Recent Progress in Gene Therapy for Ovarian Cancer




Abstract
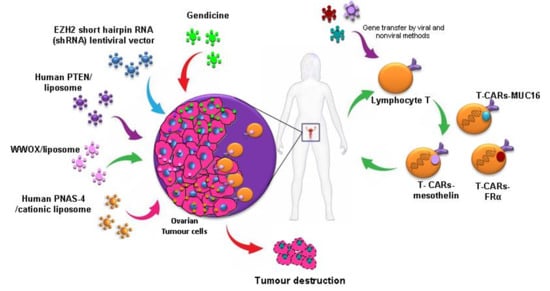
1. Introduction
1.1. Risk Factors and Prevention Strategies
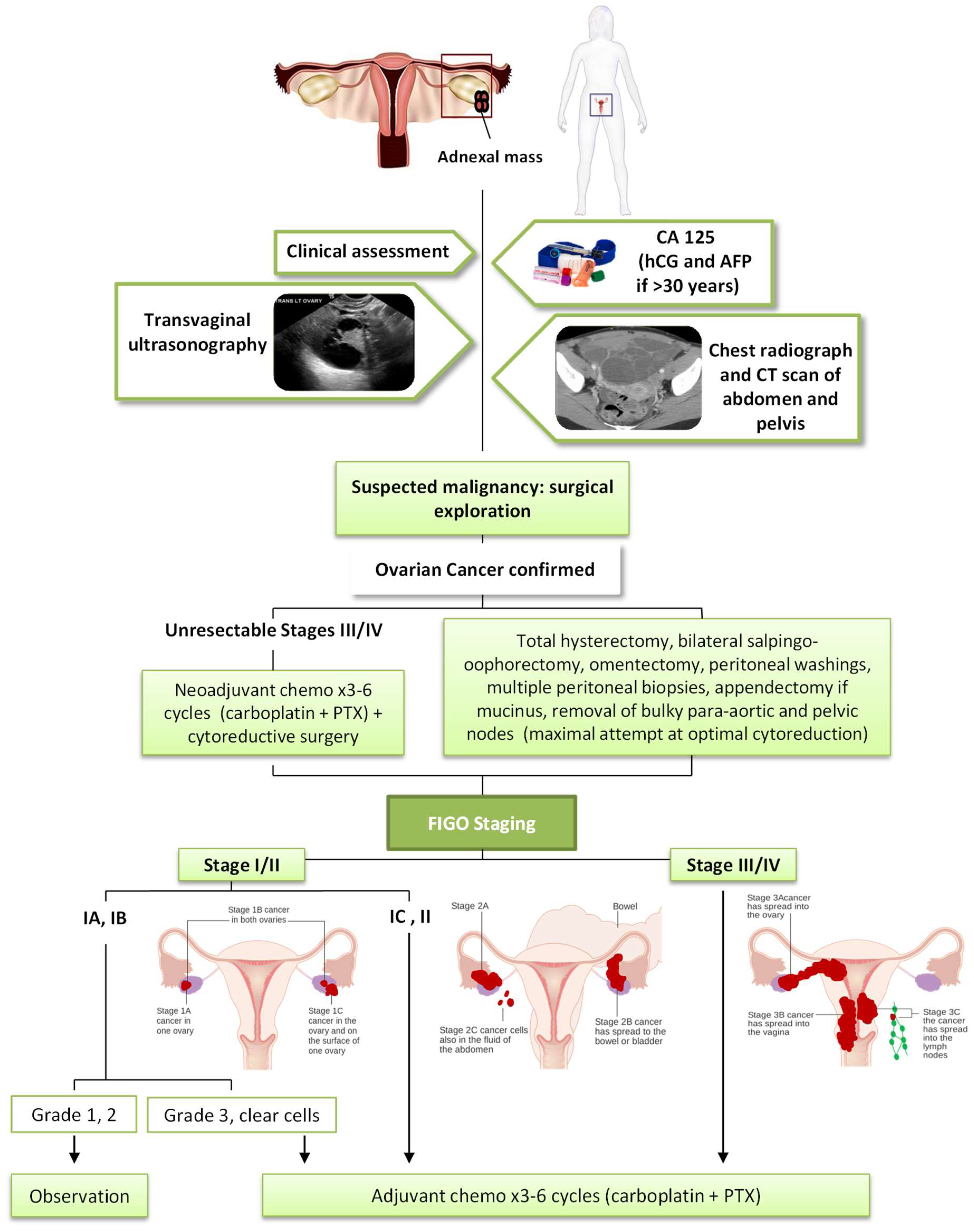
1.2. Diagnostic and Early Detection
1.3. Current Treatments of Ovarian Cancer
2. Gene Therapy in Ovarian Cancer
2.1. Improved Vectors for Gene Delivery
2.1.1. Viral Vectors
Multiple Viral Vectors Have Been Evaluated in OC
2.1.2. Non-Viral Vectors
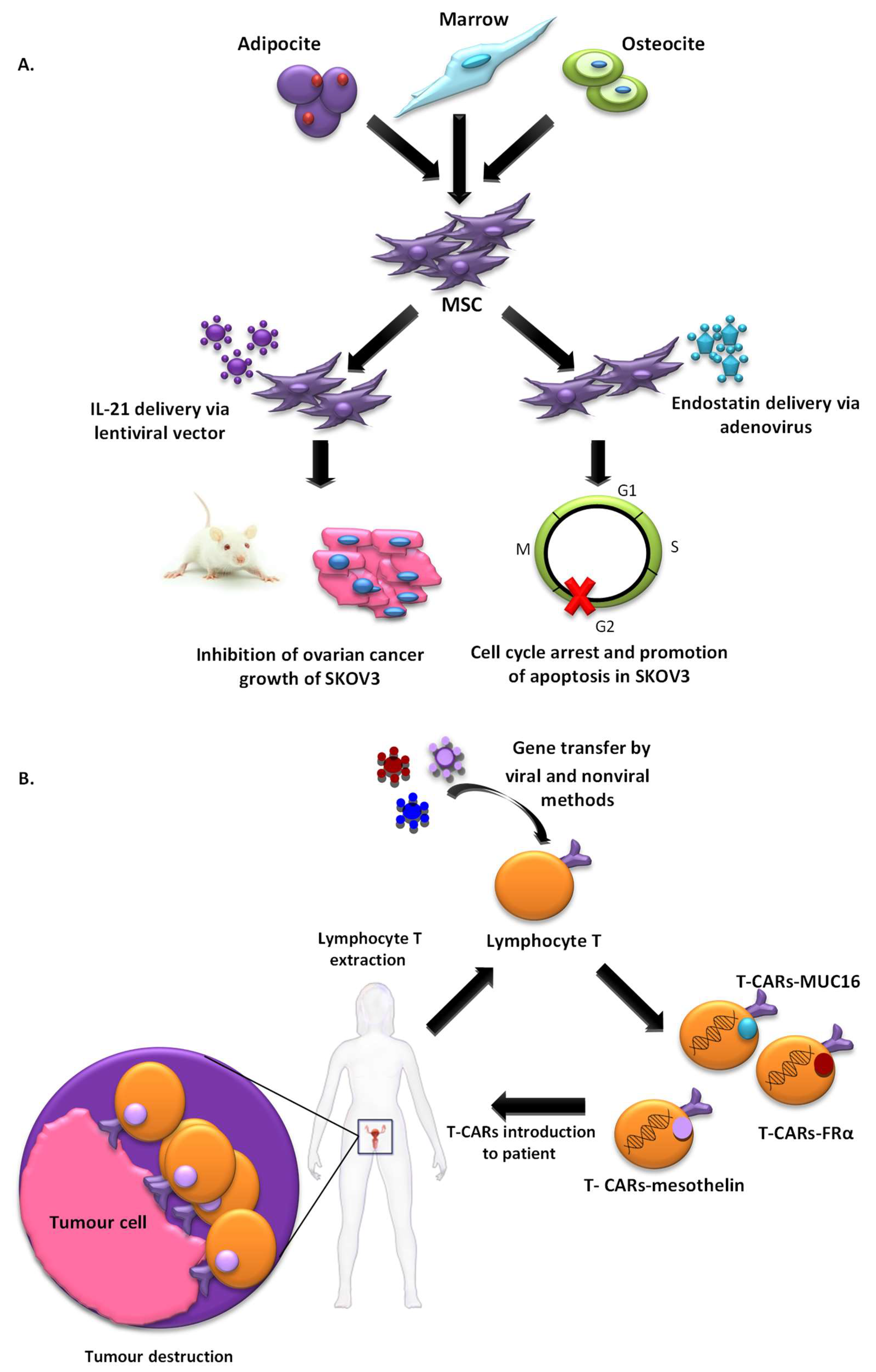
2.1.3. Cell-Based Vectors
2.1.4. Targeted Vectors for Ovarian Cancer Gene Therapy
2.2. Gene Therapy for Ovarian Cancer Treatment
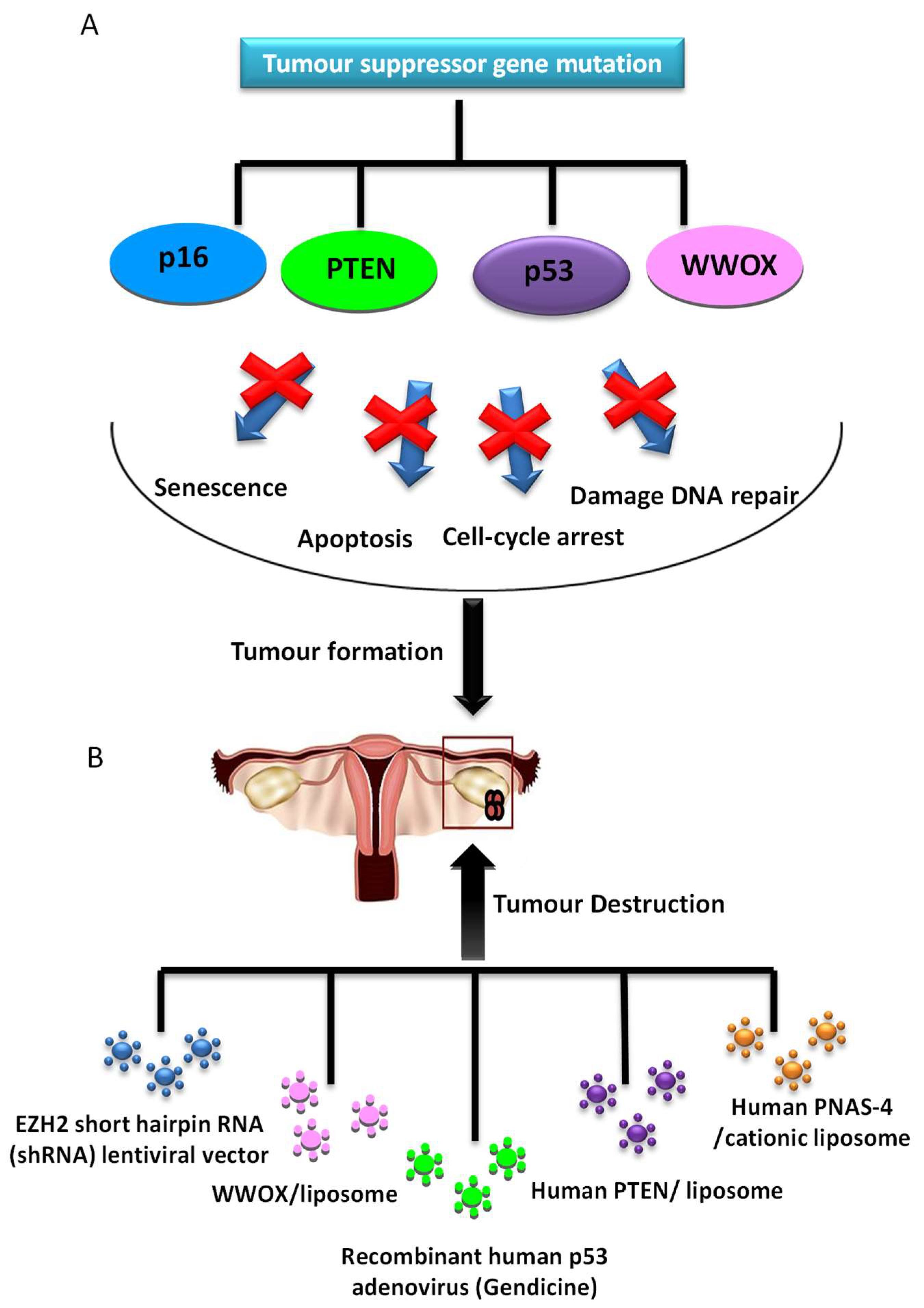
2.2.1. Tumor Suppressor Gene Therapy
2.2.2. Oncofactor Inhibition Strategies
2.2.3. Suicide Gene Therapy
2.2.4. Antiangiogenic Gene Therapy
2.2.5. Immunopotentiation
2.2.6. Multi-Drug Resistance (MDR)
2.2.7. Oncolytic Virotherapy
3. Clinical Trials
4. Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Crum, C.; Friedlander, M. Cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2015, 131, S111–S122. [Google Scholar] [CrossRef] [PubMed]
- Prat, J.; FIGO Committee on Gynecologic Oncology. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynaecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.R.; Stewart, L.M.; Holman, C.D.J.; Jordan, S.; Semmens, J.; Spilsbury, K.; Threlfall, T. Value of Pathology Review in a Population-based Series of Ovarian Tumors. Int. J. Gynecol. Pathol. 2017, 36, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Hunn, J.; Rodriguez, G.C. Ovarian cancer: Etiology, risk factors, and epidemiology. Clin. Obstet. Gynecol. 2012, 55, 3–23. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force; Grossman, D.C.; Curry, S.J.; Owens, D.K.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W.; Kemper, A.R.; Krist, A.H.; et al. Screening for Ovarian Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Bellone, S.; Siegel, E.R.; Altwerger, G.; Menderes, G.; Bonazzoli, E.; Egawa-Takata, T.; Pettinella, F.; Bianchi, A.; Riccio, F.; et al. A novel multiple biomarker panel for the early detection of high-grade serous ovarian carcinoma. Gynecol. Oncol. 2018, 149, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzalez-Martin, A.; Colombo, N.; Sessa, C.; ESMO Guidelines Working Group. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24 (Suppl. 6), vi24–vi32. [Google Scholar] [CrossRef]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Huhtala, T.; Kaikkonen, M.U.; Lesch, H.P.; Viitala, S.; Ylä-Herttuala, S.; Närvänen, A. Biodistribution and antitumor effect of Cetuximab-targeted lentivirus. Nucl. Med. Biol. 2014, 41, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Rawlinson, J.W.; Vaden, K.; Hunsaker, J.; Miller, D.F.; Nephew, K.P. Adenoviral-delivered HE4-HSV-tk sensitizes ovarian cancer cells to ganciclovir. Gene Ther. Mol. Biol. 2013, 15, 120–130. [Google Scholar] [PubMed]
- Yoshihara, C.; Hamada, K.; Koyama, Y. Preparation of a novel adenovirus formulation with artificial envelope of multilayer polymer-coatings: Therapeutic effect on metastatic ovarian cancer. Oncol. Rep. 2010, 23, 733–738. [Google Scholar] [PubMed]
- Xie, Y.; Hicks, M.J.; Kaminsky, S.M.; Moore, M.A.S.; Crystal, R.G.; Rafii, A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol. Oncol. 2014, 135, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bui Nguyen, T.M.; Nguyen, T.M.B.; Subramanian, I.V.; Xiao, X.; Nguyen, P.; Ramakrishnan, S. Adeno-associated virus-mediated delivery of kringle 5 of human plasminogen inhibits orthotopic growth of ovarian cancer. Gene Ther. 2010, 17, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, L.; Su, X.; Wang, L.; Chen, X.; Li, D.; Luo, S.; Shi, H.; Chen, L.; Wang, Y. Suppression of ovarian cancer growth via systemic administration with liposome-encapsulated adenovirus-encoding endostatin. Cancer Gene Ther. 2010, 17, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Pépin, D.; Sosulski, A.; Zhang, L.; Wang, D.; Vathipadiekal, V.; Hendren, K.; Coletti, C.M.; Yu, A.; Castro, C.M.; Birrer, M.J.; et al. AAV9 delivering a modified human Mullerian inhibiting substance as a gene therapy in patient-derived xenografts of ovarian cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E4418–E4427. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-F.; Chiang, A.J.; Tsai, H.-H.; Pomper, M.G.; Kang, T.H.; Roden, R.R.; Wu, T.-C. Ovarian cancer gene therapy using HPV-16 pseudovirion carrying the HSV-tk gene. PLoS ONE 2012, 7, e40983. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhang, X.; Jiang, W.; Wu, C.; Chen, C.; Zheng, Y.; Gu, J.; Xu, C. Tumor-directed gene therapy in mice using a composite nonviral gene delivery system consisting of the piggyBac transposon and polyethylenimine. BMC Cancer 2009, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Choi, S.J.; Oh, J.H.; Chae, S.W.; Nam, K.; Park, J.S.; Lee, H.J. Nonviral gene delivery to human ovarian cancer cells using arginine-grafted PAMAM dendrimer. Drug Dev. Ind. Pharm. 2011, 37, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Du, T.; Zhang, J.; Zhao, W.; Cheng, H.; Yang, Y.; Wu, Y.; Wang, C.; Men, K.; Gou, M. Efficient inhibition of ovarian cancer by degradable nanoparticle-delivered survivin T34A gene. Int. J. Nanomed. 2016, 11, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Gou, M.; Yi, T.; Qi, X.; Liu, P.; Wei, Y.; Zhao, X. Enhanced antitumor effect of biodegradable cationic heparin-polyethyleneimine nanogels delivering FILIP1LΔC103 gene combined with low-dose cisplatin on ovarian cancer. Oncotarget 2017, 8, 76432–76442. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Gou, M.; Yi, T.; Yang, L.; Liu, L.; Lin, X.; Su, D.; Wei, Y.; Zhao, X. Efficient Inhibition of Ovarian Cancer by Gelonin Toxin Gene Delivered by Biodegradable Cationic Heparin-polyethyleneimine Nanogels. Int. J. Med. Sci. 2015, 12, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Li, T.S.C.; Yawata, T.; Honke, K. Efficient siRNA delivery and tumor accumulation mediated by ionically cross-linked folic acid-poly(ethylene glycol)-chitosan oligosaccharide lactate nanoparticles: For the potential targeted ovarian cancer gene therapy. Eur. J. Pharm. Sci. 2014, 52, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lou, B.; Zhao, P.; Lin, C. Multifunctional disulfide-based cationic dextran conjugates for intravenous gene delivery targeting ovarian cancer cells. Mol. Pharm. 2014, 11, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-C.; Zhang, L.; Feng, S.-S.; Hong, L.; Zheng, H.-L.; Chen, L.-L.; Zheng, X.-L.; Ye, Y.-Q.; Zhao, M.-D.; Wang, W.-X.; et al. Efficient delivery of Notch1 siRNA to SKOV3 cells by cationic cholesterol derivative-based liposome. Int. J. Nanomed. 2016, 11, 5485–5496. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Yang, Y.; Kang, T.; Zhao, W.; Cheng, H.; Wu, Y.; Du, T.; Liu, B.; Li, Y.; Luo, F.; et al. Ovarian Cancer Therapy by VSVMP Gene Mediated by a Paclitaxel-Enhanced Nanoparticle. ACS Appl. Mater. Interfaces 2017, 9, 39152–39164. [Google Scholar] [CrossRef] [PubMed]
- Florinas, S.; Kim, J.; Nam, K.; Janát-Amsbury, M.M.; Kim, S.W. Ultrasound-assisted siRNA delivery via arginine-grafted bioreducible polymer and microbubbles targeting VEGF for ovarian cancer treatment. J. Control. Release 2014, 183, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Guo, J.; Sun, J.; Zhu, S.; Yan, Y.; Zhu, Y.; Li, M.; Wang, Z.; Xu, R.X. Targeted microbubbles for ultrasound mediated gene transfection and apoptosis induction in ovarian cancer cells. Ultrason. Sonochem. 2013, 20, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chang, S.; Sun, J.; Zhu, S.; Pu, C.; Li, Y.; Zhu, Y.; Wang, Z.; Xu, R.X. Targeted Microbubbles for Ultrasound Mediated Short Hairpin RNA Plasmid Transfection to Inhibit Survivin Gene Expression and Induce Apoptosis of Ovarian Cancer A2780/DDP Cells. Mol. Pharm. 2015, 12, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Heymach, J.; Krilov, L.; Alberg, A.; Baxter, N.; Chang, S.M.; Corcoran, R.; Dale, W.; DeMichele, A.; Magid Diefenbach, C.S.; Dreicer, R.; et al. Clinical Cancer Advances 2018: Annual Report on Progress Against Cancer From the American Society of Clinical Oncology. J. Clin. Oncol. 2018, 36, 1020–1044. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Porter, D.L. The Promise of Chimeric Antigen Receptor T-Cell Therapy. Oncology (Williston Park) 2016, 30, 880–890. [Google Scholar] [PubMed]
- Zhu, X.; Cai, H.; Zhao, L.; Ning, L.; Lang, J. CAR-T cell therapy in ovarian cancer: From the bench to the bedside. Oncotarget 2017, 8, 64607–64621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, B.; Shi, H. Application of chimeric antigen receptor-engineered T cells in ovarian cancer therapy. Immunotherapy 2017, 9, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Reagan, M.R.; Kaplan, D.L. Concise review: Mesenchymal stem cell tumor-homing: Detection methods in disease model systems. Stem Cells 2011, 29, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Ren, M.; Li, M.; Chen, D.; Chen, J.; Shi, F.; Wang, X.; Dou, J. Gene therapy of ovarian cancer using IL-21-secreting human umbilical cord mesenchymal stem cells in nude mice. J. Ovarian Res. 2014, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chen, W.; Zhuang, R.; Song, T.; Li, P. The effect of endostatin mediated by human mesenchymal stem cells on ovarian cancer cells in vitro. J. Cancer Res. Clin. Oncol. 2010, 136, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Dembinski, J.L.; Wilson, S.M.; Spaeth, E.L.; Studeny, M.; Zompetta, C.; Samudio, I.; Roby, K.; Andreeff, M.; Marini, F.C. Tumor stroma engraftment of gene-modified mesenchymal stem cells as anti-tumor therapy against ovarian cancer. Cytotherapy 2013, 15, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Komarova, S.; Roth, J.; Alvarez, R.; Curiel, D.T.; Pereboeva, L. Targeting of mesenchymal stem cells to ovarian tumors via an artificial receptor. J. Ovarian Res. 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Kaneti, L.; Bronshtein, T.; Malkah Dayan, N.; Kovregina, I.; Letko Khait, N.; Lupu-Haber, Y.; Fliman, M.; Schoen, B.W.; Kaneti, G.; Machluf, M. Nanoghosts as a Novel Natural Nonviral Gene Delivery Platform Safely Targeting Multiple Cancers. Nano Lett. 2016, 16, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Zwacka, R. The future of mesenchymal stem cell-based therapeutic approaches for cancer—From cells to ghosts. Cancer Lett. 2018, 414, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Malecki, M.; Dahlke, J.; Haig, M.; Wohlwend, L.; Malecki, R. Eradication of Human Ovarian Cancer Cells by Transgenic Expression of Recombinant DNASE1, DNASE1L3, DNASE2, and DFFB Controlled by EGFR Promoter: Novel Strategy for Targeted Therapy of Cancer. J. Genet. Syndr. Gene Ther. 2013, 4, 152. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Hsu, J.L.; Choi, M.-G.; Xia, W.; Yamaguchi, H.; Chen, C.-T.; Trinh, B.Q.; Lu, Z.; Ueno, N.T.; Wolf, J.K.; et al. A novel hTERT promoter-driven E1A therapeutic for ovarian cancer. Mol. Cancer Ther. 2009, 8, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-C.; Kim, B.-G.; Lee, J.-H. Thymosin β10 expression driven by the human TERT promoter induces ovarian cancer-specific apoptosis through ROS production. PLoS ONE 2012, 7, e35399. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Zugates, G.T.; Peng, W.; Holtz, D.; Dunton, C.; Green, J.J.; Hossain, N.; Chernick, M.R.; Padera, R.F.; Langer, R.; et al. Nanoparticle-delivered suicide gene therapy effectively reduces ovarian tumor burden in mice. Cancer Res. 2009, 69, 6184–6191. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Deng, Y.; Shapiro, E.M.; Bortolomai, I.; Lopez, S.; Lin, K.; Bellone, S.; Cui, J.; Menderes, G.; Black, J.D.; et al. Dual-Targeting Nanoparticles for in Vivo Delivery of Suicide Genes to Chemotherapy-Resistant Ovarian Cancer Cells. Mol. Cancer Ther. 2017, 16, 323–333. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-Y.; Deng, F.; Wei, X.-W.; Ma, C.-C.; Luo, M.; Zhang, P.; Sang, Y.-X.; Liang, X.; Liu, L.; Qin, H.-X.; et al. Ovarian cancer treatment with a tumor-targeting and gene expression-controllable lipoplex. Sci. Rep. 2016, 6, 23764. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-Y.; Wei, X.-W.; Luo, M.; Luo, S.-T.; Yang, Y.; Yu, Y.-Y.; Chen, Y.; Ma, C.-C.; Liang, X.; Guo, F.-C.; et al. Folate-linked lipoplexes for short hairpin RNA targeting claudin-3 delivery in ovarian cancer xenografts. J. Control. Release 2013, 172, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Hallaj-Nezhadi, S.; Dass, C.R.; Lotfipour, F. Intraperitoneal delivery of nanoparticles for cancer gene therapy. Future Oncol. 2013, 9, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Collinet, P.; Vereecque, R.; Sabban, F.; Vinatier, D.; Leblanc, E.; Narducci, F.; Querleu, D.; Quesnel, B. In vivo expression and antitumor activity of p53 gene transfer with naked plasmid DNA in an ovarian cancer xenograft model in nude mice. J. Obstet. Gynaecol. Res. 2006, 32, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Kigawa, J.; Sato, S.; Shimada, M.; Kanamori, Y.; Itamochi, H.; Terakawa, N. Effect of p53 gene transfer and cisplatin in a peritonitis carcinomatosa model with p53-deficient ovarian cancer cells. Gynecol. Oncol. 2002, 84, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Quist, S.R.; Wang-Gohrke, S.; Köhler, T.; Kreienberg, R.; Runnebaum, I.B. Cooperative effect of adenoviral p53 gene therapy and standard chemotherapy in ovarian cancer cells independent of the endogenous p53 status. Cancer Gene Ther. 2004, 11, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, S.; Ylikomi, T. Concomitant exposure of ovarian cancer cells to docetaxel, CPT-11 or SN-38 and adenovirus-mediated p53 gene therapy. Anticancer Drugs 2009, 20, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Sui, R.; Li, R.; Miao, J.; Liu, J. Biological characteristics of Taxol-resistant ovarian cancer cells and reversal of Taxol resistance by adenovirus expressing p53. Mol. Med. Rep. 2015, 11, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Zeimet, A.G.; Marth, C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 2003, 4, 415–422. [Google Scholar] [CrossRef]
- Zhang, W.-W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, Z.; Deng, H.; Yang, H.; Yan, F.; Qian, Z.; Chen, L.; Wei, Y.; Zhao, X. Efficient inhibition of ovarian cancer growth and prolonged survival by transfection with a novel pro-apoptotic gene, hPNAS-4, in a mouse model. In vivo and in vitro results. Oncology 2008, 75, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, S.; Weng, D.; Xing, H.; Song, X.; Zhu, T.; Xia, X.; Weng, Y.; Xu, G.; Meng, L.; et al. Reversal of the malignant phenotype of ovarian cancer A2780 cells through transfection with wild-type PTEN gene. Cancer Lett. 2008, 271, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Choi, B.Y.; Cho, Y.-Y.; Lee, S.-Y.; Huang, Z.; Kundu, J.K.; Kim, M.O.; Kim, D.J.; Bode, A.M.; Surh, Y.-J.; et al. Tumor suppressor p16(INK4a) inhibits cancer cell growth by downregulating eEF1A2 through a direct interaction. J. Cell Sci. 2013, 126, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Xu, H.; Wang, Q.; Li, M.; Meng, J.; Kuang, Y. Inhibition of enhancer of zeste homolog 2 increases the expression of p16 and suppresses the proliferation and migration of ovarian carcinoma cells in vitro and in vivo. Oncol. Lett. 2018, 15, 3233–3239. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Hu, S.; Wang, Z. Cloning of WWOX gene and its growth-inhibiting effects on ovarian cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Tong, J.; Lin, X.; Han, Q.; Huang, H. Effect of the WWOX gene on the regulation of the cell cycle and apoptosis in human ovarian cancer stem cells. Mol. Med. Rep. 2015, 12, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, E.B.; Blackburn, W.H.; Smith, M.H.; Kapa, L.B.; Lyon, L.A.; McDonald, J.F. Chemosensitization of cancer cells by siRNA using targeted nanogel delivery. BMC Cancer 2010, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Peng, S.; Yu, H.; Teng, H.; Cui, M. RNAi-mediated downregulation of NOB1 suppresses the growth and colony-formation ability of human ovarian cancer cells. Med. Oncol. 2012, 29, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shi, H.; Chen, Z.; Wu, Q.; Ren, F.; Huang, H. Effects of metastasis-associated in colon cancer 1 inhibition by small hairpin RNA on ovarian carcinoma OVCAR-3 cells. J. Exp. Clin. Cancer Res. 2011, 30, 83. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Ji, M.; Chen, C.; Shi, H. Effect of siRNA targeting MTA1 on metastasis malignant phenotype of ovarian cancer A2780 cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cui, M.; Xu, T.; Yu, W.; Zhang, L. Silencing of cyclooxygenase-2 inhibits the growth, invasion and migration of ovarian cancer cells. Mol. Med. Rep. 2014, 9, 2499–2504. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-J.; Tian, J.-Y.; Jin, Y.-M.; Wang, L.; Yang, R.-Q.; Cui, M.-H. Effects of cyclooxygenase-2 gene silencing on the biological behavior of SKOV3 ovarian cancer cells. Mol. Med. Rep. 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Ren, L.; Shang, L.; Wang, X.; Wang, J. Effect of WT1 antisense mRNA on the induction of apoptosis in ovarian carcinoma SKOV3 cells. Eur. J. Gynaecol. Oncol. 2011, 32, 651–656. [Google Scholar] [PubMed]
- Jiang, Q.; Dai, L.; Cheng, L.; Chen, X.; Li, Y.; Zhang, S.; Su, X.; Zhao, X.; Wei, Y.; Deng, H. Efficient inhibition of intraperitoneal ovarian cancer growth in nude mice by liposomal delivery of short hairpin RNA against STAT3. J. Obstet. Gynaecol. Res. 2013, 39, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Bao, Y.; Peng, W.; Goldberg, M.; Love, K.; Bumcrot, D.A.; Cole, G.; Langer, R.; Anderson, D.G.; Sawicki, J.A. Claudin-3 gene silencing with siRNA suppresses ovarian tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 3426–3430. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yi, T.; Song, X.; Li, S.; Qi, X.; Chen, X.; Lin, H.; He, X.; Li, Z.; Wei, Y.; et al. Efficient inhibition of ovarian cancer by short hairpin RNA targeting claudin-3. Oncol. Rep. 2011, 26, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.X.; Zhang, B.; Zang, J.L.; Wang, G.Y.; Gao, M.H. CD59 silencing via retrovirus-mediated RNA interference enhanced complement-mediated cell damage in ovary cancer. Cell. Mol. Immunol. 2009, 6, 61–66. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-Y.; Zhang, Y.-G.; Yang, Y.-H.; Ma, C.-C.; Wang, P.; Du, W.; Li, L.; Xiang, R.; Song, X.-R.; Zhao, X.; et al. In Vivo Ovarian Cancer Gene Therapy Using CRISPR-Cas9. Hum. Gene Ther. 2018, 29, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.A.; Carrillo, E.; Griñán-Lisón, C.; Martín, A.; Perán, M.; Marchal, J.A.; Boulaiz, H. Cancer suicide gene therapy: A patent review. Expert Opin. Ther. Pat. 2016, 26, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Zhao, X.-Y.; Li, S.; Jia, C.-J.; Jiang, L.; Shi, T.-M.; Ren, W.-D. A novel plasmid and SonoVue formulation plus ultrasound sonication for effective gene delivery in nude mice. Life Sci. 2013, 93, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-L.; Shi, Y.-L.; Li, X. Inhibitory effects of the ultrasound-targeted microbubble destruction-mediated herpes simplex virus-thymidine kinase/ganciclovir system on ovarian cancer in mice. Exp. Ther. Med. 2014, 8, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Sher, Y.-P.; Chang, C.-M.; Juo, C.-G.; Chen, C.-T.; Hsu, J.L.; Lin, C.-Y.; Han, Z.; Shiah, S.-G.; Hung, M.-C. Targeted endostatin-cytosine deaminase fusion gene therapy plus 5-fluorocytosine suppresses ovarian tumor growth. Oncogene 2013, 32, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Menghistu, T.; Twigger, K.R.; Searle, P.F.; Bhide, S.A.; Vile, R.G.; Melcher, A.A.; Pandha, H.S.; Harrington, K.J. Escherichia coli nitroreductase plus CB1954 enhances the effect of radiotherapy in vitro and in vivo. Gene Ther. 2008, 15, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Joshi, S.; Russell, P.J.; Nair, S.; Khatri, A. Purine Nucleoside Phosphorylase mediated molecular chemotherapy and conventional chemotherapy: A tangible union against chemoresistant cancer. BMC Cancer 2011, 11, 368. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Yokoyama, Y.; Osawa, Y.; Miura, R.; Mizunuma, H. Gene therapy for ovarian cancer using carbonyl reductase 1 DNA with a polyamidoamine dendrimer in mouse models. Cancer Gene Ther. 2016, 23, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Mizukami, H.; Saga, Y.; Yoshimura, I.; Hasumi, Y.; Takayama, T.; Kohno, T.; Matsushita, T.; Okada, T.; Kume, A.; et al. Suppression of ovarian cancer by muscle-mediated expression of soluble VEGFR-1/Flt-1 using adeno-associated virus serotype 1-derived vector. Int. J. Cancer 2007, 120, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Hofmann, J.; Holash, J.; Yancopoulos, G.D.; Sood, A.K.; Jaffe, R.B. Vascular endothelial growth factor trap combined with paclitaxel strikingly inhibits tumor and ascites, prolonging survival in a human ovarian cancer model. Clin. Cancer Res. 2005, 11, 6966–6971. [Google Scholar] [CrossRef] [PubMed]
- Sallinen, H.; Anttila, M.; Narvainen, J.; Koponen, J.; Hamalainen, K.; Kholova, I.; Heikura, T.; Toivanen, P.; Kosma, V.-M.; Heinonen, S.; et al. Antiangiogenic gene therapy with soluble VEGFR-1, -2, and -3 reduces the growth of solid human ovarian carcinoma in mice. Mol. Ther. 2009, 17, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Sopo, M.; Anttila, M.; Sallinen, H.; Tuppurainen, L.; Laurema, A.; Laidinen, S.; Hamalainen, K.; Tuunanen, P.; Koponen, J.K.; Kosma, V.-M.; et al. Antiangiogenic gene therapy with soluble VEGF-receptors -1, -2 and -3 together with paclitaxel prolongs survival of mice with human ovarian carcinoma. Int. J. Cancer 2012, 131, 2394–2401. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, L.; Sallinen, H.; Kokki, E.; Koponen, J.; Anttila, M.; Pulkkinen, K.; Heikura, T.; Toivanen, P.; Hämäläinen, K.; Kosma, V.-M.; et al. Preclinical safety, toxicology, and biodistribution study of adenoviral gene therapy with sVEGFR-2 and sVEGFR-3 combined with chemotherapy for ovarian cancer. Hum. Gene Ther. Clin. Dev. 2013, 24, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, L.; Sallinen, H.; Karvonen, A.; Valkonen, E.; Laakso, H.; Liimatainen, T.; Hytönen, E.; Hämäläinen, K.; Kosma, V.-M.; Anttila, M.; et al. Combined Gene Therapy Using AdsVEGFR2 and AdsTie2 With Chemotherapy Reduces the Growth of Human Ovarian Cancer and Formation of Ascites in Mice. Int. J. Gynecol. Cancer 2017, 27, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Hampl, M.; Tanaka, T.; Albert, P.S.; Lee, J.; Ferrari, N.; Fine, H.A. Therapeutic effects of viral vector-mediated antiangiogenic gene transfer in malignant ascites. Hum. Gene Ther. 2001, 12, 1713–1729. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.V.; Ghebre, R.; Ramakrishnan, S. Adeno-associated virus-mediated delivery of a mutant endostatin suppresses ovarian carcinoma growth in mice. Gene Ther. 2005, 12, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Wang, Y.; Wang, J.; Zhao, F.; Li, Y.; Cao, M.; Hu, W.; Hu, K.; He, X.F.; Chu, L.; et al. Antitumor efficacy induced by human ovarian cancer cells secreting IL-21 alone or combination with GM-CSF cytokines in nude mice model. Immunobiology 2009, 214, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, J.; Dou, J.; He, X.; Zhao, F.; Jiang, C.; Yu, F.; Hu, K.; Chu, L.; Li, X.; et al. Augmenting therapy of ovarian cancer efficacy by secreting IL-21 human umbilical cord blood stem cells in nude mice. Cell Transplant. 2011, 20, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Fewell, J.G.; Matar, M.M.; Rice, J.S.; Brunhoeber, E.; Slobodkin, G.; Pence, C.; Worker, M.; Lewis, D.H.; Anwer, K. Treatment of disseminated ovarian cancer using nonviral interleukin-12 gene therapy delivered intraperitoneally. J. Gene Med. 2009, 11, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Pilgrim, P.; Zhou, W.; Gagliano, N.; Frezza, E.E.; Jenkins, M.; Weidanz, J.A.; Lustgarten, J.; Cannon, M.; Bumm, K.; et al. rAAV/Her-2/neu loading of dendritic cells for a potent cellular-mediated MHC class I restricted immune response against ovarian cancer. Viral Immunol. 2008, 21, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tang, Y.; Liu, Y.; Guo, H.; Wang, Y.; Cai, L.; Li, Y.; Wang, B. Murine double minute 2 siRNA and wild-type p53 gene therapy enhances sensitivity of the SKOV3/DDP ovarian cancer cell line to cisplatin chemotherapy in vitro and in vivo. Cancer Lett. 2014, 343, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Iyer, A.K.; Singh, A.; Milane, L.; Choy, E.; Hornicek, F.J.; Amiji, M.M.; Duan, Z. Cluster of Differentiation 44 Targeted Hyaluronic Acid Based Nanoparticles for MDR1 siRNA Delivery to Overcome Drug Resistance in Ovarian Cancer. Pharm. Res. 2015, 32, 2097–2109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Garbuzenko, O.B.; Reuhl, K.R.; Rodriguez-Rodriguez, L.; Minko, T. Two-in-one: Combined targeted chemo and gene therapy for tumor suppression and prevention of metastases. Nanomedicine 2012, 7, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Rein, D.T.; Volkmer, A.; Bauerschmitz, G.; Beyer, I.M.; Janni, W.; Fleisch, M.C.; Welter, A.K.; Bauerschlag, D.; Schöndorf, T.; Breidenbach, M. Combination of a MDR1-targeted replicative adenovirus and chemotherapy for the therapy of pretreated ovarian cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Vivas-Mejia, P.E.; Rodriguez-Aguayo, C.; Han, H.-D.; Shahzad, M.M.K.; Valiyeva, F.; Shibayama, M.; Chavez-Reyes, A.; Sood, A.K.; Lopez-Berestein, G. Silencing survivin splice variant 2B leads to antitumor activity in taxane--resistant ovarian cancer. Clin. Cancer Res. 2011, 17, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Luo, R.-Y.; Yang, J.; Cheng, Y.-X. Knockdown of survivin contributes to antitumor activity in cisplatin-resistant ovarian cancer cells. Mol. Med. Rep. 2013, 7, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Salzano, G.; Navarro, G.; Trivedi, M.S.; De Rosa, G.; Torchilin, V.P. Multifunctional Polymeric Micelles Co-loaded with Anti-Survivin siRNA and Paclitaxel Overcome Drug Resistance in an Animal Model of Ovarian Cancer. Mol. Cancer Ther. 2015, 14, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Sun, Y.; Shen, M.; Song, K.; Yin, X.; Di, W.; Duan, Y. Enhanced Chemotherapeutic Efficacy of Paclitaxel Nanoparticles Co-delivered with MicroRNA-7 by Inhibiting Paclitaxel-Induced EGFR/ERK pathway Activation for Ovarian Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 7821–7831. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, L.; Sui, Y.-X.; Han, L.-L.; Liu, J.-H. Circadian Gene CLOCK Affects Drug-Resistant Gene Expression and Cell Proliferation in Ovarian Cancer SKOV3/DDP Cell Lines Through Autophagy. Cancer Biother. Radiopharm. 2017, 32, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-Y.; Li, P.-L.; Xu, A.; Zhang, X.-C. Involvement of GRP78 in the Resistance of Ovarian Carcinoma Cells to Paclitaxel. Asian Pac. J. Cancer Prev. 2015, 16, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guo, Y.; Han, L.; Duan, Y.; Fang, F.; Niu, S.; Ba, Q.; Zhu, H.; Kong, F.; Lin, C.; et al. In vitro and in vivo growth inhibition of drug-resistant ovarian carcinoma cells using a combination of cisplatin and a TRAIL-encoding retrovirus. Oncol. Lett. 2012, 4, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, J.R.; Gottschlich, L.; Riehl, D.; Rusch, T.; Koch, V.; Friedrich, K.; Hutzler, S.; Prüfer, S.; Friedel, T.; Hanschmann, K.-M.; et al. Enhanced lysis by bispecific oncolytic measles viruses simultaneously using HER2/neu or EpCAM as target receptors. Mol. Ther. Oncol. 2016, 3, 16003. [Google Scholar] [CrossRef] [PubMed]
- Jennings, V.A.; Ilett, E.J.; Scott, K.J.; West, E.J.; Vile, R.; Pandha, H.; Harrington, K.; Young, A.; Hall, G.D.; Coffey, M.; et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int. J. Cancer 2014, 134, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing oncolytic herpes simplex virus promotes anti-tumor activity and immunologic control of metastatic ovarian cancer in mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Goshima, F.; Esaki, S.; Luo, C.; Kamakura, M.; Kimura, H.; Nishiyama, Y. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int. J. Cancer 2014, 134, 2865–2877. [Google Scholar] [CrossRef] [PubMed]
- Dold, C.; Rodriguez Urbiola, C.; Wollmann, G.; Egerer, L.; Muik, A.; Bellmann, L.; Fiegl, H.; Marth, C.; Kimpel, J.; von Laer, D. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol. Ther. Oncol. 2016, 3, 16021. [Google Scholar] [CrossRef] [PubMed]
- Nounamo, B.; Liem, J.; Cannon, M.; Liu, J. Myxoma Virus Optimizes Cisplatin for the Treatment of Ovarian Cancer In Vitro and in a Syngeneic Murine Dissemination Model. Mol. Ther. Oncol. 2017, 6, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shu, J.; Chen, L.; Chen, X.; Zhao, J.; Li, S.; Mou, X.; Tong, X. Synergistic suppression effect on tumor growth of ovarian cancer by combining cisplatin with a manganese superoxide dismutase-armed oncolytic adenovirus. OncoTargets Ther. 2016, 9, 6381–6388. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Bossow, S.; Lampe, J.; Zimmermann, M.; Taran, F.-A.; Wallwiener, D.; Fehm, T.; Bitzer, M.; Lauer, U.M. Enhanced killing of ovarian carcinoma using oncolytic measles vaccine virus armed with a yeast cytosine deaminase and uracil phosphoribosyltransferase. Gynecol. Oncol. 2013, 130, 362–368. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 29 June 2018).
- Anwer, K.; Barnes, M.N.; Fewell, J.; Lewis, D.H.; Alvarez, R.D. Phase-I clinical trial of IL-12 plasmid/lipopolymer complexes for the treatment of recurrent ovarian cancer. Gene Ther. 2010, 17, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Anwer, K.; Kelly, F.J.; Chu, C.; Fewell, J.G.; Lewis, D.; Alvarez, R.D. Phase I trial of a formulated IL-12 plasmid in combination with carboplatin and docetaxel chemotherapy in the treatment of platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol. 2013, 131, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.D.; Sill, M.W.; Davidson, S.A.; Muller, C.Y.; Bender, D.P.; DeBernardo, R.L.; Behbakht, K.; Huh, W.K. A phase II trial of intraperitoneal EGEN-001, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: A gynecologic oncology group study. Gynecol. Oncol. 2014, 133, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Thaker, P.H.; Brady, W.E.; Lankes, H.A.; Odunsi, K.; Bradley, W.H.; Moore, K.N.; Muller, C.Y.; Anwer, K.; Schilder, R.J.; Alvarez, R.D.; et al. A phase I trial of intraperitoneal GEN-1, an IL-12 plasmid formulated with PEG-PEI-cholesterol lipopolymer, administered with pegylated liposomal doxorubicin in patients with recurrent or persistent epithelial ovarian, fallopian tube or primary peritoneal. Gynecol. Oncol. 2017, 147, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Dmitriev, I.; O’Malley, J.P.; Wang, M.; Saddekni, S.; You, Z.; Preuss, M.A.; Harris, R.D.; Aurigemma, R.; Siegal, G.P.; et al. A phase I clinical trial of Ad5.SSTR/TK.RGD, a novel infectivity-enhanced bicistronic adenovirus, in patients with recurrent gynecologic cancer. Clin. Cancer Res. 2012, 18, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Dmitriev, I.P.; Saddekni, S.; Kashentseva, E.A.; Harris, R.D.; Aurigemma, R.; Bae, S.; Singh, K.P.; Siegal, G.P.; Curiel, D.T.; et al. A phase I clinical trial of Ad5/3-Δ24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecol. Oncol. 2013, 130, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Kimball, K.J.; Preuss, M.A.; Barnes, M.N.; Wang, M.; Siegal, G.P.; Wan, W.; Kuo, H.; Saddekni, S.; Stockard, C.R.; Grizzle, W.E.; et al. A phase I study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin. Cancer Res. 2010, 16, 5277–5287. [Google Scholar] [CrossRef] [PubMed]
- Ozga, M.; Aghajanian, C.; Myers-Virtue, S.; McDonnell, G.; Jhanwar, S.; Hichenberg, S.; Sulimanoff, I. A systematic review of ovarian cancer and fear of recurrence. Palliat. Support. Care 2015, 13, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Klemba, A.; Purzycka-Olewiecka, J.K.; Wcisło, G.; Czarnecka, A.M.; Lewicki, S.; Lesyng, B.; Szczylik, C.; Kieda, C. Surface markers of cancer stem-like cells of ovarian cancer and their clinical relevance. Contemp. Oncol. 2018, 22, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Lupia, M.; Cavallaro, U. Ovarian cancer stem cells: Still an elusive entity? Mol. Cancer 2017, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wu, G. Could ALDH2*2 be the reason for low incidence and mortality of ovarian cancer for East Asia women? Oncotarget 2018, 9, 12503–12512. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medcine 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Sajdak, S.; Markowska, J.; Huczyński, A. Angiogenesis and cancer stem cells: New perspectives on therapy of ovarian cancer. Eur. J. Med. Chem. 2017, 142, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, T.; Cui, M. Are ovarian cancer stem cells the target for innovative immunotherapy? OncoTargets Ther. 2018, 11, 2615–2626. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.; Yang, R.; Lu, W.; Zhu, W.; Zhou, J.; Zheng, C.; Zhou, D.; Yu, L.; Wu, J. Adenovirus-mediated truncated Bid overexpression induced by the Cre/LoxP system promotes the cell apoptosis of CD133+ ovarian cancer stem cells. Oncol. Rep. 2017, 37, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.; Jiang, L.; Liang, S.; Kwong, J.; Yang, L.; Li, Y.; PingYin; Deng, Q.; Liang, Z. Nanog interaction with the androgen receptor signaling axis induce ovarian cancer stem cell regulation: Studies based on the CRISPR/Cas9 system. J. Ovarian Res. 2018, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Kenda Suster, N.; Virant-Klun, I.; Frkovic Grazio, S.; Smrkolj, S. The significance of the pluripotency and cancer stem cell-related marker NANOG in diagnosis and treatment of ovarian carcinoma. Eur. J. Gynaecol. Oncol. 2016, 37, 604–612. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Gene/System | Strong Points | Weak Points |
|---|---|---|---|
| Tumor Suppressor gene | p53 | Altered gene in a high percentage of OC. The use of Gendicine (a recombinant human Ad-p53) has been approved in the treatment of OC in China. Several clinical trials are currently ongoing. | Not useful in cells with a normal p53 gene. No therapeutic benefit in the first clinical trials due to the use of a wrong delivery system. |
| WWOX | Promising results in vivo Inhibition of proliferation and promotion of apoptosis in OCSC. | It has not been evaluated in clinical trials in OC. | |
| Oncofactor inhibition strategies | EGFR | Gene widely studied in cancer. Its use increases the sensitivity to chemotherapy | It has not been evaluated in vivo or in clinical trials in OC. |
| CLDN3 | Several strategies of silencing have been studied in OC (siRNA and shRNA). Promising results in vivo, with inhibition of malignant ascites formation. | It has not been evaluated in clinical trials in OC. | |
| Suicide gene therapy | HVS-TK | Strategy widely studied in gene therapy for cancer. Several clinical trials are currently ongoing. Promising results in vivo and in clinical trials. Bystander effect. | Not phase 2 or 3 clinical trials published. |
| DT-A | Promising results in vivo, with minimal cytotoxicity. | It has not been evaluated in clinical trials in OC. | |
| Antiangiogenic gene therapy | VEGFRs | Promising results in vivo, with inhibition of ascities formation. Synergist effect in combination with chemotherapy. | It has not been evaluated in clinical trials in OC. The work showed increased proliferation of tumour cells with the use of VEGFR2 and Ti2 in combination with PTX and carboplatin. |
| Endostatin | Promising results in vivo. Clinical trials have been carried out in other types of cancer. | Transient expression due to humoral immune response if adenovirus is used as delivery system. It has not been evaluated in clinical trials in OC. | |
| Genetic immunopotentiation | IL-12 | Potent immune-modulatory properties and ability to inhibit tumour angiogenesis. Promising results in vivo. Several clinical trials are currently ongoing or have been completed. It has showed to be safe and feasible in phase I trials. | Poor clinical benefits in the completed phase II trial. |
| CAR-T cell | Possibility of target any specific tumour antigen. Promising preliminary results in clinical trials in OC. | Important side effects caused in patients. | |
| Multi-Drug Resistance | MDR1 | Several strategies have been developed to knockdown it. Its silencing enhances sensitivity to anticancer drugs. Promising results in vivo. | It has not been evaluated in clinical trials in OC. |
| Survivin | Several strategies targeting this gene have been developed, including combined therapy with anticancer drugs. Promising results in vivo. | It has not been evaluated in clinical trials in OC. | |
| Oncolytic virotherapy | VSV | Can be genetically designed to deliver therapeutic genes. A phase I trial is currently ongoing. | Neurotoxicity and Induction of neutralizing antibodies. IFN response intact in most OC cells, blocking virus replication. |
| Gene | Function in Ovarian Cancer | Silencing Strategy | Model | Ref. |
|---|---|---|---|---|
| EGFR | Cell migration, proliferation and differentiation | siRNA carried in nanogels | In vitro | [64] |
| NOB1 | Protein degradation through ubiquitin proteasome pathway (maturation of the 20S proteasome) | shRNA carried in a lentiviral system | In vitro | [65] |
| MACC1 | Regulation of MET, which is involved in cellular growth and migration, angiogenesis, invasion and metastasis | shRNA plasmid | In vitro | [66] |
| MTA1 | Component of histone deacetylase 1 involved in transcriptional regulation. May enhance cell invasion, migration, adhesion and anoikis-resistance. | siRNA plasmid | In vitro | [67] |
| COX2 | Prostaglandin synthesis, involved in stimulation of proliferation and angiogenesis in cancer | siRNA and shRNA plasmids | In vitro and in vivo | [68,69] |
| WT1 | Proliferation and differentiation of the urogenital system | ASODN carried in liposomes | In vitro | [70] |
| STAT3 | Regulation of multiple oncogenes and suppressor gene expressions involved in cell proliferation and apoptosis and angiogenesis | shRNA carried in DOTAP-cholesterol liposomes | In vitro and in vivo | [71] |
| H1F-1α | Transcriptional regulator of the adaptive response to hypoxia by activation of genes involved in cell proliferation and migration, angiogenesis, apoptosis and glucose metabolism | siRNA through FA-PEG-COL nanoparticles | In vitro | [25] |
| CLDN3 | Component of tight junction (TJ) of epithelial cells and cancer cells, so is involved in invasion and metastasis | siRNA carried in lipidoid molecules, shRNA carried in PLGA-NPs, shRNA carried in F-P-LP | In vitro and in vivo | [72,73] |
| NOTCH1 | Cell development, proliferation, differentiation and apoptosis. | siRNA carried in cationic cholesterol derivative-based liposomes | In vitro | [27] |
| CD59 | Inhibition of cytolytic activity of complement | shRNA carried by a recombinant retrovirus | In vitro and in vivo | [74] |
| gDNMT1 | DNA methylation, involved in tumorigenesis, relapse and resistance of ovarian cancer. | CRISPR-Cas9 delivered by F-LP | In vitro and in vivo | [75] |
| Therapeutic Strategy | Intervention | Clinical Trial Reference | Phase | Year (First–Last Posted) |
|---|---|---|---|---|
| Suicide gene therapy | HSV-TK + GCV Vector: Adenovirus (Ad5.SSTR/TK.RGD) | NCT00964756 | Phase 1 | 2009–2013 |
| HSV-TK + GCV Vector: Vector producer cells (VPC) | NCT00005025 | Phase 2 | 2003–2013 | |
| CD + 5-FC Vector: Toca 511, a purified retroviral replicating vector encoding a modified yeast CD gene | NCT02576665 | Phase 1 | 2015–2018 | |
| Tumour suppressor gene | Inserting the p53 gene Vector: Adenovirus (Ad5CMV-p53) | NCT00003450 | Phase 1 | 2003–2009 |
| Inserting the p53 gene Vector: Adenovirus (Ad5CMV-p53) | NCT00003588 | Phase 1 | 2004–2013 | |
| Inserting the p53 gene + chemotherapy (PTX and carboplatin) Vector: Adenovirus (SCH-58500) | NCT00003880 | Phase 2 Phase 3 | 2004–2015 | |
| Inserting the p53 gene + chemotherapy (cisplatin and PTX) | NCT02435186 | Phase 2 | 2015–2015 | |
| Oncolytic virotherapy | LOAd703 (an oncolytic adenovirus serotype 5/35 encoding immunostimulatory transgenes: TMZ-CD40L and 41BBL) + chemotherapy or gemcitabine | NCT03225989 | Phase 1 Phase 2 | 2017–2018 |
| Recombinant carcinoembryonic antigen (CEA)-expressing measles virus (MV-CEA) and oncolytic measles virus encoding thyroidal sodium iodide symporter (MV-NIS) | NCT00408590 | Phase 1 | 2006–2018 | |
| Vesicular Stomatitis Virus expressing Human Interferon Beta and Sodium-Iodide Symporter (VSV-hIFNbeta-NIS) | NCT03120624 | Phase 1 | 2017–2018 | |
| Immunopotentiation | EGEN-001 (IL-12 Plasmid Formulated With PEG-PEI-Cholesterol Lipopolymer) + chemotherapy | NCT00473954 | Phase 1 | 2007–2013 |
| EGEN-001 (IL-12 Plasmid Formulated With PEG-PEI-Cholesterol Lipopolymer) | NCT00137865 | Phase 1 | 2005–2013 | |
| GEN-1 (IL-12 Plasmid Formulated With PEG-PEI-Cholesterol Lipopolymer) + chemotherapy (PTX and carboplatin) | NCT02480374 | Phase 1 | 2015–2018 | |
| NYESO-1(C259) transduced autologous T cells | NCT01567891 | Phase 1 Phase 2 | 2012–2018 | |
| TBI-1301 (Autologous T cells engineered to express a T cell receptor (TCR) targeting NY-ESO-1) + cyclophosphamide | NCT02869217 | Phase 1 | 2016–2017 | |
| TBI-1301 (Autologous T cells engineered to express a T cell receptor (TCR) targeting NY-ESO-1) + cyclophosphamide ± fludarabine | NCT02366546 | Phase 1 | 2015–2017 | |
| Autologous T cells engineered to express a T cell receptor (TCR) targeting NY-ESO-1 + cyclophosphamide + fludarabine | NCT02457650 | Phase 1 | 2015–2016 | |
| TBI-1201 (MAGE-A4-specific TCR gene transduced T lymphocytes) + cyclophosphamide ± fludarabine | NCT02096614 | Phase 1 | 2014–2017 | |
| Gen modified lymphocytes with MOv-gamma chimeric receptor gene (MOv-PBL) + IL-2 | NCT00019136 | Phase 1 | 2003–2015 | |
| TCR-Transduced PBL (T-Cells Genetically Engineered to Express T-Cell Receptors Reactive Against Mutated Neoantigens) | NCT03412877 | Phase 2 | 2018–2018 | |
| Anti-mesothelin CAR transduced PBL (retroviral vector that contains a chimeric T cell receptor (CAR) that recognizes mesothelin) + cyclophosphamide, fludarabine and aldesleukin | NCT01583686 | Phase 1 Phase 2 | 2012–2018 | |
| Anti-hCD70 CAR PBL (Transducing PBL with a chimeric antigen receptor (CAR) that engages CD70) + cyclophosphamide, fludarabine and aldesleukin | NCT02830724 | Phase 1 Phase 2 | 2016–2018 | |
| ZYC300 (vaccine which encodes the cytochrome P450 family member, CYP1B1, a known human tumor-associated antigen) + cyclophosphamide Vector: PGL-encapsulated plasmid DNA | NCT00381173 | Phase 1 | 2006–2013 | |
| Vigil (vaccine composed of autologous tumor cells which are transfected extracorporeally with a plasmid encoding for the gene for GM-CSF, an immune-stimulatory cytokine, and a bifunctional short hairpin RNA that targets furin, convertase responsible for activation of both TGβ1 and β2) + Atezolizumab | NCT03073525 | Phase 2 | 2017–2018 | |
| ALVAC(2)-NY-ESO-1 (M)/TRICOM vaccine + IDO1 inhibitor | NCT01982487 | Phase 1 Phase 2 | 2013–2013 | |
| ALVAC(2)-NY-ESO-1 (M)/TRICOM vaccine + sirolimus + GM-CSF | NCT01536054 | Phase 1 | 2012–2018 | |
| ALVAC(2)-NY-ESO-1 (M)/TRICOM vaccine + sargramostim | NCT00803569 | Phase 1 | 2008–2011 | |
| atezolizumab ± guadecitabine ± CDX-1401 vaccine (a vaccine composed of a human mAb specific for DEC-205 fused to the full-length tumor antigen NY-ESO-1) | NCT03206047 | Phase 1 Phase 2 | 2017–2018 | |
| p53MVA vaccine (modified vaccinia virus ankara vaccine expressing tumor protein p53) + gemcitabine hydrochloride | NCT02275039 | Phase 1 | 2014–2018 | |
| p53MVA vaccine + Pembrolizumab | NCT03113487 | Phase 2 | 2017–2018 | |
| p53 peptide vaccine + ISA-51 + IL-2 ± GM-CSF | NCT00001827 | Phase 2 | 1999–2017 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Áyen, Á.; Jiménez Martínez, Y.; Marchal, J.A.; Boulaiz, H. Recent Progress in Gene Therapy for Ovarian Cancer. Int. J. Mol. Sci. 2018, 19, 1930. https://doi.org/10.3390/ijms19071930
Áyen Á, Jiménez Martínez Y, Marchal JA, Boulaiz H. Recent Progress in Gene Therapy for Ovarian Cancer. International Journal of Molecular Sciences. 2018; 19(7):1930. https://doi.org/10.3390/ijms19071930
Chicago/Turabian StyleÁyen, Ángela, Yaiza Jiménez Martínez, Juan A. Marchal, and Houria Boulaiz. 2018. "Recent Progress in Gene Therapy for Ovarian Cancer" International Journal of Molecular Sciences 19, no. 7: 1930. https://doi.org/10.3390/ijms19071930
APA StyleÁyen, Á., Jiménez Martínez, Y., Marchal, J. A., & Boulaiz, H. (2018). Recent Progress in Gene Therapy for Ovarian Cancer. International Journal of Molecular Sciences, 19(7), 1930. https://doi.org/10.3390/ijms19071930