.; Davies, J.C.; Gill, D.R.; Griesenbach, U.; Harrison, P.T.; Harrison, P.T.; Henig, N.; Higgins, T.; Hyde, S.C.; et al. Genetic medicines for CF: Hype versus reality. Pediatr. Pulmonol. 2016, 51, S5–S17. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.; Southern, K.W. Topical cystic fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease. Cochrane Database Syst. Rev. 2013, 26, CD005599. [Google Scholar]
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Q.; Ertl, H.C.; Wilson, J.M. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J. Virol. 1995, 69, 2004–2015. [Google Scholar] [PubMed]
- Moss, R.B.; Milla, C.; Colombo, J.; Accurso, P.L.; Zeitlin, P.L.; Clancy, J.P.; Spencer, L.T.; Pilewiski, J.; Waltz, D.A.; Dorkin, H.L.; et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: A randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007, 18, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Mitomo, K.; Griesenbach, U.; Inoue, M.; Somerton, L.; Meng, C.; Akiba, E.; Tabata, T.; Ueda, Y.; Franke, G.M.; Farley, R.; et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol. Ther. 2010, 18, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Sun, X.; Feng, Z.; Li, G.; Fisher, J.T.; Stewart, Z.A.; Engelhardt, J.F. Optimization of recombinant adeno-associated virus-mediated expression for large transgenes, using a synthetic promoter and tandem array enhancers. Hum. Gene Ther. 2015, 26, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A. Mechanisms governing lentivirus integration site selection. Curr. Gene Ther. 2008, 8, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Nienhuis, A.W.; Dunbar, C.E.; Sorrentino, B.P. Genotoxicity of retroviral integration in hematopoietic cells. Mol. Ther. 2006, 13, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Bartholomae, C.C.; Arens, A.; Balaggan, K.S.; Yáňez-Muňoz RJ Montini, E.; Howe, S.J.; Paruzynski, A.; Korn, B.; Appelt, J.U.; Macneil, A.; et al. Lentiviral vector integration profiles differ in rodent postmitotic tissues. Mol. Ther. 2011, 19, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Wang, X.; Liu, J.; Hu, X.; Shi, L.; Shen, X.; Ying, W.; Sun, X.; Wang, X.; Huang, P.; et al. CRISPR/Cas9—Mediated precise atrgeted integration in vivo using a double cut donor with short homology arms. EBioMedicine 2017, 20, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, C.; Wong, S.P.; Harbottle, R.; Lipps, H.J. Scaffold/matrix attached region-based nonviral episomal vectors. Hum. Gene Ther. 2011, 22, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Piechaczek, C.; Fetzer, C.; Baiker, A.; Bode, J.; Lipps, H.J. A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucl. Acids Res. 1999, 27, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hagedorn, C.; Schulz, E.; Lipps, H.J.; Ehrhardt, A. Viral hybrid-vectors for delivery of autonomous replicons. Curr. Gene Ther. 2014, 14, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, C.; Gogol-Döring, A.; Schreiber, S.; Epplen, J.T.; Lipps, H.J. Genome-wide profiling of S/MAR-based replicon contact sites. Nucl. Acids Res. 2017, 45, 7841–7854. [Google Scholar] [CrossRef] [PubMed]
- Manzini, S.; Vargiolu, A.; Stehle, I.M.; Bacci, M.L.; Cerrito, M.G.; Giovannoni, R.; Zannoni, A.; Bianco, M.R.; Forni, M.; Donini, P.; et al. Genetically modified pigs produced with a nonviral episomal vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17672–17677. [Google Scholar] [CrossRef] [PubMed]
- Argyros, O.; Wong, S.P.; Niceta, M.; Waddington, S.N.; Howe, S.J.; Coutelle, C.; Miller, A.D.; Harbottle, R.P. Persistent episomal transgene expression in liver following delivery of a scaffold/matrix attachment region containing non-viral vector. Gene Ther. 2008, 15, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Koirala, A.; Makkia, R.S.; Conley, S.M.; Cooper, M.J.; Naash, M.I. S/MAR-containing DNA nanoparticles promote persistent RPE gene expression and improvement in RPE65-associated LCA. Hum. Mol. Genet. 2013, 22, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Koirala, A.; Conley, S.M.; Naash, M.I. Episomal maintenance of S/MAR-containing non-viral vectors for RPE-based diseases. Adv. Exp. Med. Biol. 2014, 801, 703–709. [Google Scholar] [PubMed]
- Verghese, S.C.; Goloviznina, N.A.; Skinner, A.M.; Lipps, H.J.; Kurre, P. S/MAR sequence confers long-term mitotic stability on non-integrating lentiviral vector episomes without selection. Nucl. Acids Res. 2014, 42, e53. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Dho, S.; Bear, C.E.; Kartner, N.; Kennedy, D.; Riordan, J.R.; Tsui, L.C.; Soskett, J.K. cAMP-inducible chloride conductance in mouse fibroblast lines stably expressing the human cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 1991, 88, 7500–7504. [Google Scholar] [CrossRef] [PubMed]
- LeSimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010, 588, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Stehle, J.M.; Postberg, J.; Rupprecht, S.; Cremer, T.; Jackson, D.A.; Lipps, H.J. Establishment and mitotic stability of an extra-chromosomal mammalian replicon. BMC Cell Biol. 2007, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.J.; Ott, E.; Papior, P.; Schepers, A. The latent origin of replication of Epstein-Barr virus directs viral genomes to active regions of the nucleus. J. Virol. 2010, 84, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Ziros, P.G.; Micheva, I.D.; Zoumbos, N.C.; Athanassiadou, A. Gene transfer into human hematopoietic progenitor cells with an episomal vector carrying an S/MAR element. Gene Ther. 2006, 13, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Auriche, C.; Di Domenico, E.G.; Pierandrei, S.; Lucarelli, M.; Castellani, S.; Conese, M.; Melani, R.; Zegarra-Moran, O.; Ascenzioni, F. CFTR expression and activity from the human CFTR locus in BAC vectors, with regulatory regions, isolated by a single-step procedure. Gene Ther. 2010, 17, 1341–1354. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, L.; Braz, C.; Cattani, S.; Ramalho, A.; Christan, S.; Edlinger, M.; Ascenzioni, F.; Laner, A.; Kraner, S.; Amaral, M.; et al. Escherichia coli-cloned CFTR Loci relevant for human artificial chromosome therapy. Hum. Gene Ther. 2010, 21, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established cell lines used in cystic fibrosis research. J. Cyst. Fibros. 2004, 3, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Hirt, B. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 1967, 26, 365–369. [Google Scholar] [CrossRef]
- Del Porto, P.; Cifani, N.; Guarnieri, S.; Di Domenico, E.G.; Mariggiò, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar]
- Cifani, N.; Pompili, B.; Anile, M.; Patella, M.; Diso, D.; Venuta, F.; Cimino, G.; Quattrucci, S.; Di Domenico, E.G.; Ascenzioni, F.; et al. Reactive-oxygen-species-mediated P. aeruginosa killing is functional in human cystic fibrosis macrophages. PLoS ONE 2013, 8, e71717. [Google Scholar] [CrossRef] [PubMed]
- Castellani, S.; Orlando, C.; Carbone, A.; Di Gioia, S.; Conese, M. Magnetofection Enhances Lentiviral-Mediated Transduction of Airway Epithelial Cells through Extracellular and Cellular Barriers. Genes 2016, 7, E103. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Rocco, D.; Pompili, B.; Castellani, S.; Morini, E.; Cavinato, L.; Cimino, G.; Mariggiò, M.A.; Guarnieri, S.; Conese, M.; Del Porto, P.; et al. Assembly and Functional Analysis of an S/MAR Based Episome with the Cystic Fibrosis Transmembrane Conductance Regulator Gene. Int. J. Mol. Sci. 2018, 19, 1220. https://doi.org/10.3390/ijms19041220
De Rocco D, Pompili B, Castellani S, Morini E, Cavinato L, Cimino G, Mariggiò MA, Guarnieri S, Conese M, Del Porto P, et al. Assembly and Functional Analysis of an S/MAR Based Episome with the Cystic Fibrosis Transmembrane Conductance Regulator Gene. International Journal of Molecular Sciences. 2018; 19(4):1220. https://doi.org/10.3390/ijms19041220
Chicago/Turabian StyleDe Rocco, Davide, Barbara Pompili, Stefano Castellani, Elena Morini, Luca Cavinato, Giuseppe Cimino, Maria A Mariggiò, Simone Guarnieri, Massimo Conese, Paola Del Porto, and et al. 2018. "Assembly and Functional Analysis of an S/MAR Based Episome with the Cystic Fibrosis Transmembrane Conductance Regulator Gene" International Journal of Molecular Sciences 19, no. 4: 1220. https://doi.org/10.3390/ijms19041220
APA StyleDe Rocco, D., Pompili, B., Castellani, S., Morini, E., Cavinato, L., Cimino, G., Mariggiò, M. A., Guarnieri, S., Conese, M., Del Porto, P., & Ascenzioni, F. (2018). Assembly and Functional Analysis of an S/MAR Based Episome with the Cystic Fibrosis Transmembrane Conductance Regulator Gene. International Journal of Molecular Sciences, 19(4), 1220. https://doi.org/10.3390/ijms19041220