Stress-Activated Protein Kinases in Spinal Cord Injury: Focus on Roles of p38


{kind=link}
Abstract
1. Introduction
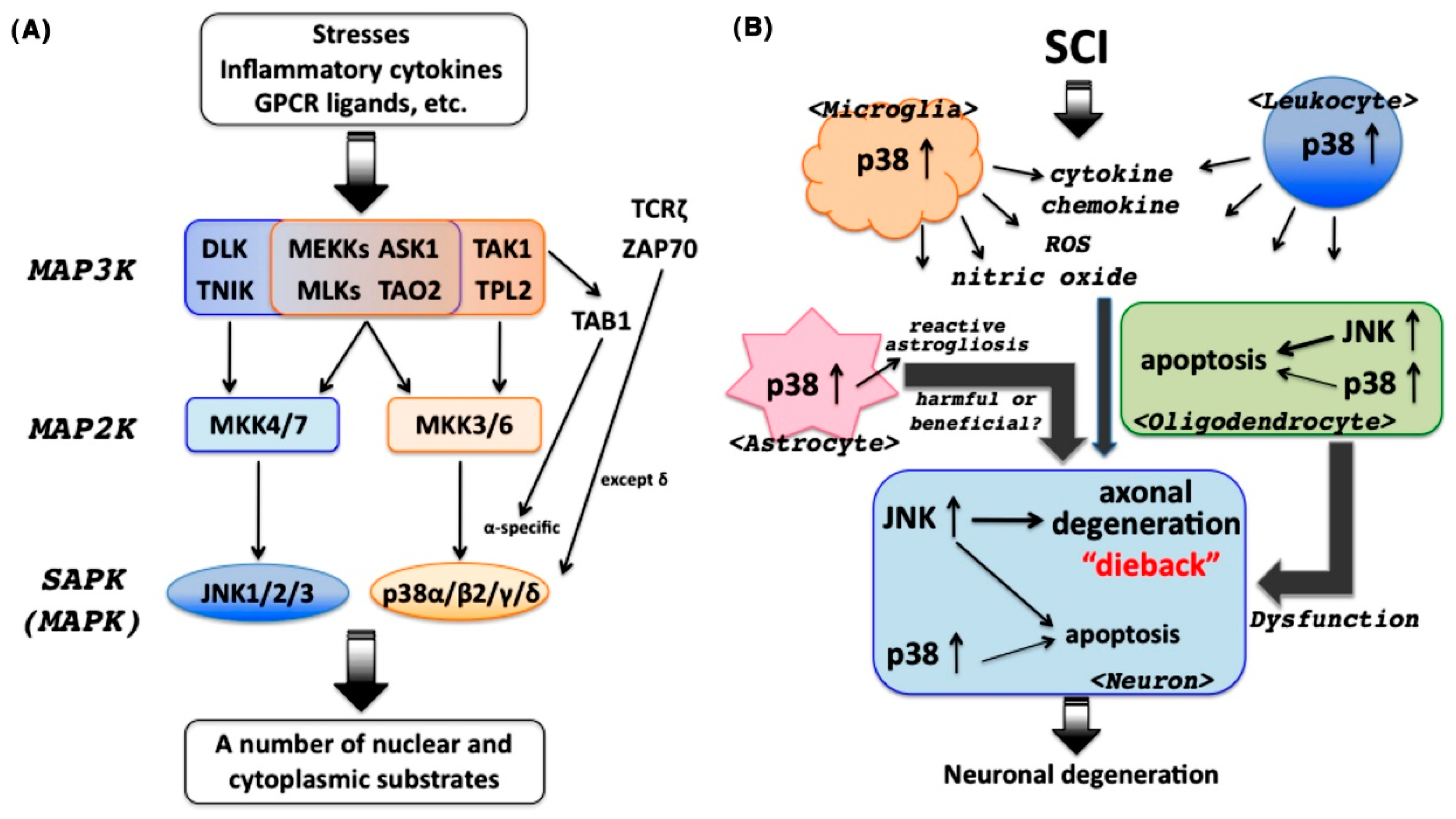
2. SAPKs in the CNS
3. Involvement of JNK in the Pathogenesis of SCI
4. Involvement of p38 in the Pathogenesis of SCI
4.1. p38 as a Central Player in Inflammatory Responses
4.2. Spatial Activation of p38 after SCI
4.3. Correlation between p38 Pathway Inhibition and Functional Recovery after SCI
4.3.1. p38 Inhibitors
4.3.2. Minocycline
4.3.3. Plant-Derived Agents
4.4. p38 as a Promising Target for Therapeutic Intervention in SCI?
5. Closing Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β |
| AD | Alzheimer’s disease |
| ARE | AU-rich element |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| ATF2 | Activating transcription factor 2 |
| BDA | Biotinylated dextran amine |
| CNS | Central nervous system |
| CREB | cAMP response element binding protein |
| CSAID | Cytokine-suppressive anti-inflammatory drug |
| CSF | Cerebrospinal fluid |
| CST | Corticospinal tract |
| DCs | Dendritic cells |
| DLK | Dual leucine zipper-bearing kinase |
| EAE | Experimental autoimmune encephalomyelitis |
| eIF4E | Eukaryotic translation initiation factor 4E |
| HPAIV | Highly pathogenic avian influenza virus |
| IL | Interleukin |
| IFN-γ | Interferon-γ |
| iNOS | Inducible nitric oxide synthase |
| JNK | c-Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MAP2K | MAPK kinase |
| MAP3K | MAPKK kinase |
| Mcl-1 | Myeloid cell leukemia sequence-1 |
| miR | MicroRNA |
| MIP | Macrophage inflammatory protein |
| MK2 | MAPK-activated protein kinase 2 |
| MLK | Mixed lineage kinase |
| Mnk | MAPK-interacting serine/threonine kinase |
| MPTP | 1-Methyl-4-phenyl-1,2,4,6-tetrahydropyridine |
| MSK | Mitogen- and stress-activated kinase |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NGF | Nerve growth factor |
| p75NTR | p75 Neurotrophin receptor |
| SAPK | Stress-activated protein kinase |
| SCI | Spinal cord injury |
| SCG10 | Superior cervical ganglion 10 |
| TAB1 | TAK1-binding protein 1 |
| TAK1 | TGF-β-activated kinase 1 |
| TAO2 | Thousand-and-one amino acid kinase 2 |
| TGF-β | Transforming growth factor-β |
| Th | T-helper |
| TNF-α | Tumor necrosis factor-α |
| TNIK | TNF receptor-associated factor 2- and NCK-interacting protein kinase |
| TLR | Toll-like receptor |
| TPL2 | Tumor progression locus 2 |
| TrkA | Tyrosine receptor kinase A |
| TWY | Tiptoe-walking Yoshimura |
| UV | Ultraviolet |
| ZAP70 | ζ chain-associated protein kinase |
References
- Horner, P.J.; Gage, F.H. Regenerating the damaged central nervous system. Nature 2000, 407, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Hilton, B.J.; Moulson, A.J.; Tetzlaff, W. Neuroprotection and secondary damage following spinal cord injury: Concepts and methods. Neurosci. Lett. 2017, 652, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; He, X.; Ren, Y. Function of microglia and macrophages in secondary damage after spinal cord injury. Neural Regen. Res. 2014, 9, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Sadowsky, C.L.; McDonald, J.W. Restoring function after spinal cord injury. Neurologist 2003, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rincón, M.; Davis, R.J. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 2009, 228, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Le-Niculescu, H.; Bonfoco, E.; Kasuya, Y.; Claret, F.-X.; Green, D.R.; Karin, M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 1999, 19, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [PubMed]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Young, J.; Ulevitch, R.J.; Han, J. The primary structure of p38γ: A new member of p38 group of MAP kinases. Biochem. Biophys. Res. Commun. 1996, 228, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38δ. J. Biol. Chem. 1997, 272, 30122–30128. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; McDonnell, P.C.; Gum, R.J.; Hand, A.T.; Lee, J.C.; Young, P.R. Novel homologues of CSBP/p38 MAP kinase: Activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem. Biophys. Res. Commun. 1997, 235, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Ichijo, H. Neuronal p38 MAPK signalling: An emerging regulator of cell fate and function in the nervous system. Genes Cells 2002, 7, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Gram, H.; Di Padova, F.; Huang, B.; New, L.; Ulevitch, R.J.; Luo, Y.; Han, J. MAPKK-Independent Activation of p38α Mediated by TAB1-Dependent Autophosphorylation of p38α. Science 2002, 295, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.M.; Mittelstadt, P.R.; Guszczynski, T.; Copeland, T.D.; Yamaguchi, H.; Appella, E.; Fornace, A.J., Jr.; Ashwell, J.D. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat. Immunol. 2005, 6, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Carboni, L.; Carletti, R.; Tacconi, S.; Corti, C.; Ferraguti, F. Differential expression of SAPK isoforms in the rat brain. An in situ hybridisation study in the adult rat brain and during post-natal development. Mol. Brain Res. 1998, 60, 57–68. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Kuan, C.; Whitmarsh, A.J.; Rinócn, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Brecht, S.; Kirchhof, R.; Chromik, A.; Willesen, M.; Nicolaus, T.; Raivich, G.; Wessig, J.; Waetzig, V.; Goetz, M.; Claussen, M.; et al. Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 2005, 21, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.Y.; Yang, D.D.; Samanta Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Sudo, T.; Senftleben, U.; Dadak, A.M.; Johnson, R.; Karin, M. Requirement for p38α in erythropoietin expression: A role for stress kinases in erythropoiesis. Cell 2000, 102, 221–231. [Google Scholar] [CrossRef]
- Adams, R.H.; Porras, A.; Alonso, G.; Jones, M.; Vintersten, K.; Panelli, S.; Valladares, A.; Perez, L.; Klein, R.; Nebreda, A.R. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 2000, 6, 109–116. [Google Scholar] [CrossRef]
- Beardmore, V.A.; Hinton, H.J.; Eftychi, C.; Apostolaki, M.; Armaka, M.; Darragh, J.; McIlrath, J.; Carr, J.M.; Armit, L.J.; Clacher, C.; et al. Generation and characterization of p38β (MAPK11) gene-targeted mice. Mol. Cell. Biol. 2005, 25, 10454–10464. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.A.; Le Grand, F.; Scimè, A.; Kuang, S.; von Maltzahn, J.; Seale, V.; Cuenda, A.; Ranish, J.A.; Rudnicki, M.A. p38-γ-dependent gene silencing restricts entry into the myogenic differentiation program. J. Cell Biol. 2009, 187, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Schindler, E.M.; Hindes, A.; Gribben, E.L.; Burns, C.J.; Yin, Y.; Lin, M.; Owen, R.J.; Longmore, G.D.; Kissling, G.E.; Arthur, J.S.C.; et al. p38δ mitogen-activated protein kinase is essential for skin tumor development in mice. Cancer Res. 2004, 69, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.; Huang, P.; Qi, Z.; Zhang, N.; Han, S.; Fang, L.; Li, J. Cell type-specific activation of p38 MAPK in the brain regions of hypoxic preconditioned mice. Neurochem. Int. 2007, 51, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Sudo, T.; Kasuya, Y.; Siga, T.; Hu, B.; Osada, H. Immunolocalization of p38 MAPK in mouse brain. Brain Res. 2000, 887, 350–358. [Google Scholar] [CrossRef]
- Bruchas, M.R.; Macey, T.A.; Lowe, J.D.; Chavkin, C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.R.; Ryou, M.; Poteet, E.; Wen, Y.; He, R.; Sun, F.; Yuan, F.; Jin, K.; Yang, S. Involvement of p38 MAPK in reactive astrogliosisinduced by ischemic stroke. Brain Res. 2014, 1551, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Uemura, K.; Kuzuya, A.; Maesako, M.; Asada-Utsugi, M.; Kubota, M.; Aoyagi, N.; Yoshioka, K.; Okawa, K.; Inoue, H.; et al. N-cadherin regulates p38 MAPK signaling via association with JNK-associated leucine zipper protein: Implications for neurodegeneration in Alzheimer disease. J. Biol. Chem. 2011, 286, 7619–7628. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Harada, C.; Namekata, K.; Matsuzawa, A.; Camps, M.; Ji, H.; Swinnen, D.; Jorand-Lebrun, C.; Muzerelle, M.; Vitte, P.A.; et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol. Med. 2010, 12, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Wang, Y.; Vogel, P.; Kanneganti, T.-D.; Otsu, K.; Chi, H. Signaling via the kinase p38α programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat. Immunol. 2012, 13, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Jirmanova, L.; Torchia, M.L.G.; Sarma, N.D.; Mittelstadt, P.R.; Ashwell, J.D. Lack of the T-cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood 2011, 118, 3280–3289. [Google Scholar] [CrossRef] [PubMed]
- Noubade, R.; Krementsov, D.N.; Rio, R.; Thornton, T.; Nagaleekar, V.; Saligrama, N.; Spitzack, A.; Spach, K.; Sabio, G.; Davis, R.J.; et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood 2011, 118, 3290–3300. [Google Scholar] [CrossRef] [PubMed]
- Namiki, K.; Matsunaga, H.; Yoshioka, K.; Tanaka, K.; Murata, K.; Ishida, J.; Sakairi, A.; Kim, J.; Tokuhara, N.; Shibakawa, N.; et al. Mechanism for p38α-mediated experimental autoimmune encephalomyelitis. J. Biol. Chem. 2012, 287, 24228–24238. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ranaivo, H.R.; Roy, S.M.; Hu, W.; Craft, J.M.; McNamara, L.K.; Chico, L.W.; Van Eldik, L.J.; Watterson, D.M. A novel p38α MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflamm. 2007, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Yone, K.; Sakou, T.; Wada, S.; Nagamine, T.; Niiyama, T.; Ichijo, H. Induction of apoptosis signal regulating kinase 1 (ASK1) after spinal cord injury in rats: Possible involvement of ASK1-JNK and -p38 pathways in neuronal apoptosis. J. Neuropathol. Exp. Neurol. 1999, 58, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Setoguchi, T.; Yone, K.; Komiy, S. Expression of apoptosis signal-regulating kinase 1 in mouse spinal cord under chronic mechanical compression: Possible involvement of the stress-activated mitogen-activated protein kinase pathways in spinal cord cell apoptosis. Spine 2008, 33, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.M.; Tep, C.; Yune, T.Y.; Zhou, X.Z.; Uchida, T.; Lu, K.P.; Yoon, S.O. Opposite regulation of oligodendrocyte apoptosis by JNK3 and Pin1 after spinal cord injury. J. Neurosci. 2007, 27, 8395–8404. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, V.; Kujala, P.; Klumperman, J.; Goldstein, L.S.B. Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 2005, 168, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Miller, B.R.; Babetto, E.; Cho, Y.; Sasaki, Y.; Qayum, S.; Russler, E.V.; Cavalli, V.; Milbrandt, J.; DiAntonio, A. SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. USA 2012, 109, E3696–E3705. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Ueno, M.; Lee, S.; Nakamura, Y.; Sato, A.; Yoshimura, K.; Kishima, H.; Yoshimine, T.; Yamashita, T. C-Jun N-terminal kinase induces axonal degeneration and limits motor recovery after spinal cord injury in mice. Neurosci. Res. 2011, 71, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Chen, X.; Morel, M.-P.; Doulazmi, M.; Sclip, A.; Cannaya, V.; Veglianese, P.; Kraftsik, R.; Mariani, J.; Borsello, T.; et al. Specific inhibition of the JNK pathway promotes locomotor recovery and neuroprotection after mouse spinal cord injury. Neurobiol. Dis. 2012, 46, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Barnat, M.; Enslen, H.; Propst, F.; Davis, R.J.; Soares, S.; Nothias, F. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J. Neurosci. 2010, 30, 7804–7816. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, D.C.; Oh, T.H.; Yune, T.Y. Analgesic effect of acupuncture is mediated via inhibition of JNK activation in astrocytes after spinal cord injury. PLoS ONE 2013, 8, e73948. [Google Scholar] [CrossRef] [PubMed]
- Martini, A.C.; Forner, S.; Koepp, J.; Rae, G.A. Inhibition of spinal c-Jun-NH2-terminal kinase (JNK) improves locomotor activity of spinal cord injured rats. Neurosci. Lett. 2016, 621, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S. Inflammation and apoptosis: Linked therapeutic targets in spinal cord injury. Trends Mol. Med. 2004, 10, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Okada, S. The pathophysiological role of acute inflammation after spinal cord injury. Inflamm. Regen. 2016, 36, 20. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.E.; Sully, G.; Clark, A.R.; Saklatvala, J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilization. Cell. Signal. 2004, 16, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, M.; Kirkwood, K.L. p38α stabilizes interleukin-6 mRNA via multipleAU-rich elements. J. Biol. Chem. 2008, 283, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Chen, J.; Otsuka, M.; Mols, J.; Ren, S.; Wang, Y.; Han, J. Macrophage deletion of p38α partially impairs lipopolysaccharide-induced cellular activation. J. Immunol. 2008, 180, 5075–5082. [Google Scholar] [CrossRef] [PubMed]
- Krementsov, D.N.; Thornton, T.M.; Teuscher, C.; Rinócn, M. The emerging role of p38 mitogen-activated protein kinase in multiple sclerosis and its models. Mol. Cell. Biol. 2013, 33, 3728–3734. [Google Scholar] [CrossRef] [PubMed]
- Nagaleekar, V.K.; Sabio, G.; Aktan, I.; Chant, A.; Howe, I.W.; Thornton, T.M.; Benoit, P.J.; Davis, R.J.; Rincon, M.; Boyson, J.E. Translational control of NKT cell cytokine production by p38 MAPK. J. Immunol. 2011, 186, 4140–4146. [Google Scholar] [CrossRef] [PubMed]
- Börgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, B.R.; Wang, X.; Kuang, F.; Duan, X.L.; Jiao, X.Y.; Ju, G. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006, 79, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Stirling, D.P.; Liu, J.; Plunet, W.; Steeves, J.D.; Tetzlaff, W. SB203580, a p38 mitogen-activated protein kinase inihbitor, fails to improve functional outcome following a moderate spinal cord injury in rat. Neuroscience 2008, 155, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Kong, K.M.; Qi, W.L.; Ye, W.L.; Song, P.S. Interleukin-1 beta induction of neuron apoptosis depends on p38 mitogen-activated protein kinase activity after spinal cord injury. Acta Pharmacol. Sin. 2005, 26, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, H.; Ogata, T.; Morino, T.; Chuai, M.; Yamamoto, H. Continuous intrathecal infusion of SB203580, a selective inhibitor of p38 mitogen-activated protein kinase, reduces the damage of hind-limb function after thoracic spinal cord injury in rat. Neurosci. Res. 2003, 47, 209–217. [Google Scholar] [CrossRef]
- Umezawa, H.; Naito, Y.; Tanaka, K.; Yoshioka, K.; Suzuki, K.; Sudo, T.; Hagihara, M.; Hatano, M.; Tatsumi, K.; Kasuya, Y. Genetic and pharmacological inhibition of p38α improves locomotor recovery after spinal cord injury. Front. Pharmacol. 2017, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Tikka, T.M.; Koistinaho, J.E. Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J. Immunol. 2001, 166, 7527–7533. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, Y.; Dodel, R.; Farlow, M.R.; Paul, S.M.; Du, Y. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci. Lett. 2011, 315, 61–64. [Google Scholar] [CrossRef]
- Hua, X.; Svensson, C.I.; Matsui, T.; Fitzsimmons, B.; Yaksh, T.L.; Webb, M. Intrathecal minocycline attenuates peripheral inflammationinduced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur. J. Neurosci. 2005, 22, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Yune, T.Y.; Lee, J.Y.; Jung, G.Y.; Kim, S.J.; Jiang, M.H.; Kim, Y.C.; Oh, Y.J.; Markelonis, G.J.; Oh, T.H. Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J. Neurosci. 2007, 27, 7751–7761. [Google Scholar] [CrossRef] [PubMed]
- Beattie, M.S.; Harrington, A.W.; Lee, R.; Kim, J.Y.; Boyce, S.L.; Longo, F.M.; Bresnahan, J.C.; Hempstead, B.L.; Yoon, S.O. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 2002, 36, 375–386. [Google Scholar] [CrossRef]
- Stirling, D.P.; Koochesfahani, K.M.; Steeves, J.D.; Tetzlaff, W. Minocycline as a neuroprotective agent. Neuroscientist 2005, 11, 308–322. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Bachstetter, A.D.; Van Eldik, L.J. Deficiency in p38β MAPK fails to inhibit cytokine production or protect neurons against inflammatory insult in in vitro and in vivo mouse models. PLoS ONE 2015, 8, e56852. [Google Scholar] [CrossRef] [PubMed]
- Lawson, S.K.; Dobrikova, E.Y.; Shveygert, M.; Gromeier, M. p38α mitogen-activated protein kinase depletion and repression of signal transduction to translation machinery by miR-124 and -128 in neurons. Mol. Cell. Biol. 2013, 33, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Casha, S.; Zygun, D.; McGowan, M.D.; Bains, I.; Yong, V.W.; Hurlbert, R.J. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 2012, 135, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Urdzikova, L.M.; Karova, K.; Ruzicka, J.; Kloudova, A.; Shannon, C.; Dubisova, J.; Murali, R.; Kubinova, S.; Sykova, E.; Jhanwar-Uniyal, M.; et al. The anti-inflammatory compound curcumin enhances locomotor and sensory recovery after spinal cord injury in rats by immunomodulation. Int. J. Mol. Sci. 2016, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.; Wang, C.; Yang, C. NF-kappaB signaling pathways in neurological inflammation. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Bracchi-Ricard, V.; Hu, W.; Frydel, B.; Bramwell, A.; Karmally, S.; Green, E.J.; Bethea, J.R. Inhibition of astroglial nuclear factor κB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 2005, 202, 145–156. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, G.; Ye, J.; Yang, L.; Hong, Y.; Liu, G.; Zhong, H.; Cai, X. Effect of curcumin on acute spinal cord injury in mice via inhibition of inflammation and TAK1 pathway. Pharmacol. Rep. 2017, 69, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Su, B.; Zhu, H.; Chen, C.; Zhao, G. Protective effect of geraniol inhibits inflammatory response, oxidative stress and apoptosis in traumatic injury of the spinal cord through modulation of NF-κB and p38 MAPK. Exp. Ther. Med. 2016, 12, 3607–3613. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Fu, C.; Wang, Z.; Zhang, Z.; Wang, H.; Liu, Y. Asiaticoside attenuates the effects of spinal cord injury through antioxidant and anti-inflammatory effects, and inhibition of the p38-MAPK mechanism. Mol. Med. Rep. 2015, 12, 8294–8300. [Google Scholar] [CrossRef] [PubMed]
- Takanami-Ohnishi, Y.; Amano, S.; Kimura, S.; Asada, S.; Utani, A.; Maruyama, M.; Osada, H.; Tsunoda, H.; Irukayama-Tomobe, Y.; Goto, K.; et al. Essential role of p38 mitogen-activated protein kinase in contact hypersensitivity. J. Biol. Chem. 2002, 277, 37896–37903. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Amano, S.; Furuya, M.; Namiki, K.; Sakurai, K.; Nishiyama, M.; Sudo, T.; Tatsumi, K.; Kuriyama, T.; Kimura, S.; et al. Involvement of p38α mitogen-activated protein kinase in lung metastasis of tumor cells. J. Biol. Chem. 2006, 281, 36767–36775. [Google Scholar] [CrossRef] [PubMed]
- Namiki, K.; Nakamura, A.; Furuya, M.; Mizuhashi, S.; Matsuo, Y.; Tokuhara, N.; Sudo, T.; Hama, H.; Kuwaki, T.; Yano, S.; et al. Involvement of p38α in kainate-induced seizure and neuronal cell damage. J. Recept. Signal Transduct. 2007, 27, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Namiki, K.; Sudo, T.; Kasuya, Y. p38α controls self-renewal and fate decision of neurosphere-forming cells in adult hippocampus. FEBS Open Bio 2015, 5, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Van Eldik, L.J. The p38 MAP kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 2010, 1, 199–211. [Google Scholar] [PubMed]
- Godl, K.; Wissing, J.; Kurtenbach, A.; Habenberger, P.; Blencke, S.; Gutbrod, H.; Salassidis, K.; Stein-Gerlach, M.; Missio, A.; Cotton, M. An efficient proteomics method to identify the cellular targets of protein kinase inhibitors. Proc. Natl. Acad. Sci. USA 2003, 100, 15434–15439. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.X.; Zhuang, Z.Y.; Woolf, C.J.; Ji, R.R. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 2003, 23, 4017–4022. [Google Scholar] [PubMed]
- Ji, R.R.; Suter, M.R. p38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain 2007, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Johannesen, I.L.; Sindrup, S.H.; Bach, F.W.; Jensen, T.S. Pain and dysesthesia in patients with spinal cord injury: A postal survey. Spinal Cord 2001, 39, 256–262. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasuya, Y.; Umezawa, H.; Hatano, M. Stress-Activated Protein Kinases in Spinal Cord Injury: Focus on Roles of p38. Int. J. Mol. Sci. 2018, 19, 867. https://doi.org/10.3390/ijms19030867
Kasuya Y, Umezawa H, Hatano M. Stress-Activated Protein Kinases in Spinal Cord Injury: Focus on Roles of p38. International Journal of Molecular Sciences. 2018; 19(3):867. https://doi.org/10.3390/ijms19030867
Chicago/Turabian StyleKasuya, Yoshitoshi, Hiroki Umezawa, and Masahiko Hatano. 2018. "Stress-Activated Protein Kinases in Spinal Cord Injury: Focus on Roles of p38" International Journal of Molecular Sciences 19, no. 3: 867. https://doi.org/10.3390/ijms19030867
APA StyleKasuya, Y., Umezawa, H., & Hatano, M. (2018). Stress-Activated Protein Kinases in Spinal Cord Injury: Focus on Roles of p38. International Journal of Molecular Sciences, 19(3), 867. https://doi.org/10.3390/ijms19030867