PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basic Overview of the Molecular Features of PPARβ/δ
3. PPARβ/δ as a Major Regulator of Metabolic Disorders
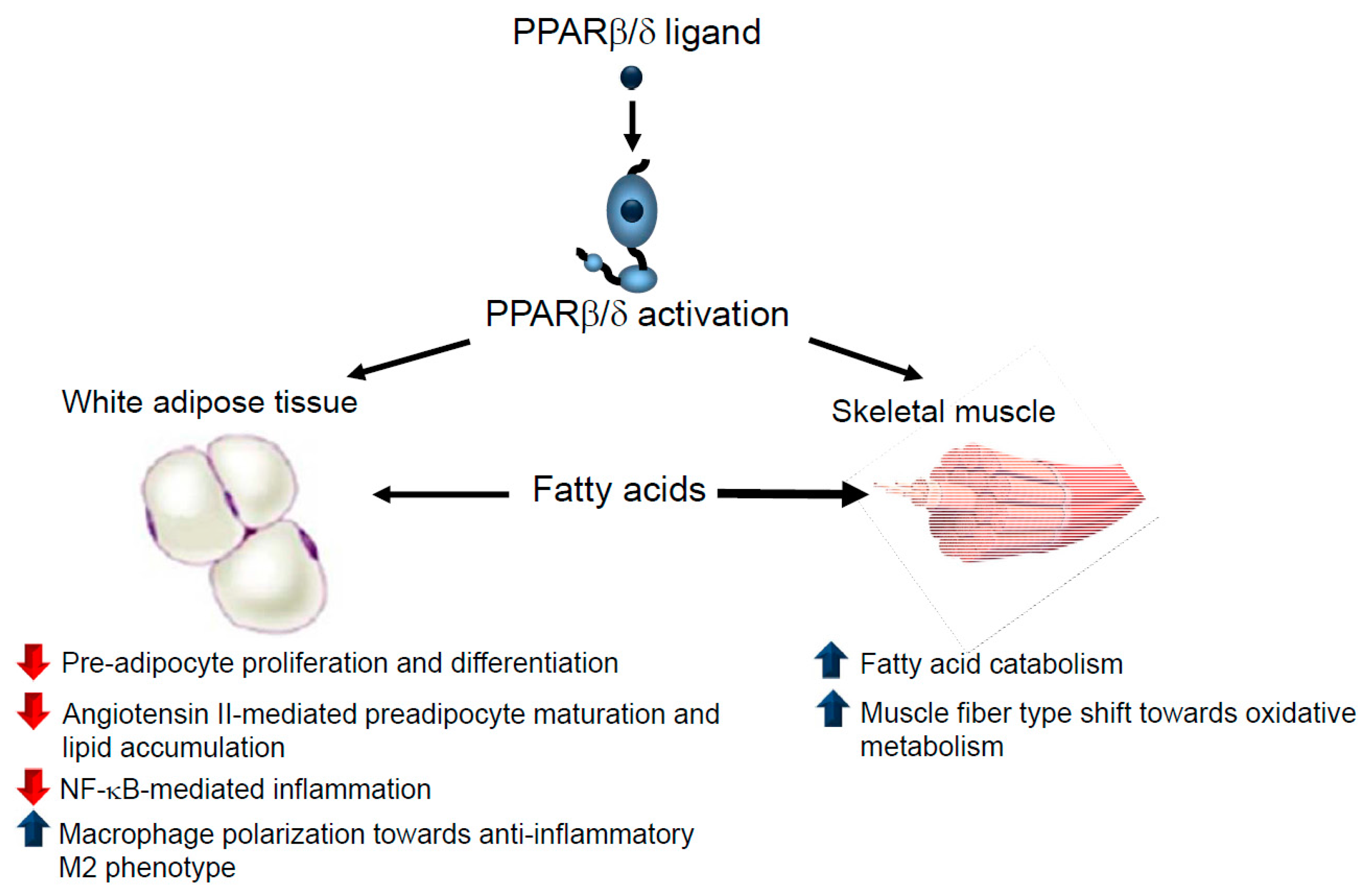
3.1. Obesity
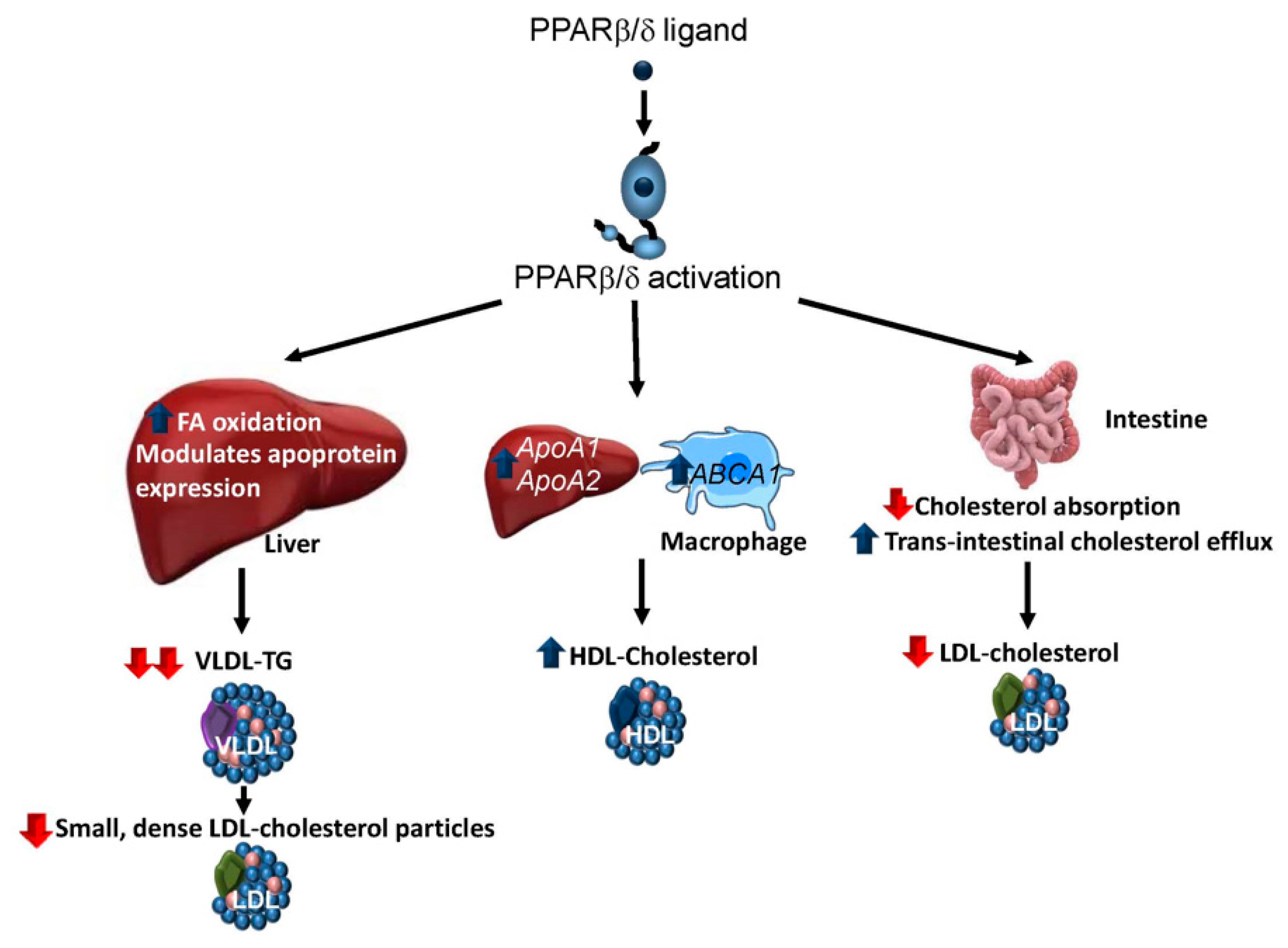
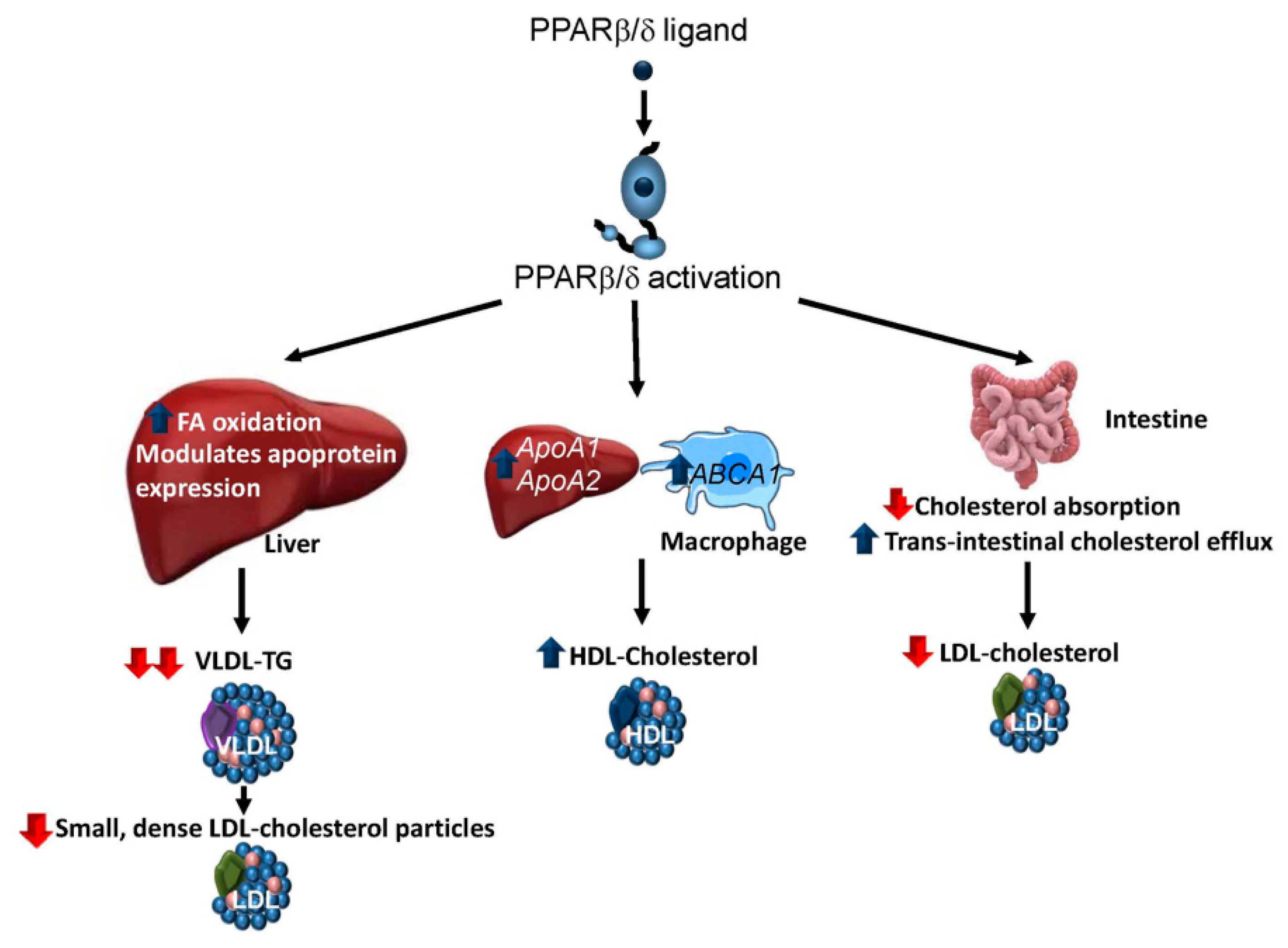
3.2. Dyslipidaemia
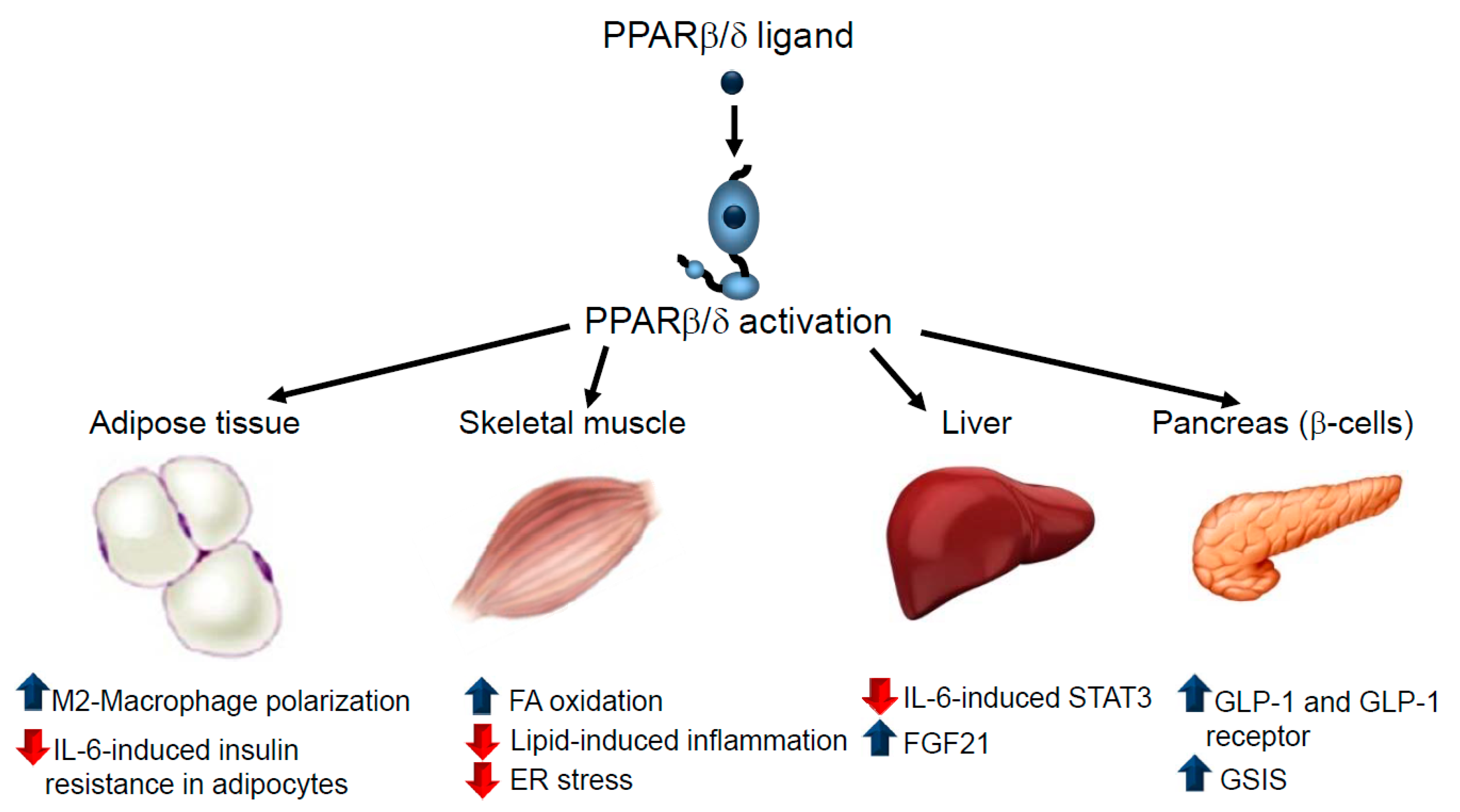
3.3. Type 2 Diabetes Mellitus
3.4. Non-Alcoholic Fatty Liver Disease (NAFLD)
4. Safety of PPARβ/δ Agonists
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Birsoy, K.; Festuccia, W.T.; Laplante, M. A comparative perspective on lipid storage in animals. J. Cell Sci. 2013, 126, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The global obesity pandemic: Shaped by global drivers and local environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Wyatt, S.B.; Winters, K.P.; Dubbert, P.M. Overweight and obesity: Prevalence, consequences, and causes of a growing public health problem. Am. J. Med. Sci. 2006, 331, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.U.; Hu, F.B.; Schulze, M.B. Metabollically healthy obesity: Epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 2013, 1, 152–162. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Després, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 2008, 6, e237. [Google Scholar] [CrossRef] [PubMed]
- Giordiano Attianese, G.M.P.; Desvergne, B. Integrative and systemic approaches for evaluating PPARβ/δ (PPARD) function. Nucl. Recept. Signal. 2015, 13, e001. [Google Scholar]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor β. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Vázquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARβ/δ. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Carrera, M. Unraveling the Effects of PPARβ/δ on Insulin Resistance and Cardiovascular Disease. Trends Endocrinol. Metab. 2016, 27, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Bishop-Bailey, D.; Bystrom, J. Emerging roles of peroxisome proliferator-activated receptor-β/δ in inflammation. Pharmacol. Ther. 2009, 124, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Borland, M.G.; Foreman, J.E.; Girroir, E.E.; Zolfaghari, R.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Ross, A.C.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-β/δ inhibits cell proliferation in human HaCaT keratinocytes. Mol. Pharmacol. 2008, 74, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Rieck, M.; Meissner, W.; Ries, S.; Müller-Brüsselbach, S.; Müller, R. Ligand-mediated regulation of peroxisome proliferator-activated receptor (PPAR)β/δ: A comparative analysis of PPAR-selective agonists and all-trans retinoic acid. Mol. Pharmacol. 2008, 74, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Billin, A.N. PPAR-β/δ agonists for type 2 diabetes and dyslipidemia: An adopted orphan still looking for a home. Expert Opin. Investig. Drugs 2008, 17, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Haimerl, M.; Paik, Y.H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc. Natl. Acad. Sci. USA 2012, 109, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Liao, D.; He, W.; Ong, E.S.; Nelson, M.C.; Olefsky, J.M.; Boland, R.; Evans, R.M. Effects of peroxisome proliferator-activated receptor δ on placentation, adiposity, and colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doktorova, M.; Zwarts, I.; Zutphen, T.V.; Dijk, T.H.; Bloks, V.W.; Harkema, L.; Bruin, A.; Downes, M.; Evans, R.M.; Verkade, H.J.; et al. Intestinal PPARδ protects against diet-induced obesity, insulin resistance and dyslipidemia. Sci. Rep. 2017, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Bastie, C.; Holst, D.; Gaillard, D.; Jehl-Pietri, C.; Grimaldi, P.A. Expression of peroxisome proliferator-activated receptor PPARδ promotes induction of PPARgamma and adipocyte differentiation in 3T3C2 fibroblasts. J. Biol. Chem. 1999, 274, 21920–21925. [Google Scholar] [CrossRef] [PubMed]
- Bastie, C.; Luquet, S.; Holst, D.; Jehl-Pietri, C.; Grimaldi, P.A. Alterations of peroxisome proliferator-activated receptor δ activity affect fatty acid-controlled adipose differentiation. J. Biol. Chem. 2000, 275, 38768–38773. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.B.; Zhang, H.; Rasmussen, T.H.; Petersen, R.K.; Flindt, E.N.; Kristiansen, K. Peroxisome proliferator-activated receptor δ (PPARδ)-mediated regulation of preadipocyte proliferation and gene expression is dependent on cAMP signaling. J. Biol. Chem. 2001, 276, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Puri, N.; Kim, D.H.; Hinds, T.D.; Stechschulte, L.A.; Favero, G.; Rodella, L.; Shapiro, J.I.; Jude, D.; Abraham, N.G. PPARδ binding to heme oxygenase 1 promoter prevents angiotensin II-induced adipocyte dysfunction in Goldblatt hypertensive rats. Int. J. Obes. 2014, 38, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Bortolotto, J.W.; Margis, R.; Ferreira, A.C.; Padoin, A.V.; Mottin, C.C.; Guaragna, R.M. Adipose tissue distribution and quantification of PPARβ/δ and PPARgamma1-3 mRNAs: Discordant gene expression in subcutaneous, retroperitoneal and visceral adipose tissue of morbidly obese patients. Obes. Surg. 2007, 17, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, R.; Serrano, L.; Coll, T.; Moullan, N.; Sánchez, R.M.; Merlos, M.; Palomer, X.; Laguna, J.C.; Michalik, L.; Wahli, W.; et al. Activation of peroxisome proliferator- activated receptor β/δ inhibits lipopolysaccharide-induced cytokine production in adipocytes by lowering nuclear factor-κB activity via extracellular signal-related kinase 1/2. Diabetes 2008, 57, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–895. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Schwartz, S.; Littlejohn, T., 3rd; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, a novel peroxisome proliferator receptor-δ agonist: Lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Borén, J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis 2015, 239, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Barroso, E.; Rodríguez-Calvo, R.; Serrano-Marco, L.; Astudillo, A.M.; Balsinde, J.; Palomer, X.; Vázquez-Carrera, M. The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1/–lipin 1–PPAR/pathway leading to increased fatty acid oxidation. Endocrinology 2011, 152, 1848–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risérus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Müller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARβ/δ in regulation of gene expression in mouse liver. Physiol. Genomics 2010, 41, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.D.; Fiévet, C.; Hennuyer, N.; Peinado-Onsurbe, J.; Duez, H.; Bergera, J.; Cullinan, C.A.; Sparrow, C.P.; Baffic, J.; Berger, G.D.; et al. Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett. 2000, 473, 333–336. [Google Scholar] [CrossRef]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor δ agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, D.L.; Massien, C.; Pearce, G.; Billin, A.N.; Perlstein, I.; Willson, T.M.; Hassall, D.G.; Ancellin, N.; Patterson, S.D.; Lobe, D.C.; et al. Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor δ agonist. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Chehaibi, K.; Cedó, L.; Metso, J.; Palomer, X.; Santos, D.; Quesada, H.; Naceur Slimane, M.; Wahli, W.; Julve, J.; Vázquez-Carrera, M.; et al. PPAR-β/δ activation promotes phospholipid transfer protein expression. Biochem. Pharmacol. 2015, 94, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, J.N.; Kruit, J.K.; Havinga, R.; Baller, J.F.; Chimini, G.; Lestavel, S.; Staels, B.; Groot, P.H.; Groen, A.K.; Kuipers, F. Reduced cholesterol absorption upon PPARδ activation coincides with decreased intestinal expression of NPC1L1. J. Lipid Res. 2005, 46, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Vrins, C.L.; van der Velde, A.E.; van den Oever, K.; Levels, J.H.; Huet, S.; Oude Elferink, R.P.; Kuipers, F.; Groen, A.K. Peroxisome proliferator-activated receptor δ activation leads to increased transintestinal cholesterol efflux. J. Lipid Res. 2009, 50, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Ehrenborg, E.; Skogsberg, J. Peroxisome proliferator-activated receptor δ and cardiovascular disease. Atherosclerosis 2013, 231, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.J.; Pearce, G.L.; Jones, N.P.; Sprecher, D.L. Lipid effects of peroxisome proliferator-activated receptor-δ agonist GW501516 in subjects with low high-density lipoprotein cholesterol: Characteristics of metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Roberts, B.K.; Wang, X.; Geaney, J.C.; Naim, S.; Wojnoonski, K.; Karpf, D.B.; Krauss, R.M. Effects of the PPAR-δ agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis 2012, 220, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Olefsky, J.M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012, 15, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Bojic, L.A.; Sawyez, C.G.; Telford, D.E.; Edwards, J.Y.; Hegele, R.A.; Huff, M.W. Activation of peroxisome proliferator-activated receptor δ inhibits human macrophage foam cell formation and the inflammatory response induced by very low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2919–2928. [Google Scholar] [CrossRef] [PubMed]
- Korns, D.; Frasch, S.C.; Fernandez-Boyanapalli, R.; Henson, P.M.; Bratton, D.L. Modulation of Macrophage Efferocytosis in Inflammation. Front. Immunol. 2011, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, L.; Odegaard, J.I.; Morel, C.R.; Heredia, J.E.; Mwangi, J.W.; Ricardo-Gonzalez, R.R.; Goh, Y.P.; Eagle, A.R.; Dunn, S.E.; Awakuni, J.U. PPAR-δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 2009, 15, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Liu, J.; Lau, C.W.; Huang, Y. IL-6 in diabetes and cardiovascular complications. Br. J. Pharmacol. 2014, 171, 3595–3603. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Marco, L.; Rodríguez-Calvo, R.; El Kochairi, I.; Palomer, X.; Michalik, L.; Wahli, W.; Vázquez-Carrera, M. Activation of peroxisome proliferator-activated receptor-β/-δ (PPAR-β/-δ) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes. Diabetes 2011, 60, 1990–1999. [Google Scholar] [CrossRef] [PubMed]
- Coll, T.; Alvarez-Guardia, D.; Barroso, E.; Gómez-Foix, A.M.; Palomer, X.; Laguna, J.C.; Vázquez-Carrera, M. Activation of peroxisome proliferator-activated receptor-δ by GW501516 prevents fatty acid-induced nuclear factor-kB activation and insulin resistance in skeletal muscle cells. Endocrinology 2010, 151, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, S.; Nguyen-Tran, V.; Baré, O.; Huang, X.; Spiegelman, B.; Wu, Z. PPARδ agonism activates fatty acid oxidation via PGC-1α but does not increase mitochondrial gene expression and function. J. Biol. Chem. 2009, 284, 18624–18633. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Burkart-Hartman, E.M.; Han, D.H.; Finck, B.; Leone, T.C.; Smith, E.Y.; Ayala, J.E.; Holloszy, J.; Kelly, D.P. The nuclear receptor PPARβ/δ programs muscle glucose metabolism in cooperation with AMPK and MEF2. Genes Dev. 2011, 25, 2619–2630. [Google Scholar] [CrossRef] [PubMed]
- Salvadó, L.; Barroso, E.; Gómez-Foix, A.M.; Palomer, X.; Michalik, L.; Wahli, W.; Vázquez-Carrera, M. PPARβ/δ prevents endoplasmic reticulum stress-associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2014, 57, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Marco, L.; Barroso, E.; El Kochairi, I.; Palomer, X.; Michalik, L.; Wahli, W.; Vázquez-Carrera, M. The peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia 2012, 55, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.; Ham, S.A.; Lee, W.J.; Hwang, S.I.; Park, J.A.; Hwang, J.S.; Hur, J.; Shin, H.C.; Han, S.G.; Lee, C.H.; et al. Ligand-dependent Interaction of PPARδ with T Cell Protein Tyrosine Phosphatase 45 Enhances Insulin Signaling. Diabetes 2017, 67, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [PubMed]
- Christodoulides, C.; Dyson, P.; Sprecher, D.; Tsintzas, K.; Karpe, F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J. Clin. Endocrinol. Metab. 2009, 94, 3594–3601. [Google Scholar] [CrossRef] [PubMed]
- Vetere, A.; Choudhary, A.; Burns, S.M.; Wagner, B.K. Targeting the pancreatic β-cell to treat diabetes. Nat. Rev. Drug Discov. 2014, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Daoudi, M.; Hennuyer, N.; Borland, M.G.; Touche, V.; Duhem, C.; Gross, B.; Caiazzo, R.; Kerr-Conte, J.; Pattou, F.; Peters, J.M.; et al. PPARβ/δ activation induces enteroendocrine L cell GLP-1 production. Gastroenterology 2011, 140, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tong, Y.; Gong, M.; Lu, Y.; Wang, C.; Zhou, M.; Yang, Q.; Mao, T.; Tong, N. Activation of PPARβ/δ protects pancreatic β cells from palmitate-induced apoptosis by upregulating the expression of GLP-1 receptor. Cell Signal. 2014, 26, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Ravnskjaer, K.; Frigerio, F.; Boergesen, M.; Nielsen, T.; Maechler, P.; Mandrup, S. PPARδ is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J. Lipid Res. 2010, 51, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Abbott, M.J.; Ahmadian, M.; Lopes, A.B.; Wang, Y.; Sul, H.S. Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet β cells. Cell Metab. 2013, 18, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Inada, Y.; Nakano, S.; Tamura, T.; Takahashi, T.; Maruyama, K.; Yamazaki, Y.; Kuroda, J.; Shibata, N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARδ agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur. J. Pharmacol. 2006, 536, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Chen, C.T.; Cheng, K.C.; Li, Y.X.; Yeh, C.H.; Cheng, J.T. Pharmacological activation of peroxisome proliferator-activated receptor δ improves insulin resistance and hepatic steatosis in high fat diet-induced diabetic mice. Horm. Metab. Res. 2011, 43, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.E.; Steinberg, G.R.; et al. PPARδ activation attenuates hepatic steatosis in Ldlr−/− mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Xie, X.; Fan, Y.; Tian, J.; Guan, Y.; Wang, X.; Zhu, Y.; Wang, N. Peroxisome proliferator-activated receptor-δ induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology 2008, 48, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Garbacz, W.G.; Huang, J.T.; Higgins, L.G.; Wahli, W.; Palmer, C.N. PPARα Is Required for PPARδ Action in Regulation of Body Weight and Hepatic Steatosis in Mice. PPAR Res. 2015, 2015, 927057. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor δ/β in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Choi, R.; Kim, H.M.; Cho, E.J.; Kim, B.H.; Choi, Y.S.; Naowaboot, J.; Lee, E.Y.; Yang, Y.C.; Shin, J.Y.; et al. Peroxisome proliferator-activated receptor δ agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp. Mol. Med. 2012, 44, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Kostadinova, R.; Montagner, A.; Gouranton, E.; Fleury, S.; Guillou, H.; Dombrowicz, D.; Desreumaux, P.; Wahli, W. GW501516-activated PPARβ/δ promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Wang, D.; Katkuri, S.; Wang, H.; Dey, S.K.; DuBois, R.N. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat. Med. 2004, 10, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Gonzalez, F.J.; Müller, R. Establishing the Role of PPARβ/δ in Carcinogenesis. Trends Endocrinol. Metab. 2015, 26, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Müller, R. PPARβ/δ in human cancer. Biochimie 2017, 136, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; Sheridan, D.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Peña, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vázquez-Carrera, M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. https://doi.org/10.3390/ijms19030913
Palomer X, Barroso E, Pizarro-Delgado J, Peña L, Botteri G, Zarei M, Aguilar D, Montori-Grau M, Vázquez-Carrera M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. International Journal of Molecular Sciences. 2018; 19(3):913. https://doi.org/10.3390/ijms19030913
Chicago/Turabian StylePalomer, Xavier, Emma Barroso, Javier Pizarro-Delgado, Lucía Peña, Gaia Botteri, Mohammad Zarei, David Aguilar, Marta Montori-Grau, and Manuel Vázquez-Carrera. 2018. "PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders" International Journal of Molecular Sciences 19, no. 3: 913. https://doi.org/10.3390/ijms19030913
APA StylePalomer, X., Barroso, E., Pizarro-Delgado, J., Peña, L., Botteri, G., Zarei, M., Aguilar, D., Montori-Grau, M., & Vázquez-Carrera, M. (2018). PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. International Journal of Molecular Sciences, 19(3), 913. https://doi.org/10.3390/ijms19030913