The Role of miRNAs in Virus-Mediated Oncogenesis


Abstract

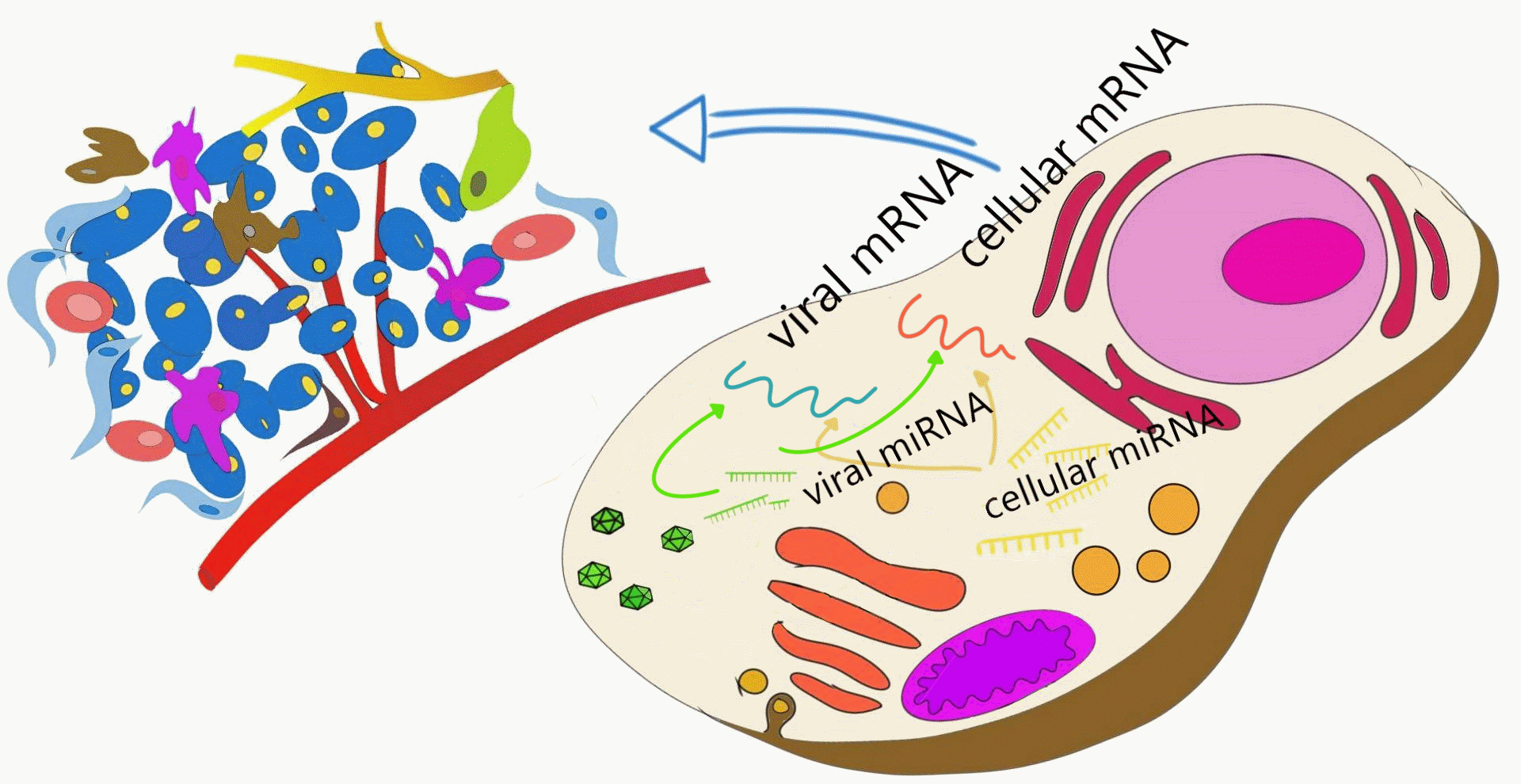
1. Introduction
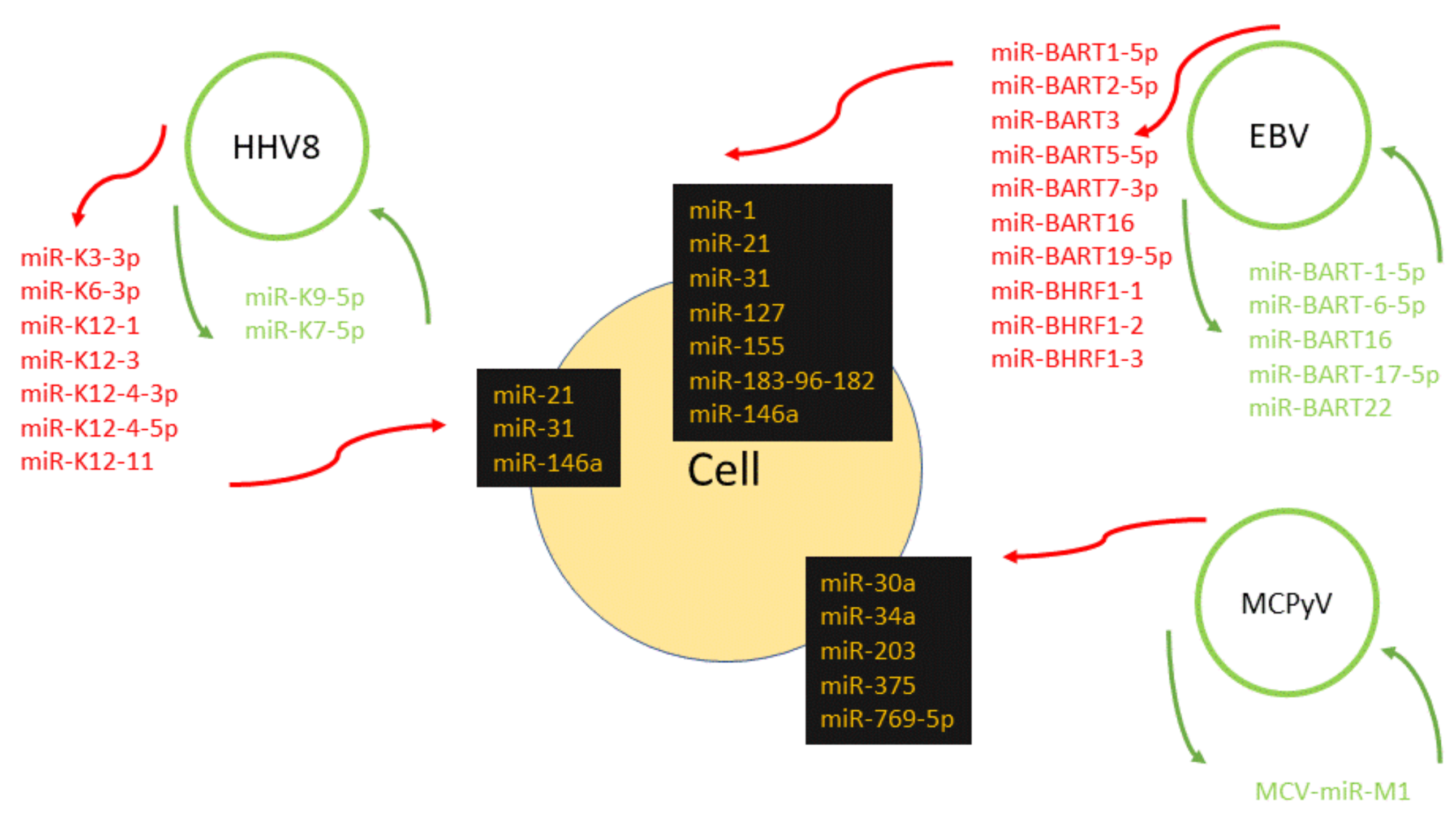
2. Epstein–Barr Virus
3. HHV-8
4. Hepatitis B Virus
5. Human Papillomavirus
6. Merkel Cell Polyomavirus
7. Hepatitis C Virus
8. Retroviruses
9. Controversial Oncoviral-Encoded miRNAs
10. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Glossary
| Immortalization of the host cell | continual proliferation of the cell mostly caused by mutation or by the activity of the viral oncogenes |
| Transformation of the host cell | morphologic, physiologic and genetic changes of the cell initiating a process of tumor development and progression |
References
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burdenf cancers attributable tonfectionsn 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarksf cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarksf cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A reviewf human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networksn cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viruses and microRNAs: RISCynteractions with serious consequences. Genes Dev. 2011, 25, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particlesn Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Mui, U.N.; Haley, C.T.; Tyring, S.K. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, A.; Glavina Durdov, M.; Capkun, V.; Jakelic Pitesa, J.; Bozic Sakic, M. Classical Hodgkin Lymphoma with Positive Epstein-Barr Virus Statuss Associated with More FOXP3 Regulatory T Cells. Med. Sci. Monit. 2016, 22, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Dotti, G. EBV-associated lymphoproliferativeisorders: Classification and treatment. Oncologist 2008, 13, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Jacome, A.A.; Lima, E.M.; Kazzi, A.I.; Chaves, G.F.; Mendonca, D.C.; Maciel, M.M.; Santos, J.S. Epstein-Barr virus-positive gastric cancer: Aistinct molecular subtypef theisease? Rev. Soc. Bras. Med. Trop. 2016, 49, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Chen, K.; Young, K.H. Epstein-Barr virus-positive T/NK-cell lymphoproliferativeisorders. Exp. Mol. Med. 2015, 47, e133. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2As a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Swanson-Mungerson, M.; Bultema, R.; Longnecker, R. Epstein-Barr virus LMP2Amposes sensitivity to apoptosis. J. Gen. Virol. 2010, 91, 2197–2202. [Google Scholar] [CrossRef] [PubMed]
- Incrocci, R.; Barse, L.; Stone, A.; Vagvala, S.; Montesano, M.; Subramaniam, V.; Swanson-Mungerson, M. Epstein-Barr Virus Latent Membrane Protein 2A (LMP2A) enhances IL-10 production through the activationf Bruton’s tyrosine kinase and STAT3. Virology 2017, 500, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Sivachandran, N.; Wang, X.; Frappier, L. Functionsf the Epstein-Barr virus EBNA1 proteinn viral reactivation and lyticnfection. J. Virol. 2012, 86, 6146–6158. [Google Scholar] [CrossRef] [PubMed]
- Sivachandran, N.; Sarkari, F.; Frappier, L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma throughisruptionf PML nuclear bodies. PLoS Pathog. 2008, 4, e1000170. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The proto-oncogene c-mycs airect target genef Epstein-Barr virus nuclear antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [PubMed]
- Bhattacharjee, S.; Ghosh Roy, S.; Bose, P.; Saha, A. Rolef EBNA-3 Family Proteinsn EBV Associated B-cell Lymphomagenesis. Front. Microbiol. 2016, 7, 457. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identificationf virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Sullivan, C.S. Virus-encoded microRNAs: Anverview and a look to the future. PLoS Pathog. 2012, 8, e1003018. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulationf LMP1 protein expression by EBV-encoded microRNAs. Proc. Natl. Acad. Sci. USA 2007, 104, 16164–16169. [Google Scholar] [CrossRef] [PubMed]
- Lung, R.W.; Tong, J.H.; Sung, Y.M.; Leung, P.S.; Ng, D.C.; Chau, S.L.; Chan, A.W.; Ng, E.K.; Lo, K.W.; To, K.F. Modulationf LMP2A expression by a newlydentified Epstein-Barr virus-encoded microRNA miR-BART22. Neoplasia 2009, 11, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jaker, C.; Hock, J.; Meister, G.; Grasser, F.A. Epstein-Barr virus-encoded microRNA miR-BART2own-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; et al. Editingf Epstein-Barr virus-encoded BART6 microRNAs controls theiricer targeting and consequently affects viral latency. J. Biol. Chem. 2010, 285, 33358–33370. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.M.; Lyu, X.M.; Luo, W.R.; Cui, X.F.; Ye, Y.F.; Yuan, C.C.; Peng, Q.X.; Wu, D.H.; Liu, T.F.; Wang, E.; et al. EBV-miR-BART7-3p promotes the EMT and metastasisf nasopharyngeal carcinoma cells by suppressing the tumor suppressor PTEN. Oncogene 2015, 34, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Vereide, D.T.; Seto, E.; Chiu, Y.F.; Hayes, M.; Tagawa, T.; Grundhoff, A.; Hammerschmidt, W.; Sugden, B. Epstein-Barr virus maintains lymphomas viats miRNAs. Oncogene 2014, 33, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Yuen, K.S.; Tsao, S.W.; Chen, H.; Kok, K.H.; Jin, D.Y. Perturbationf biogenesis and targetingf Epstein-Barr virus-encoded miR-BART3 microRNA by adenosine-to-inosine editing. J. Gen. Virol. 2013, 94, 2739–2744. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV microRNAsn primary lymphomas and targetingf CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Haar, J.; Bernhardt, K.; Linnstaedt, S.D.; Bannert, H.; Lips, H.; Cullen, B.R.; Delecluse, H.J. The membersf an Epstein-Barr virus microRNA cluster cooperate to transform B lymphocytes. J. Virol. 2011, 85, 9801–9810. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; Linnstaedt, S.D.; Esoda, C.; Krisko, J.F.; Martinez-Torres, F.; Delecluse, H.J.; Cullen, B.R.; Garcia, J.V. A clusterf virus-encoded microRNAs accelerates acute systemic Epstein-Barr virusnfection butoes not significantly enhance virus-inducedncogenesisn vivo. J. Virol. 2013, 87, 5437–5446. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-inducedmmune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.W.; Choi, Y.; Kwon, O.K.; Lee, S.S.; Chung, H.Y.; Yu, W.; Bae, H.I.; Seo, A.N.; Kang, H.; Lee, S.K.; et al. High levelf viral microRNA-BART20-5p expressions associated with worse survivalf patients with Epstein-Barr virus-associated gastric cancer. Oncotarget 2017, 8, 14988–14994. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Li, Z.H.; Chen, S.; Chan, J.Y.; Yin, M.; Zhang, M.J.; Wong, T.S. Epstein-Barr virus encoded microRNA BART7 regulates radiation sensitivityf nasopharyngeal carcinoma. Oncotarget 2017, 8, 20297–20308. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Godshalk, S.E.; Bhaduri-McIntosh, S.; Slack, F.J. Epstein-Barr virus-mediatedysregulationf human microRNA expression. Cell Cycle 2008, 7, 3595–3600. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.C.; Chung, G.T.; Lun, S.W.; To, K.F.; Choy, K.W.; Lau, K.M.; Siu, S.P.; Guan, X.Y.; Ngan, R.K.; Yip, T.T.; et al. miR-31s consistentlynactivatedn EBV-associated nasopharyngeal carcinoma and contributes tots tumorigenesis. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, X.; Wang, Y.; Sun, Y.; Zheng, J.; Zhu, D. MiR-155 up-regulation by LMP1 DNA contributes toncreased nasopharyngeal carcinoma cell proliferation and migration. Eur. Arch. Otorhinolaryngol. 2014, 271, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Linnstaedt, S.D.; Gottwein, E.; Skalsky, R.L.; Luftig, M.A.; Cullen, B.R. Virallynduced cellular microRNA miR-155 plays a key rolen B-cellmmortalization by Epstein-Barr virus. J. Virol. 2010, 84, 11670–11678. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, Q.; Hu, C.M. Epstein-Barr virus-encoded LMP1ncreases miR-155 expression, which promotes radioresistancef nasopharyngeal carcinoma via suppressing UBQLN1. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4507–4515. [Google Scholar] [PubMed]
- Du, Z.M.; Hu, L.F.; Wang, H.Y.; Yan, L.X.; Zeng, Y.X.; Shao, J.Y.; Ernberg, I. Upregulationf MiR-155n nasopharyngeal carcinomas partlyriven by LMP1 and LMP2A andownregulates a negative prognostic marker JMJD1A. PLoS ONE 2011, 6, e19137. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Sekizuka, T.; Uehara, T.; Hishima, T.; Mine, S.; Fukumoto, H.; Sato, Y.; Hasegawa, H.; Kuroda, M.; Katano, H. Next-generation sequencingf miRNAsn clinical samplesf Epstein-Barr virus-associated B-cell lymphomas. Cancer Med. 2017, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Onnis, A.; Cocco, M.; De Falco, G.; Imperatore, F.; Giuseppina, A.; Costanzo, V.; Cerino, G.; Mannucci, S.; Cantisani, R.; et al. B-cellifferentiationn EBV-positive Burkitt lymphomasmpaired at posttranscriptional level by miRNA-altered expression. Int. J. Cancer 2010, 126, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.; Anastasiadou, E.; Garg, N.; Lenze, D.; Boccellato, F.; Vincenti, S.; Severa, M.; Coccia, E.M.; Bigi, R.; Cirone, M.; et al. Differential regulationf miR-21 and miR-146a by Epstein-Barr virus-encoded EBNA2. Leukemia 2012, 26, 2343–2352. [Google Scholar] [CrossRef] [PubMed]
- Oussaief, L.; Fendri, A.; Chane-Woon-Ming, B.; Poirey, R.; Delecluse, H.J.; Joab, I.; Pfeffer, S. Modulationf MicroRNA Cluster miR-183-96-182 Expression by Epstein-Barr Virus Latent Membrane Protein 1. J. Virol. 2015, 89, 12178–12188. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, J.; Zhong, J.; Huang, Z.; Luo, X.; Huang, Y.; Feng, S.; Shao, J.; Liu, D. miR-1, regulated by LMP1, suppresses tumour growth and metastasis by targeting K-rasn nasopharyngeal carcinoma. Int. J. Exp. Pathol. 2015, 96, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identificationf herpesvirus-like DNA sequencesn AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: Aistinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L. Kaposi’s sarcoma-associated herpesvirus-like DNA sequencesn multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an arrayf viral microRNAsn latentlynfected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Bellare, P.; Ganem, D. Regulationf KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, D.; He, Z.; Deng, Q.; Robertson, E.S.; Lan, K. miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS ONE 2011, 6, e16224. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Li, Z.; Chu, C.Y.; Feng, J.; Feng, J.; Sun, R.; Rana, T.M. MicroRNAs encoded by Kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 2010, 11, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jia, X.; Shen, C.; Zhang, M.; Xu, J.; Shang, Y.; Zhu, K.; Hu, M.; Yan, Q.; Qin, D.; et al. A KSHV microRNA enhances viral latency andnduces angiogenesis by targeting GRK2 to activate the CXCR2/AKT pathway. Oncotarget 2016, 7, 32286–32305. [Google Scholar] [CrossRef] [PubMed]
- Plaisance-Bonstaff, K.; Choi, H.S.; Beals, T.; Krueger, B.J.; Boss, I.W.; Gay, L.A.; Haecker, I.; Hu, J.; Renne, R. KSHV miRNAsecrease expressionf lytic genesn latentlynfected PEL and endothelial cells by targeting host transcription factors. Viruses 2014, 6, 4005–4023. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, V.; Nicot, C.; Gessain, A.; Valensi, F.; Gabarre, J.; Matta, H.; Chaudhary, P.M.; Mahieux, R. In primary effusion lymphoma cells, MYB transcriptional repressions associated with v-FLIP expressionuring latent KSHVnfection while both v-FLIP and v-GPCR becomenvolveduring the lytic cycle. Br. J. Haematol. 2007, 138, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Gao, Y.; Lin, X.; He, Z.; Zhao, Q.; Deng, Q.; Lan, K. A human herpesvirus miRNA attenuatesnterferon signaling and contributes to maintenancef viral latency by targeting IKKepsilon. Cell Res. 2011, 21, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Stedman, W.; Yousef, M.; Renne, R.; Lieberman, P.M. Epigenetic regulationf Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 2010, 84, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Skalsky, R.L.; Maldonado, A.M.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Identificationf cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yan, Q.; Ding, X.; Shen, C.; Hu, M.; Zhu, Y.; Qin, D.; Lu, H.; Krueger, B.J.; Renne, R.; et al. The SH3BGR/STAT3 Pathway Regulates Cell Migration and Angiogenesis Induced by a Gammaherpesvirus MicroRNA. PLoS Pathog. 2016, 12, e1005605. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, W.; Qin, J.; Lu, C.; Fan, W. Kaposi’s sarcoma-associated herpesvirus (KSHV)-encoded microRNAs promote matrix metalloproteinases (MMPs) expression and pro-angiogenic cytokine secretionn endothelial cells. J. Med. Virol. 2017, 89, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ramosa Silva, S.; He, M.; Liang, Q.; Lu, C.; Feng, P.; Jung, J.U.; Gao, S.J. An Oncogenic Virus Promotes Cell Survival and Cellular Transformation by Suppressing Glycolysis. PLoS Pathog. 2016, 12, e1005648. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Cullen, B.R. A human herpesvirus microRNAnhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 2010, 84, 5229–5237. [Google Scholar] [CrossRef] [PubMed]
- Suffert, G.; Malterer, G.; Hausser, J.; Viiliainen, J.; Fender, A.; Contrant, M.; Ivacevic, T.; Benes, V.; Gros, F.; Voinnet, O.; et al. Kaposi’s sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog. 2011, 7, e1002405. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Samols, M.A.; Plaisance, K.B.; Boss, I.W.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Kaposi’s sarcoma-associated herpesvirus encodes anrthologf miR-155. J. Virol. 2007, 81, 12836–12845. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as anrthologuef cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.H.; Wu, M.F.; Wu, Y.H.; Chang, S.J.; Lin, S.F.; Sharp, T.V.; Wang, H.W. The M type K15 proteinf Kaposi’s sarcoma-associated herpesvirus regulates microRNA expression viats SH2-binding motif tonduce cell migration andnvasion. J. Virol. 2009, 83, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Punj, V.; Matta, H.; Schamus, S.; Tamewitz, A.; Anyang, B.; Chaudhary, P.M. Kaposi’s sarcoma-associated herpesvirus-encoded viral FLICEnhibitory protein (vFLIP) K13 suppresses CXCR4 expression by upregulating miR-146a. Oncogene 2010, 29, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.A.M.; Akula, S.M. Profilingf cellular microRNA responsesuring the early stagesf KSHVnfection. Arch. Virol. 2017, 162, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Spada, E.; Mele, A.; Caserta, C.A.; Pulsoni, A. The associationf hepatitis B virusnfection with B-cell non-Hodgkin lymphoma—A review. Am. J. Blood Res. 2012, 2, 18–28. [Google Scholar] [PubMed]
- Ye, Y.F.; Xiang, Y.Q.; Fang, F.; Gao, R.; Zhang, L.F.; Xie, S.H.; Liu, Z.; Du, J.L.; Chen, S.H.; Hong, M.H.; et al. Hepatitis B virusnfection and riskf nasopharyngeal carcinoman southern China. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Ghosh, S.; Dasgupta, D.; Ghosh, A.; Datta, S.; Sikdar, N.; Datta, S.; Chowdhury, A.; Banerjee, S. Hepatitis B Virus X Protein Upregulates hELG1/ ATAD5 Expression through E2F1n Hepatocellular Carcinoma. Int. J. Biol. Sci. 2016, 12, 30–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geng, M.; Xin, X.; Bi, L.Q.; Zhou, L.T.; Liu, X.H. Molecular mechanismf hepatitis B virus X protein functionn hepatocarcinogenesis. World J. Gastroenterol. 2015, 21, 10732–10738. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, J.; Steel, L.F.; Bouchard, M.J. Hepatitis B virus and microRNAs: Complexnteractions affecting hepatitis B virus replication and hepatitis B virus-associatediseases. World J. Gastroenterol. 2015, 21, 7375–7399. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Cacciola, I.; Saffioti, F.; Raimondo, G. Hepatitis B virus PreS/S gene variants: Pathobiology and clinicalmplications. J. Hepatol. 2014, 61, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Lee, J. Hepatitis B virusntegration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral-human chimeric transcript predisposes risk to liver cancerevelopment and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanismsf HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.G.; Wang, C.M.; Tian, C.; Wang, Y.; Li, L.; Sun, W.S.; Li, R.F.; Liu, Y.G. miR-122nhibits viral replication and cell proliferationn hepatitis B virus-related hepatocellular carcinoma and targets NDRG3. Oncol. Rep. 2011, 26, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Wang, S.; Wu, B.; Hao, J.; Fan, H.; Ju, Y.; Ding, Y.; Chen, L.; Chu, X.; et al. Hepatitis B virus mRNA-mediated miR-122nhibition upregulates PTTG1-binding protein, which promotes hepatocellular carcinoma tumor growth and cellnvasion. J. Virol. 2013, 87, 2193–2205. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Gramantieri, L.; Giovannini, C.; Veronese, A.; Ferracin, M.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Croce, C.M.; Tavolari, S.; et al. MiR-122/cyclin G1nteraction modulates p53 activity and affectsoxorubicin sensitivityf human hepatocarcinoma cells. Cancer Res. 2009, 69, 5761–5767. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Han, C.; Zhang, J.; Lu, D.; Dash, S.; Feitelson, M.; Lim, K.; Wu, T. Epigenetic regulationf MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X proteinn hepatocellular carcinoma cells. Hepatology 2013, 58, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Zhang, J.; Zhang, S.; Shan, C.; Ye, L.; Zhang, X. Upregulated microRNA-29a by hepatitis B virus X protein enhances hepatoma cell migration by targeting PTENn cell culture model. PLoS ONE 2011, 6, e19518. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, X.J.; Shi, K.Q.; Chen, Y.P.; Ren, Y.F.; Song, Y.J.; Li, G.; Xue, Y.F.; Fang, Y.X.; Deng, Z.J.; et al. Hepatitis B surface antigennhibits MICA and MICB expression vianductionf cellular miRNAsn hepatocellular carcinoma cells. Carcinogenesis 2014, 35, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Xiang, T.; Ren, G.; Tan, C.; Liu, R.; Xu, X.; Wu, Z. miR-101sown-regulated by the hepatitis B virus x protein andnduces aberrant DNA methylation by targeting DNA methyltransferase 3A. Cell Signal. 2013, 25, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, Y.; Guo, Y.; Sun, S. Down-regulated microRNA-152nduces aberrant DNA methylationn hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology 2010, 52, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Tan, C.; Tang, C.; Ren, G.; Xiang, T.; Qiu, Z.; Liu, R.; Wu, Z. Epigenetic repressionf miR-132 expression by the hepatitis B virus x proteinn hepatitis B virus-related hepatocellular carcinoma. Cell Signal. 2013, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, Y.; Toh, S.T.; Sung, W.K.; Tan, P.; Chow, P.; Chung, A.Y.; Jooi, L.L.; Lee, C.G. Lethal-7sown-regulated by the hepatitis B virus x protein and targets signal transducer and activatorf transcription 3. J. Hepatol. 2010, 53, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RASs regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA let-7aown-regulates MYC and reverts MYC-induced growthn Burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Liu, F.; Zhang, T.; Lv, N.; Liu, Q.; Shan, C.; Du, Y.; Kong, G.; Wang, T.; Ye, L.; et al. Hepatitis B virus X protein upregulates Lin28A/Lin28B through Sp-1/c-Myc to enhance the proliferationf hepatoma cells. Oncogene 2014, 33, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28Bnhibit let-7 microRNA biogenesis byistinct mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, Y.; Mishima, T.; Yokomuro, S.; Arima, Y.; Kawahigashi, Y.; Shigehara, K.; Kanda, T.; Yoshida, H.; Uchida, E.; Tajiri, T.; et al. Sequencing and bioinformatics-based analysesf the microRNA transcriptomen hepatitis B-related hepatocellular carcinoma. PLoS ONE 2011, 6, e15304. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dong, F.; Xu, Z.; Sharma, S.; Hu, X.; Chen, D.; Zhang, L.; Zhang, J.; Dong, Q. MicroRNA profilen HBV-inducednfection and hepatocellular carcinoma. BMC Cancer 2017, 17, 805. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, L.; Ji, X.; Yang, B.; Zhang, Y.; Fu, X.D. Hepatitis B viral RNAirectly mediatesown-regulationf the tumor suppressor microRNA miR-15a/miR-16-1n hepatocytes. J. Biol. Chem. 2013, 288, 18484–18493. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Kim, J.W.; Park, S.J.; Min, B.Y.; Jang, E.S.; Kim, N.Y.; Jeong, S.H.; Shin, C.M.; Lee, S.H.; Park, Y.S.; et al. c-Myc-mediatedverexpressionf miR-17-92 suppresses replicationf hepatitis B virusn human hepatoma cells. J. Med. Virol. 2013, 85, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shen, A.; Rider, P.J.; Yu, Y.; Wu, K.; Mu, Y.; Hao, Q.; Liu, Y.; Gong, H.; Zhu, Y.; et al. A liver-specific microRNA binds to a highly conserved RNA sequencef hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J. 2011, 25, 4511–4521. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Hummel, M.; Iftner, T.; Laimins, L.A. The E8E2C protein, a negative regulatorf viral transcription and replication, is required for extrachromosomal maintenancef human papillomavirus type 31n keratinocytes. J. Virol. 2000, 74, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meyers, C.; Wang, H.K.; Chow, L.T.; Zheng, Z.M. Constructionf a full transcription mapf human papillomavirus type 18uring productive viralnfection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef] [PubMed]
- Lace, M.J.; Anson, J.R.; Thomas, G.S.; Turek, L.P.; Haugen, T.H. The E8–E2 gene productf human papillomavirus type 16 represses early transcription and replication butsispensable for viral plasmid persistencen keratinocytes. J. Virol. 2008, 82, 10841–10853. [Google Scholar] [CrossRef] [PubMed]
- Petti, L.; Nilson, L.A.; DiMaio, D. Activationf the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. EMBO J. 1991, 10, 845–855. [Google Scholar] [PubMed]
- Bravo, I.G.; Alonso, A. Mucosal human papillomaviruses encode fourifferent E5 proteins whose chemistry and phylogeny correlate with malignantr benign growth. J. Virol. 2004, 78, 13613–13626. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Herrero, R.; Desalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicityf human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6ncoprotein encoded by human papillomavirus types 16 and 18 promotes theegradationf p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Mortensen, F.; Schneider, D.; Barbic, T.; Sladewska-Marquardt, A.; Kuhnle, S.; Marx, A.; Scheffner, M. Rolef ubiquitin and the HPV E6ncoproteinn E6AP-mediated ubiquitination. Proc. Natl. Acad. Sci. USA 2015, 112, 9872–9877. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Howley, P.M. Human papillomavirusmmortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Durst, M.; Schneider, A.; von Knebel Doeberitz, M. Type-dependentntegration frequencyf human papillomavirus genomesn cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Vagenas, D.; Salazar, C.; Kenny, L.; Perry, C.; Calvopina, D.; Punyadeera, C. Salivary miRNA panel toetect HPV-positive and HPV-negative head and neck cancer patients. Oncotarget 2017, 8, 99990–100001. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.M.; Marques, J.P.; Soares, A.R.; Carreto, L.; Santos, M.A. MicroRNA expression variabilityn human cervical tissues. PLoS ONE 2010, 5, e11780. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, F.; Xu, J.; Ye, F.; Shen, Y.; Zhou, J.; Lu, W.; Wan, X.; Ma, D.; Xie, X. Progressive miRNA expression profilesn cervical carcinogenesis anddentificationf HPV-related target genes for miR-29. J. Pathol. 2011, 224, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. microRNAs are biomarkersfncogenic human papillomavirusnfections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, Y.; Zhu, M.; Liu, S.; Wang, X. miRNA Expression Profilesf HPV-Infected Patients with Cervical Cancern the Uyghur Populationn China. PLoS ONE 2016, 11, e0164701. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.I.; Hoskins, E.E.; Wells, S.I.; Ferris, R.L.; Khan, S.A. Alterationf microRNA profilesn squamous cell carcinomaf the head and neck cell lines by human papillomavirus. Head Neck 2011, 33, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Lajer, C.B.; Nielsen, F.C.; Friis-Hansen, L.; Norrild, B.; Borup, R.; Garnaes, E.; Rossing, M.; Specht, L.; Therkildsen, M.H.; Nauntofte, B.; et al. Different miRNA signaturesfral and pharyngeal squamous cell carcinomas: A prospective translational study. Br. J. Cancer 2011, 104, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Lajer, C.B.; Garnaes, E.; Friis-Hansen, L.; Norrild, B.; Therkildsen, M.H.; Glud, M.; Rossing, M.; Lajer, H.; Svane, D.; Skotte, L.; et al. The rolef miRNAsn human papilloma virus (HPV)-associated cancers: Bridging between HPV-related head and neck cancer and cervical cancer. Br. J. Cancer 2012, 106, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.B.; Lin, A.; Xu, W.; Waldron, L.; Perez-Ordonez, B.; Weinreb, I.; Shi, W.; Bruce, J.; Huang, S.H.; O’Sullivan, B.; et al. Potentially prognostic miRNAsn HPV-associatedropharyngeal carcinoma. Clin. Cancer Res. 2013, 19, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Davis, J.W.; Taylor, K.H.; Johnson, J.; Shi, Z.; Williams, R.; Atasoy, U.; Lewis, J.S., Jr.; Stack, M.S. Identificationf a human papillomavirus-associatedncogenic miRNA paneln humanropharyngeal squamous cell carcinoma validated by bioinformatics analysisf the Cancer Genome Atlas. Am. J. Pathol. 2015, 185, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Vojtechova, Z.; Sabol, I.; Salakova, M.; Smahelova, J.; Zavadil, J.; Turek, L.; Grega, M.; Klozar, J.; Prochazka, B.; Tachezy, R. Comparisonf the miRNA profilesn HPV-positive and HPV-negative tonsillar tumors and a model systemf human keratinocyte clones. BMC Cancer 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- Nasman, A.; Attner, P.; Hammarstedt, L.; Du, J.; Eriksson, M.; Giraud, G.; Ahrlund-Richter, S.; Marklund, L.; Romanitan, M.; Lindquist, D.; et al. Incidencef human papillomavirus (HPV) positive tonsillar carcinoman Stockholm, Sweden: An epidemicf viral-induced carcinoma? Int. J. Cancer 2009, 125, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulationf microRNA-mRNA Target Pairs by Human Papillomavirus 16 Oncoproteins. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Emmrich, S.; Putzer, B.M. Checks and balances: E2F-microRNA crosstalkn cancer control. Cell Cycle 2010, 9, 2555–2567. [Google Scholar] [CrossRef] [PubMed]
- Myklebust, M.P.; Bruland, O.; Fluge, O.; Skarstein, A.; Balteskard, L.; Dahl, O. MicroRNA-15bsnduced with E2F-controlled genesn HPV-related cancer. Br. J. Cancer 2011, 105, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.K.; McCoy, J.P.; Banerjee, N.S.; Rader, J.S.; Broker, T.R.; Meyers, C.; Chow, L.T.; Zheng, Z.M. Oncogenic HPVnfectionnterrupts the expressionf tumor-suppressive miR-34a through viralncoprotein E6. RNA 2009, 15, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Au Yeung, C.L.; Tsang, T.Y.; Yau, P.L.; Kwok, T.T. Human papillomavirus type 16 E6nduces cervical cancer cell migration through the p53/microRNA-23b/urokinase-type plasminogen activator pathway. Oncogene 2011, 30, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Chen, H.; Zhou, D.; Li, D.; Bai, R.; Zheng, S.; Ge, W. MicroRNA-9 up-regulationsnvolvedn colorectal cancer metastasis via promoting cell motility. Med. Oncol. 2012, 29, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gao, G.; Hu, X.; Wang, Y.; Schwarz, J.K.; Chen, J.J.; Grigsby, P.W.; Wang, X. Activationf miR-9 by human papillomavirusn cervical cancer. Oncotarget 2014, 5, 11620–11630. [Google Scholar] [PubMed]
- Yamamoto, N.; Kinoshita, T.; Nohata, N.; Itesako, T.; Yoshino, H.; Enokida, H.; Nakagawa, M.; Shozu, M.; Seki, N. Tumor suppressive microRNA-218nhibits cancer cell migration andnvasion by targeting focal adhesion pathwaysn cervical squamous cell carcinoma. Int. J. Oncol. 2013, 42, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Hanazawa, T.; Nohata, N.; Kikkawa, N.; Enokida, H.; Yoshino, H.; Yamasaki, T.; Hidaka, H.; Nakagawa, M.; Okamoto, Y.; et al. Tumor suppressive microRNA-218nhibits cancer cell migration andnvasion through targeting laminin-332n head and neck squamous cell carcinoma. Oncotarget 2012, 3, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.M.; Phillips, B.L.; Chan, E.K. miR-375 activates p21 and suppresses telomerase activity by coordinately regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3zeta. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonalntegrationf a polyomavirusn human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Arguelles, M.E.; Melon, S.; Rojo, S.; Fernandez-Blazquez, A.; Boga, J.A.; Palacio, A.; Vivanco, B.; de Oña, M. Detection and quantificationf Merkel cell polyomavirus. Analysisf Merkel cell carcinoma cases from 1977 to 2015. J. Med. Virol. 2017, 89, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; DoRosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.M.; Sadasivam, S.; et al. Improvedetection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Investig. 2012, 122, 4645–4653. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Van Ghelue, M.; Johannessen, M. Oncogenic potentialsf the human polyomavirus regulatory proteins. Cell Mol. Life Sci. 2007, 64, 1656–1678. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.Y.; Lee, M.C.; Lai, N.S.; Lu, M.C. BK virus as a potentialncovirus for bladder cancern a renal transplant patient. J. Formos. Med. Assoc. 2015, 114, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Polz, D.; Morshed, K.; Stec, A.; Podsiadlo, L.; Polz-Dacewicz, M. Do polyomavirus hominis strains BK and JC play a rolenral squamous cell carcinoma? Ann. Agric. Environ. Med. 2015, 22, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Fung, C.Y.; Chang, F.P.; Huang, W.S.; Chen, W.C.; Wang, J.Y.; Chang, D. Prevalence and genotypedentificationf human JC virusn colon cancern Taiwan. J. Med. Virol. 2008, 80, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expressionf viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Grundhoff, A.T.; Tevethia, S.; Pipas, J.M.; Ganem, D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 2005, 435, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Fink, L.H.; O’Hara, B.; Atwood, W.J.; Sullivan, C.S. Evolutionarily conserved functionf a viral microRNA. J. Virol. 2008, 82, 9823–9828. [Google Scholar] [CrossRef] [PubMed]
- Bauman, Y.; Nachmani, D.; Vitenshtein, A.; Tsukerman, P.; Drayman, N.; Stern-Ginossar, N.; Lankry, D.; Gruda, R.; Mandelboim, O. Andentical miRNAf the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escapemmune elimination. Cell Host Microbe 2011, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validationf a novel mature microRNA encoded by the Merkel cell polyomavirusn human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Renwick, N.; Cekan, P.; Masry, P.A.; McGeary, S.E.; Miller, J.B.; Hafner, M.; Li, Z.; Mihailovic, A.; Morozov, P.; Brown, M.; et al. Multicolor microRNA FISH effectivelyifferentiates tumor types. J. Clin. Investig. 2013, 123, 2694–2702. [Google Scholar] [CrossRef] [PubMed]
- Martel-Jantin, C.; Filippone, C.; Cassar, O.; Peter, M.; Tomasic, G.; Vielh, P.; Briere, J.; Petrella, T.; Aubriot-Lorton, M.H.; Mortier, L.; et al. Genetic variability andntegrationf Merkel cell polyomavirusn Merkel cell carcinoma. Virology 2012, 426, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Cox, J.E.; Azarm, K.D.; Wylie, K.N.; Woolard, K.D.; Pesavento, P.A.; Sullivan, C.S. Identificationf a polyomavirus microRNA highly expressedn tumors. Virology 2015, 476, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Brostoff, T.; Dela Cruz, F.N., Jr.; Church, M.E.; Woolard, K.D.; Pesavento, P.A. The raccoon polyomavirus genome and tumor antigen transcription are stable and abundantn neuroglial tumors. J. Virol. 2014, 88, 12816–12824. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Lee, L.; Caramuta, S.; Hoog, A.; Browaldh, N.; Bjornhagen, V.; Larsson, C.; Lui, W.O. MicroRNA expression patterns related to merkel cell polyomavirusnfectionn human merkel cell carcinoma. J. Investig. Dermatol. 2014, 134, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Morgan, T.R. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 2006, 3, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, S.; Bacchi-Reggiani, L.; de Biase, D.; Fornelli, A.; Masetti, M.; Tura, A.; Grizzi, F.; Zanello, M.; Mastrangelo, L.; Lombardi, R.; et al. Possible association between hepatitis C virus and malignanciesifferent from hepatocellular carcinoma: A systematic review. World J. Gastroenterol. 2015, 21, 12896–12953. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.M.; Piacentini, M. Molecular mechanismsf hepatitis C virus-induced hepatocellular carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identificationf microRNAsf the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulationf hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011, 39, 7716–7729. [Google Scholar] [CrossRef] [PubMed]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5′ terminal nucleotidesf the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 2011, 108, 3193–3198. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencingf microRNA-122n primates with chronic hepatitis C virusnfection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; vaner Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatmentf HCVnfection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Aly, H.H.; Tajima, A.; Inoue, I.; Shimotohno, K. Regulationf the hepatitis C virus genome replication by miR-199a. J. Hepatol. 2009, 50, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Lin, L.; Zhou, W.; Wang, Z.; Ding, G.; Dong, Q.; Qin, L.; Wu, X.; Zheng, Y.; Yang, Y.; et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 2011, 19, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Yeh, Y.J.; Tseng, C.P.; Hsu, S.D.; Chang, Y.L.; Sakamoto, N.; Huang, H.D. Let-7bs a novel regulatorf hepatitis C virus replication. Cell Mol. Life Sci. 2012, 69, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Shrivastava, S.; Bhanja Chowdhury, J.; Ray, R.; Ray, R.B. Transcriptional suppressionf miR-181c by hepatitis C virus enhances homeobox A1 expression. J. Virol. 2014, 88, 7929–7940. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Di Bisceglie, A.M.; Ray, R.B. Hepatitis C virus-mediated enhancementf microRNA miR-373mpairs the JAK/STAT signaling pathway. J. Virol. 2015, 89, 3356–3365. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, T.; Honda, M.; Shimakami, T.; Horii, R.; Yamashita, T.; Sakai, Y.; Sakai, A.; Okada, H.; Watanabe, R.; Murakami, S.; et al. MicroRNA-27a regulates lipid metabolism andnhibits hepatitis C virus replicationn human hepatoma cells. J. Virol. 2013, 87, 5270–5286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, W.; Cheng, N.; Wang, K.; Li, B.; Jiang, X.; Sun, S. Hepatitis C virus-induced up-regulationf microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012, 56, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xie, Y.; Yang, P.; Chen, P.; Zhang, L. HCV core protein-inducedown-regulationf microRNA-152 promoted aberrant proliferation by regulating Wnt1n HepG2 cells. PLoS ONE 2014, 9, e81730. [Google Scholar] [CrossRef]
- Ishida, H.; Tatsumi, T.; Hosui, A.; Nawa, T.; Kodama, T.; Shimizu, S.; Hikita, H.; Hiramatsu, N.; Kanto, T.; Hayashi, N.; et al. Alterationsn microRNA expression profilen HCV-infected hepatoma cells:nvolvementf miR-491n regulationf HCV replication via the PI3 kinase/Akt pathway. Biochem. Biophys. Res. Commun. 2011, 412, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Banaudha, K.; Kaliszewski, M.; Korolnek, T.; Florea, L.; Yeung, M.L.; Jeang, K.T.; Kumar, A. MicroRNA silencingf tumor suppressor DLC-1 promotes efficient hepatitis C virus replicationn primary human hepatocytes. Hepatology 2011, 53, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Varnholt, H.; Drebber, U.; Schulze, F.; Wedemeyer, I.; Schirmacher, P.; Dienes, H.P.; Odenthal, M. MicroRNA gene expression profilef hepatitis C virus-associated hepatocellular carcinoma. Hepatology 2008, 47, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Ura, S.; Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Sunakozaka, H.; Sakai, Y.; Horimoto, K.; Kaneko, S. Differential microRNA expression between hepatitis B and hepatitis C leadingisease progression to hepatocellular carcinoma. Hepatology 2009, 49, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Pernot, S.; El Saghire, H.; Durand, S.C.; Thumann, C.; Crouchet, E.; Ye, T.; Fofana, I.; Oudot, M.A.; Barths, J.; et al. Hepatitis C Virus-Induced Upregulationf MicroRNA miR-146a-5pn Hepatocytes Promotes Viral Infection and Deregulates Metabolic Pathways Associated with Liver Disease Pathogenesis. J. Virol. 2016, 90, 6387–6400. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221verexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Kannian, P.; Green, P.L. Human T Lymphotropic Virus Type 1 (HTLV-1): Molecular Biology and Oncogenesis. Viruses 2010, 2, 2037–2077. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latencyn resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Hakim, S.T.; Alsayari, M.; McLean, D.C.; Saleem, S.; Addanki, K.C.; Aggarwal, M.; Mahalingam, K.; Bagasra, O. A large numberf the human microRNAs target lentiviruses, retroviruses, and endogenous retroviruses. Biochem. Biophys. Res. Commun. 2008, 369, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, K.; Corradin, A.; Zanovello, P.; Amadori, A.; Bronte, V.; Ciminale, V.; D’Agostino, D.M. Rolef microRNAsn HTLV-1nfection and transformation. Mol. Aspects Med. 2010, 31, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.T.; Nicot, C. miR-28-3ps a cellular restriction factor thatnhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virusnfection. J. Biol. Chem. 2015, 290, 5381–5390. [Google Scholar] [CrossRef] [PubMed]
- Piedade, D.; Azevedo-Pereira, J.M. MicroRNAs, HIV and HCV: A complex relation towards pathology. Rev. Med. Virol. 2016, 26, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppressionf microRNA-silencing pathway by HIV-1uring virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulationf cyclin T1 and HIV-1 Replication by microRNAsn resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.L.; Rice, A.P. miR-198nhibits HIV-1 gene expression and replicationn monocytes andts mechanismf action appears tonvolve repressionf cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.S.; Wu, T.C.; Sang, W.W.; Ruan, Z. MiR-217snvolvedn Tat-induced HIV-1 long terminal repeat (LTR) transactivation byown-regulationf SIRT1. Biochim. Biophys. Acta 2012, 1823, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Van Duyne, R.; Guendel, I.; Klase, Z.; Narayanan, A.; Coley, W.; Jaworski, E.; Roman, J.; Popratiloff, A.; Mahieux, R.; Kehn-Hall, K.; et al. Localization and sub-cellular shuttlingf HTLV-1 tax with the miRNA machinery. PLoS ONE 2012, 7, e40662. [Google Scholar] [CrossRef] [PubMed]
- Pichler, K.; Schneider, G.; Grassmann, R. MicroRNA miR-146a and furtherncogenesis-related cellular microRNAs areysregulatedn HTLV-1-transformed T lymphocytes. Retrovirology 2008, 5, 100. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Yasunaga, J.; Bennasser, Y.; Dusetti, N.; Harris, D.; Ahmad, N.; Matsuoka, M.; Jeang, K.T. Roles for microRNAs, miR-93 and miR-130b, and tumor protein 53-induced nuclear protein 1 tumor suppressorn cell growthysregulation by human T-cell lymphotrophic virus 1. Cancer Res. 2008, 68, 8976–8985. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Lepelletier, Y.; Hermine, O.; Nicot, C. Deregulationf microRNAnvolvedn hematopoiesis and themmune responsen HTLV-I adult T-cell leukemia. Blood 2009, 113, 4914–4917. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Nakano, K.; Miyake, A.; Yamochi, T.; Kagami, Y.; Tsutsumi, A.; Matsuda, Y.; Sato-Otsubo, A.; Muto, S.; Utsunomiya, A.; et al. Polycomb-mediated lossf miR-31 activates NIK-dependent NF-kappaB pathwayn adult T cell leukemia andther cancers. Cancer Cell 2012, 21, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Vernin, C.; Thenoz, M.; Pinatel, C.; Gessain, A.; Gout, O.; Delfau-Larue, M.H.; Nazaret, N.; Legras-Lachuer, C.; Wattel, E.; Mortreux, F. HTLV-1 bZIP factor HBZ promotes cell proliferation and geneticnstability by activating OncomiRs. Cancer Res. 2014, 74, 6082–6093. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.B.; Wu, F.L.; Kong, D.; Guo, A.G. HBV-encoded microRNA candidate andts target. Comput. Biol. Chem. 2007, 31, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, H.; Sun, H.; Fan, H.; Hu, Y.; Liu, M.; Li, X.; Tang, H. Hepatitis B Virus-Encoded MicroRNA Controls Viral Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Ren, Y.F.; Song, Y.J.; Xue, Y.F.; Zhang, X.J.; Cao, S.T.; Deng, Z.J.; Wu, J.; Chen, L.; Li, G.; et al. MicroRNA-581 promotes hepatitis B virus surface antigen expression by targeting Dicer and EDEM1. Carcinogenesis 2014, 35, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sun, Y.; Ruan, J.; Chen, R.; Chen, X.; Chen, C.; Kreuze, J.F.; Fei, Z.; Zhu, X.; Gao, S. Using small RNAeep sequencingata toetect human viruses. BioMed. Res. Int. 2016, 2016, 2596782. [Google Scholar] [CrossRef] [PubMed]
- Selitsky, S.R.; Dinh, T.A.; Toth, C.L.; Kurtz, L.; Honda, M.; Struck, B.R.; Kaneko, S.; Vickers, K.C.; Lemon, S.M.; Sethupathy, P. Transcriptomic analysisf chronic hepatitis B and C and liver cancer reveals microRNA-mediated controlf cholesterol synthesis programs. mBio 2015, 6, e01500-15. [Google Scholar] [CrossRef] [PubMed]
- Selitsky, S.R.; Baran-Gale, J.; Honda, M.; Yamane, D.; Masaki, T.; Fannin, E.E.; Guerra, B.; Shirasaki, T.; Shimakami, T.; Kaneko, S.; et al. Small tRNA-derived RNAs arencreased and more abundant than microRNAsn chronic hepatitis B and C. Sci. Rep. 2015, 5, 7675. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Pietila, T.; Ronty, M.; Michon, F.; Frilander, M.J.; Ritari, J.; Tarkkanen, J.; Paulin, L.; Auvinen, P.; Auvinen, E. Identification and validationf human papillomavirus encoded microRNAs. PLoS ONE 2013, 8, e70202. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, E.; Pietila, T.; Nieminen, P.; Qian, K.; Auvinen, E. Low expression levelsf putative HPV encoded microRNAsn cervical samples. Springerplus 2016, 5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weng, S.L.; Huang, K.Y.; Weng, J.T.; Hung, F.Y.; Chang, T.H.; Lee, T.Y. Genome-wideiscoveryf viral microRNAs basedn phylogenetic analysis and structural evolutionf various human papillomavirus subtypes. Brief Bioinform. 2017. [Google Scholar] [CrossRef]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR elements processed by Dicer to yield a viral micro-RNAnvolvedn chromatin remodelingf the viral LTR. BMC Mol. Biol. 2007, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Plante, I.; Landry, P.; Barat, C.; Janelle, M.E.; Flamand, L.; Tremblay, M.J.; Provost, P. Identificationf functional microRNAs released through asymmetrical processingf HIV-1 TAR element. Nucleic Acids Res. 2008, 36, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Rouha, H.; Thurner, C.; Mandl, C.W. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res. 2010, 38, 8328–8337. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Jongejan, A.; van Kampen, A.H.C.; Berkhout, B.; Das, A.T. Tat-dependent productionf an HIV-1 TAR-encoded miRNA-like small RNA. Nucleic Acids Res. 2016, 44, 4340–4353. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.A.; Zhao, H.; Yue, S.C.; Anandaiah, A.; Koziel, H.; Tachado, S.D. Novel HIV-1 miRNAs stimulate TNFalpha releasen human macrophages via TLR8 signaling pathway. PLoS ONE 2014, 9, e106006. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Feng, H.; Da, Q.; Jiang, H.; Chen, L.; Xie, L.; Huang, Q.; Xiong, H.; Luo, F.; Kang, L.; et al. Expressionf HIV-encoded microRNA-TAR andtsnhibitory effectn viral replicationn human primary macrophages. Arch. Virol. 2016, 161, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.; Winograd, R.; Davis, J.; Carpio, L.; Hildreth, R.; Heydarian, M.; Fu, S.; McCaffrey, T.; Meiri, E.; Ayash-Rashkovsky, M.; et al. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology 2009, 6. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Vigneault-Edwards, J.; Letourneau, K.; Gobeil, L.A.; Plante, I.; Burnett, J.C.; Rossi, J.J.; Provost, P. Regulationf host gene expression by HIV-1 TAR microRNAs. Retrovirology 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Ito, M.; Tsutsumi, Y.; Ichikawa, Y.; Okuyama, H.; Brisibe, E.A.; Saksena, N.K.; Fujii, Y.R. HIV-1 nef suppression by virally encoded microRNA. Retrovirology 2004, 1, 44. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Navas-Martin, S.; Martin-Garcia, J. MicroRNAs and HIV-1nfection: Antiviral activities and beyond. J. Mol. Biol. 2014, 426, 1178–1197. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.; Ahlawat, A.; Gupta, S.D. HIV-1 genome-encoded hiv1-mir-H1mpairs cellular responses tonfection. Mol. Cell. Biochem. 2009, 323, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, M.; Geng, G.; Liu, B.; Huang, Z.; Luo, H.; Zhou, J.; Guo, X.; Cai, W.; Zhang, H. A novel HIV-1-encoded microRNA enhancests viral replication by targeting the TATA box region. Retrovirology 2014, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cullen, B.R. Analysisf thenteractionf primate retroviruses with the human RNAnterference machinery. J. Virol. 2007, 81, 12218–12226. [Google Scholar] [CrossRef] [PubMed]
- Whisnant, A.W.; Bogerd, H.P.; Flores, O.; Ho, P.; Powers, J.G.; Sharova, N.; Stevenson, M.; Chen, C.; Cullen, B.R. In-depth analysisf thenteractionf HIV-1 with cellular microRNA biogenesis and effector mechanisms. mBio 2013, 4, e00193-13. [Google Scholar] [CrossRef] [PubMed]
- Schopman, N.C.; Willemsen, M.; Liu, Y.P.; Bradley, T.; van Kampen, A.; Baas, F.; Berkhout, B.; Haasnoot, J. Deep sequencingf virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012, 40, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Vongrad, V.; Imig, J.; Mohammadi, P.; Kishore, S.; Jaskiewicz, L.; Hall, J.; Günthard, H.F.; Beerenwinkel, N.; Metzner, K.J. HIV-1 RNAs are not partf the Argonaute 2 associated RNAnterference pathwayn macrophages. PLoS ONE 2015, 10, e0132127. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. RNA virus microRNA that mimics a B-cellncomiR. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targetingf microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Virus Family | Virus Species | Mature miRNAs (According to miRBase, Updated 2014) | MiRNAs Mentioned in This Review | Proposed Function | Target | Reference |
|---|---|---|---|---|---|---|
| Herpesviridae | Epstein–Barr virus (EBV) | 44 | miR-BART17-5p | cell transformation | LMP1 | [25] |
| miR-BART16 | cell transformation, anti-apoptotic role | LMP1, Casp3 | [25,31] | |||
| miR-BART1-5p | LMP1, Casp3 | [25,31] | ||||
| miR-BART5-5p | PUMA | [29] | ||||
| miR-BART19-5p | PUMA | [29] | ||||
| miR-BART22 | escape from host immune surveillance | LMP2A | [26] | |||
| miR-BART2-5p | regulation of latent–lytic switch, evasion of the host‘s immune system | BALF5, MICB | [27,36] | |||
| miR-BART6-5p | regulation of viral replication | EBNA2 | [28] | |||
| miR-BART7-3p | promotion of EMT and metastasis, regulation of radiation sensitivity | PTEN, GFPT1 | [30,38] | |||
| miR-BART3 | proliferation and cell transformation | DICE1 | [32] | |||
| miR-BHRF1-1 | immunomodulatory function | CXCL11 | [33] | |||
| miR-BHRF1-2 | CXCL11 | |||||
| miR-BHRF1-3 | CXCL11 | |||||
| Herpesvirus-8 (HHV-8)/Kaposi’s sarcoma herpesvirus (KSHV) | 25 | miR-K9-5p | regulation of lytic induction | RTA | [54,55] | |
| miR-K7-5p | ||||||
| miR-K3 | regulation of viral latency and angiogenesis | nuclear factor I/B, GRK2, THBS1 | [56,62,63] | |||
| miR-K12-11 | MYB, IKKε, THBS1 | [58,60,62] | ||||
| miR-K12-4 | regulation of viral latency, anti-apoptotic role | Rbl2, Casp3 | [61,67] | |||
| miR-K6-3p | regulation of angiogenesis | THBS1, SH3BGR | [62,63] | |||
| miR-K12-1 | anti-apoptotic role, regulation of angiogenesis | p21, Casp3, THBS1 | [62,66,67] | |||
| Polyomaviridae | Merkel cell polyomavirus (MCPyV) | 1 | MCV-miR-M1 | regulation of viral lifecycle | early viral transcripts | [145] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojtechova, Z.; Tachezy, R. The Role of miRNAs in Virus-Mediated Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1217. https://doi.org/10.3390/ijms19041217
Vojtechova Z, Tachezy R. The Role of miRNAs in Virus-Mediated Oncogenesis. International Journal of Molecular Sciences. 2018; 19(4):1217. https://doi.org/10.3390/ijms19041217
Chicago/Turabian StyleVojtechova, Zuzana, and Ruth Tachezy. 2018. "The Role of miRNAs in Virus-Mediated Oncogenesis" International Journal of Molecular Sciences 19, no. 4: 1217. https://doi.org/10.3390/ijms19041217
APA StyleVojtechova, Z., & Tachezy, R. (2018). The Role of miRNAs in Virus-Mediated Oncogenesis. International Journal of Molecular Sciences, 19(4), 1217. https://doi.org/10.3390/ijms19041217