Chondroprotective Effects and Mechanisms of Dextromethorphan: Repurposing Antitussive Medication for Osteoarthritis Treatment

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
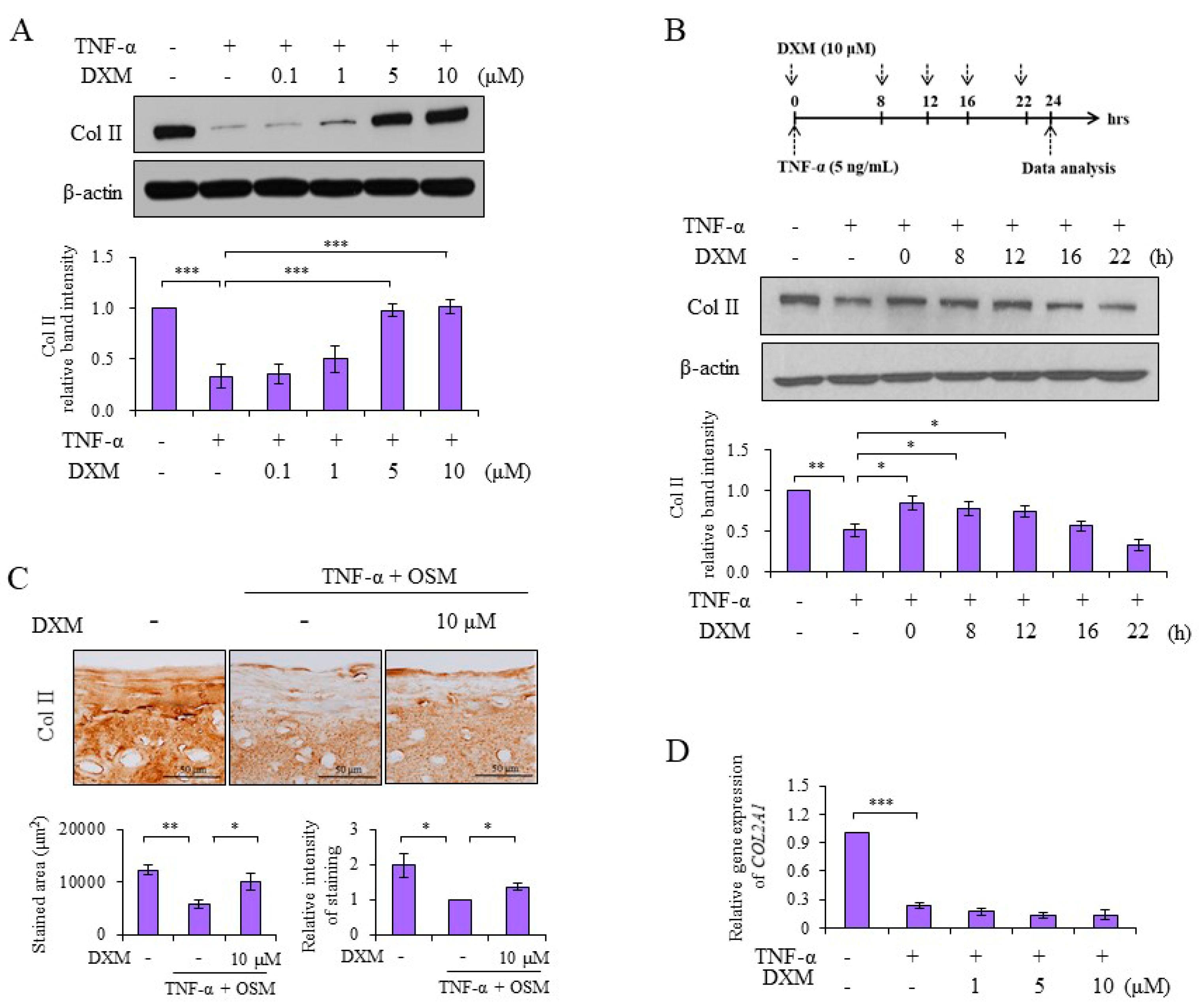
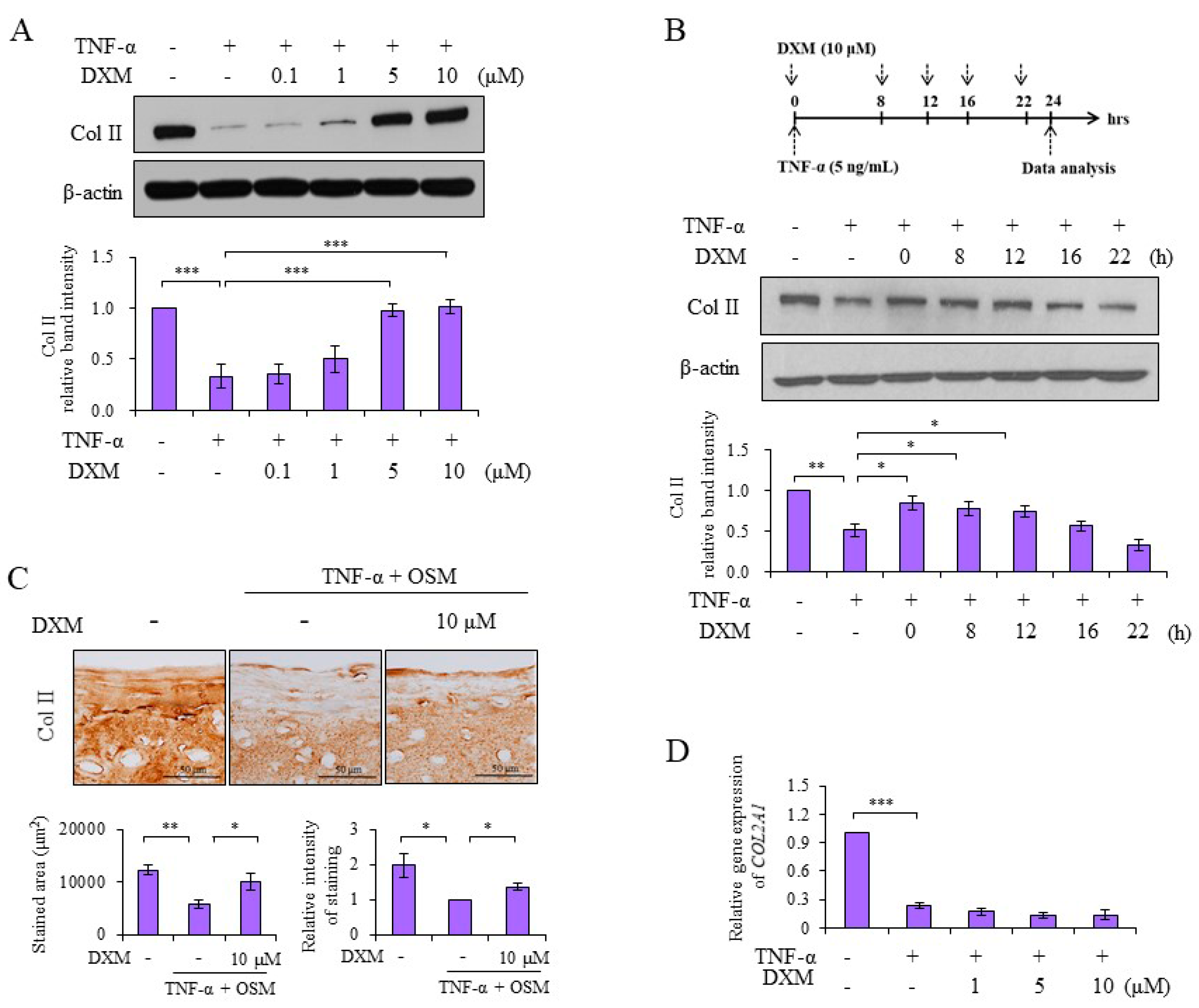
2.1. DXM Counteracted TNF-α-Mediated Reduction of Col II
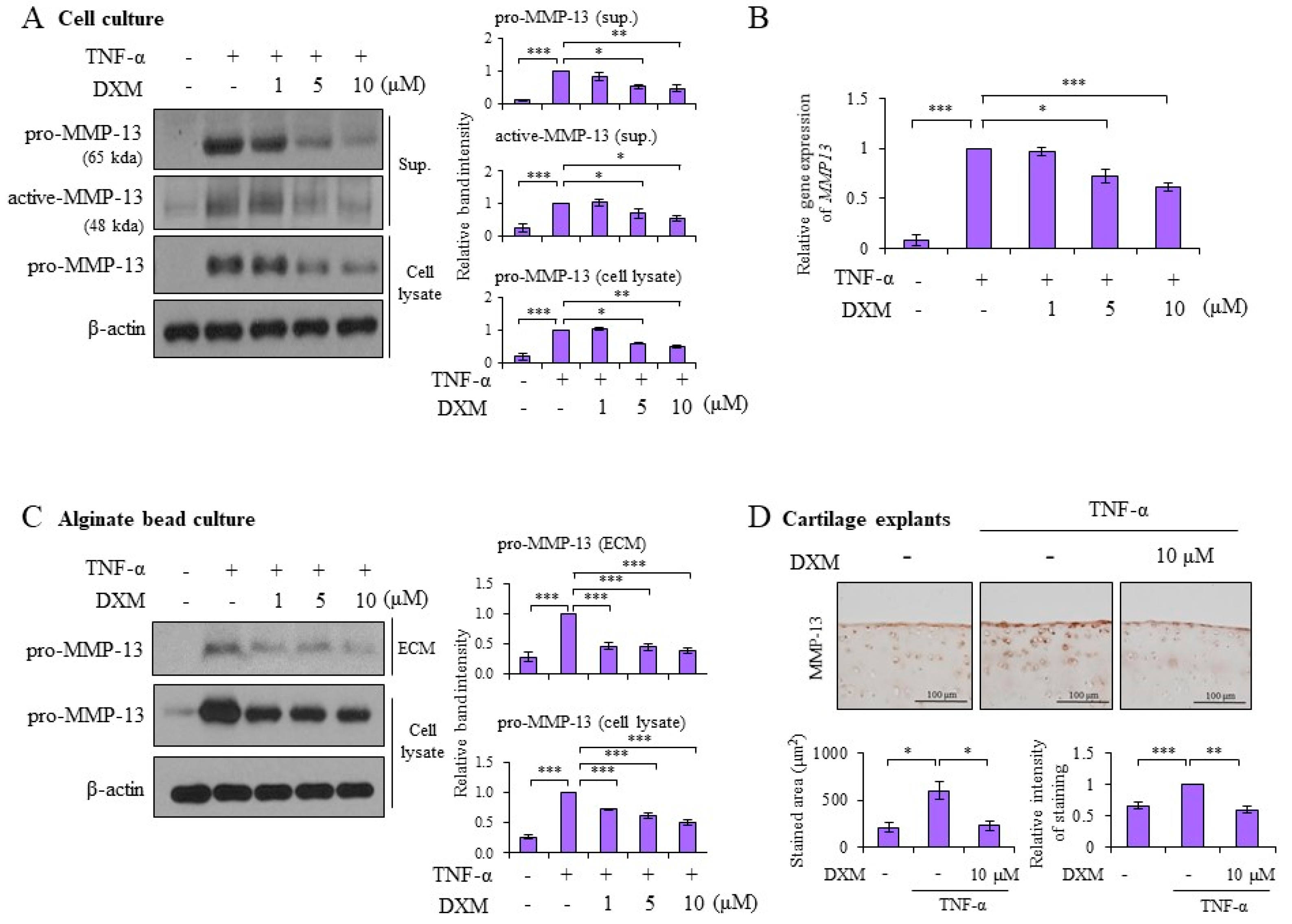
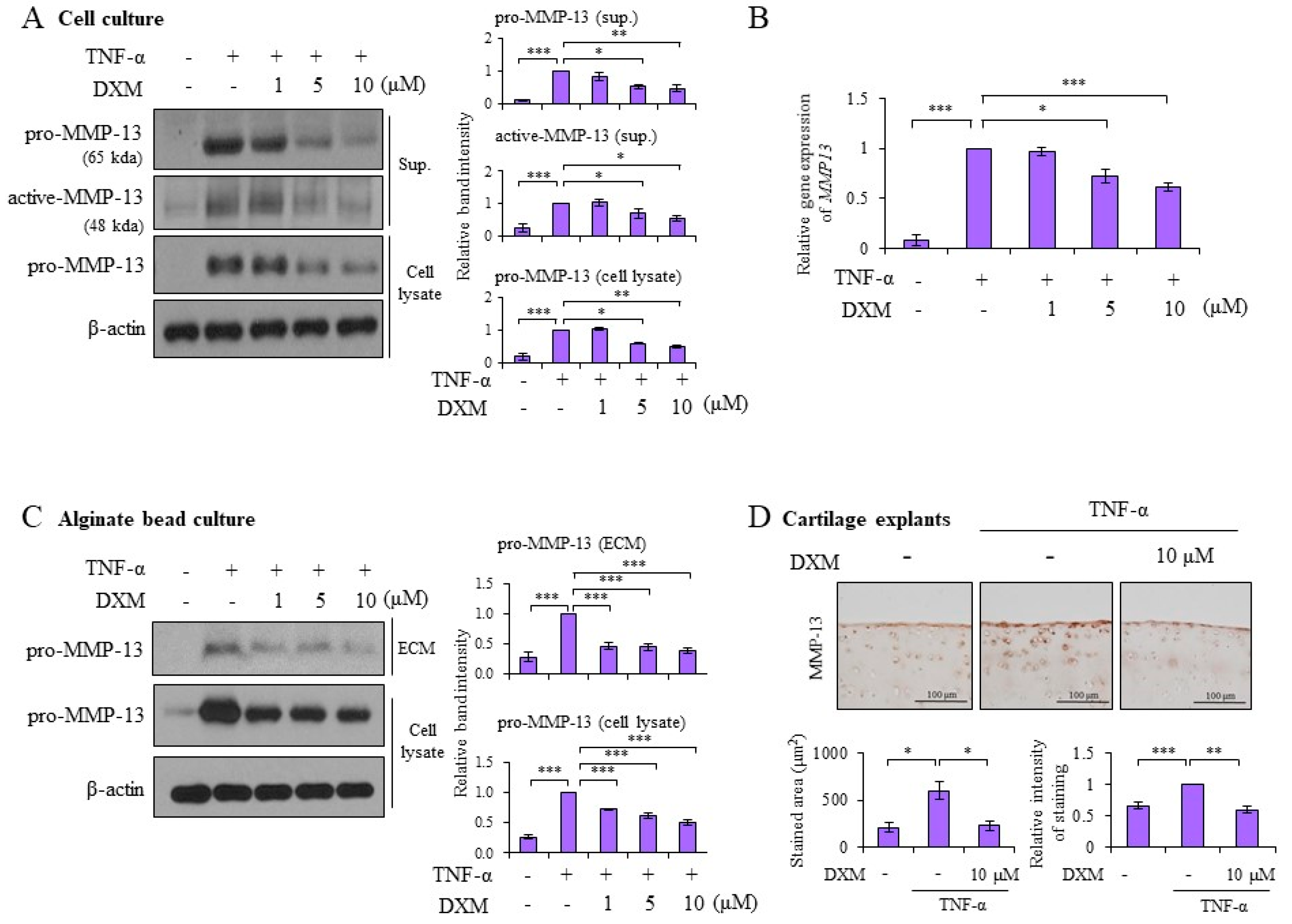
2.2. DXM Decreased TNF-α-Induced MMP-13 Expression in Chondrocyte Cultures, 3-D Alginate Beads, and Cartilage Explants
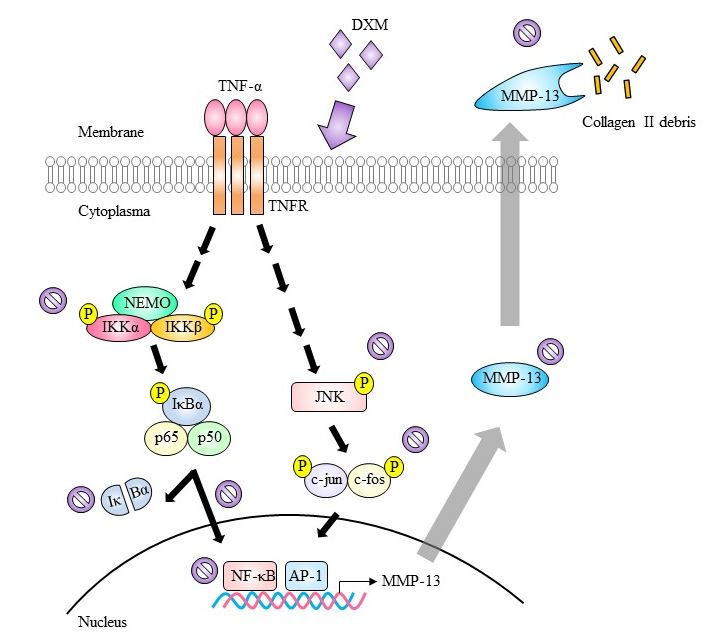
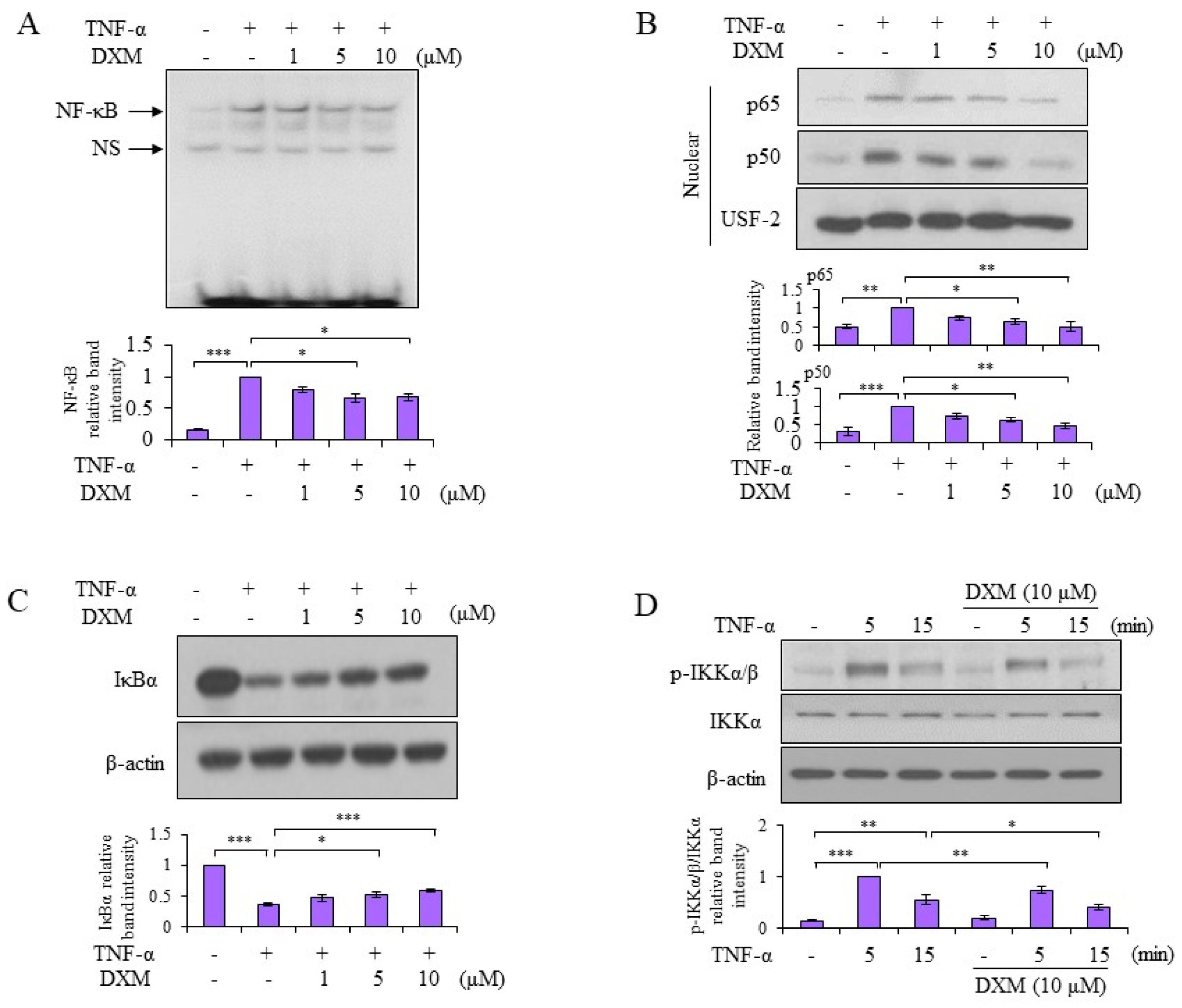
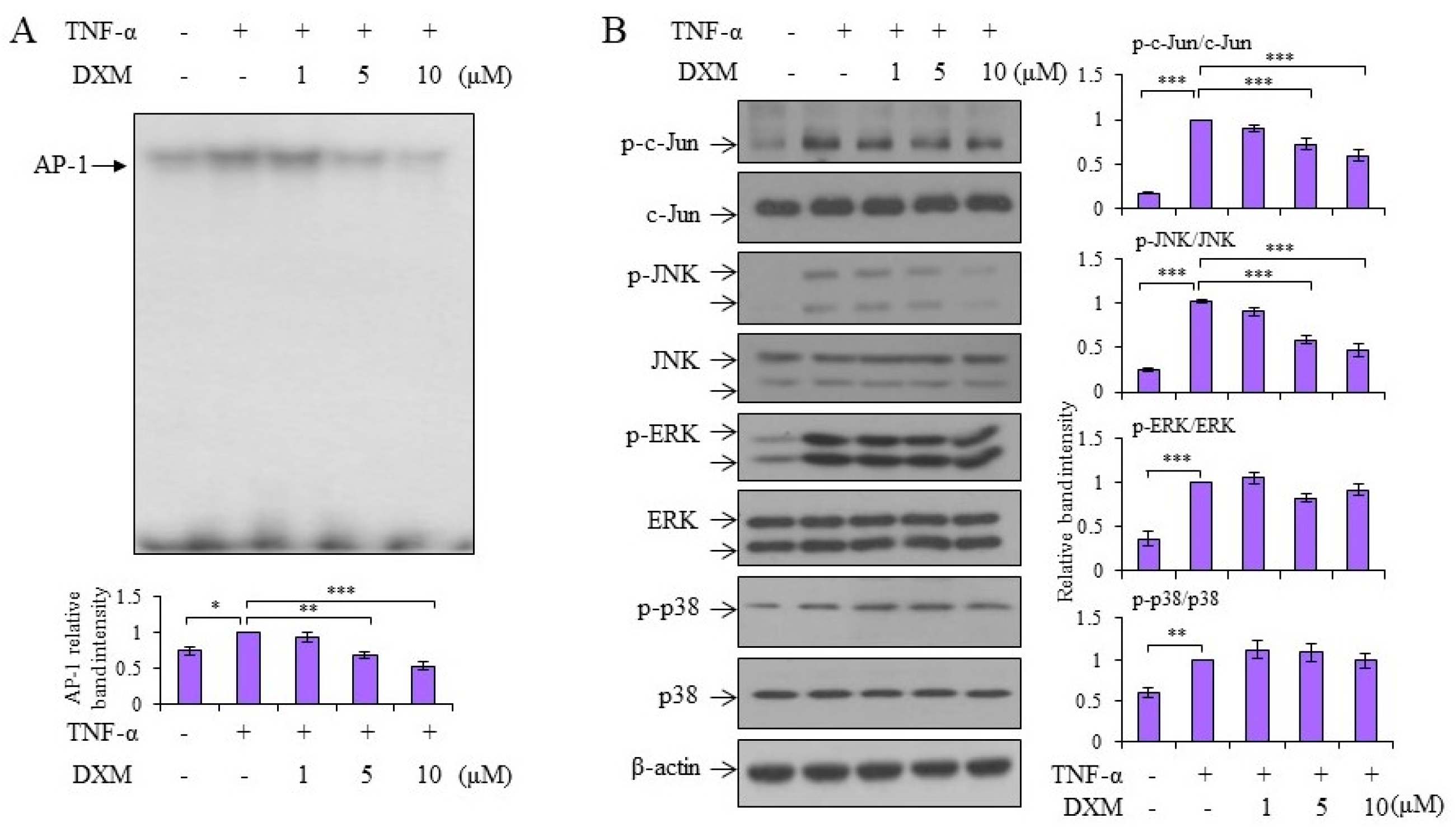
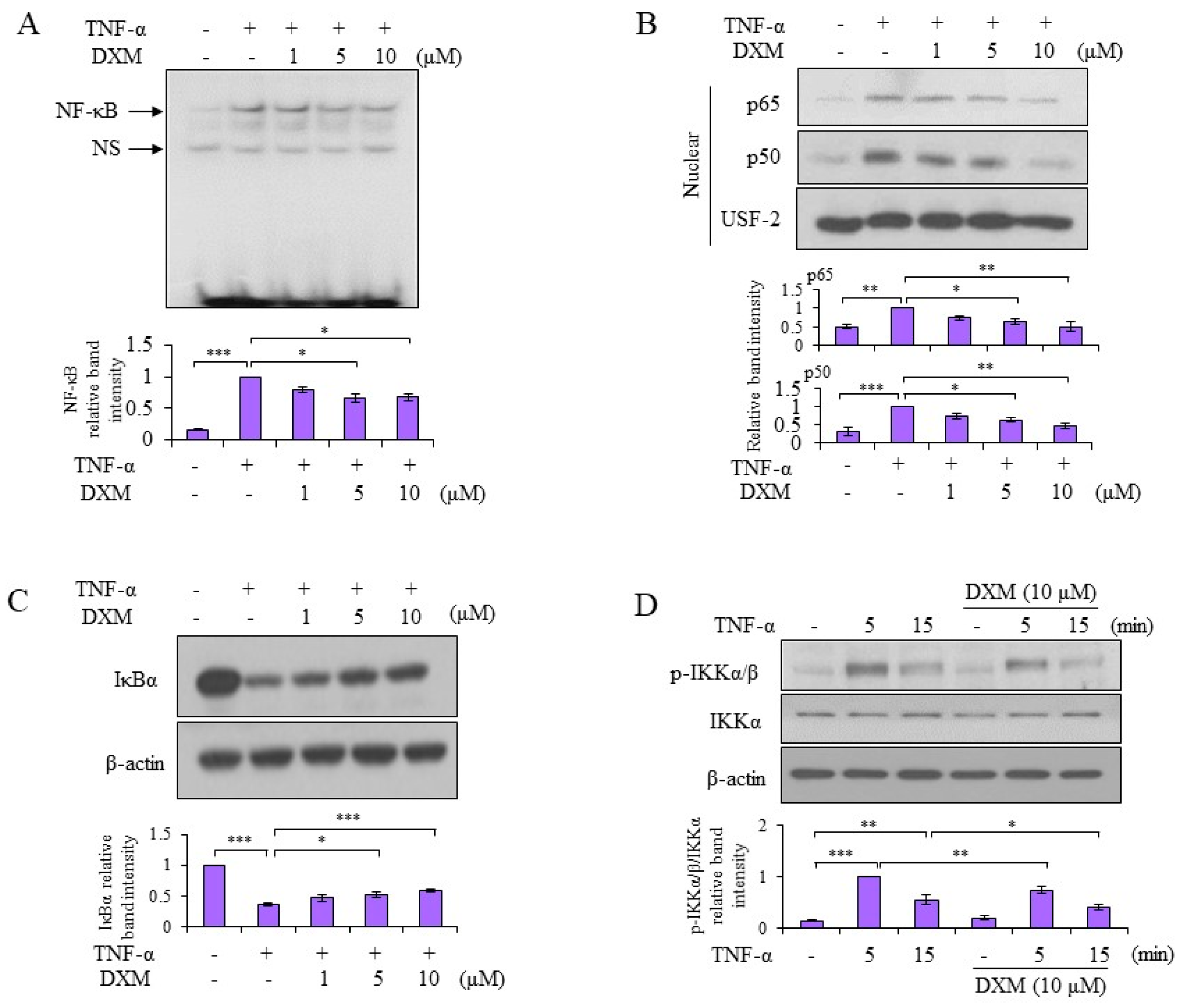
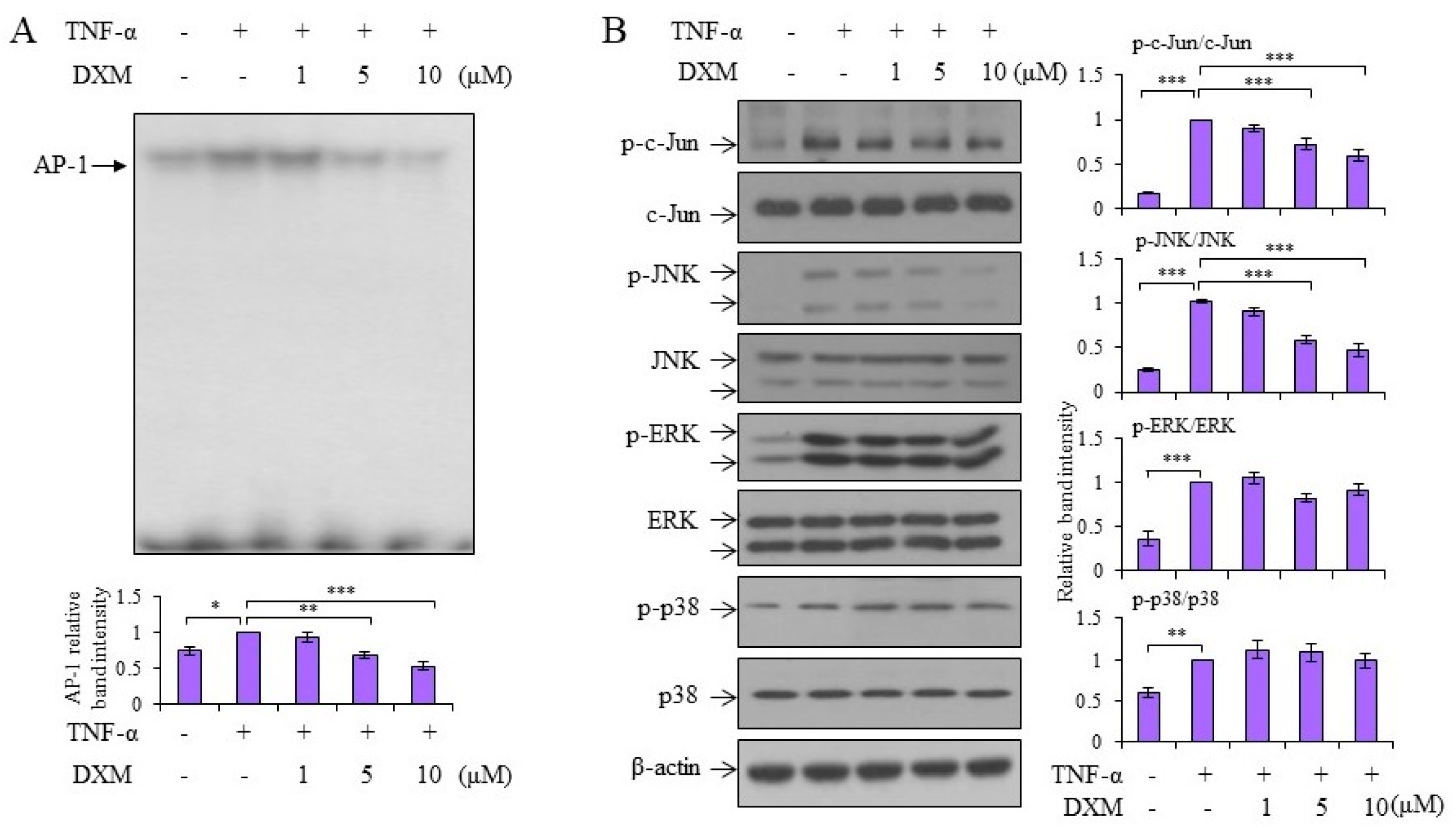
2.3. Signaling Pathway Targeted by DXM
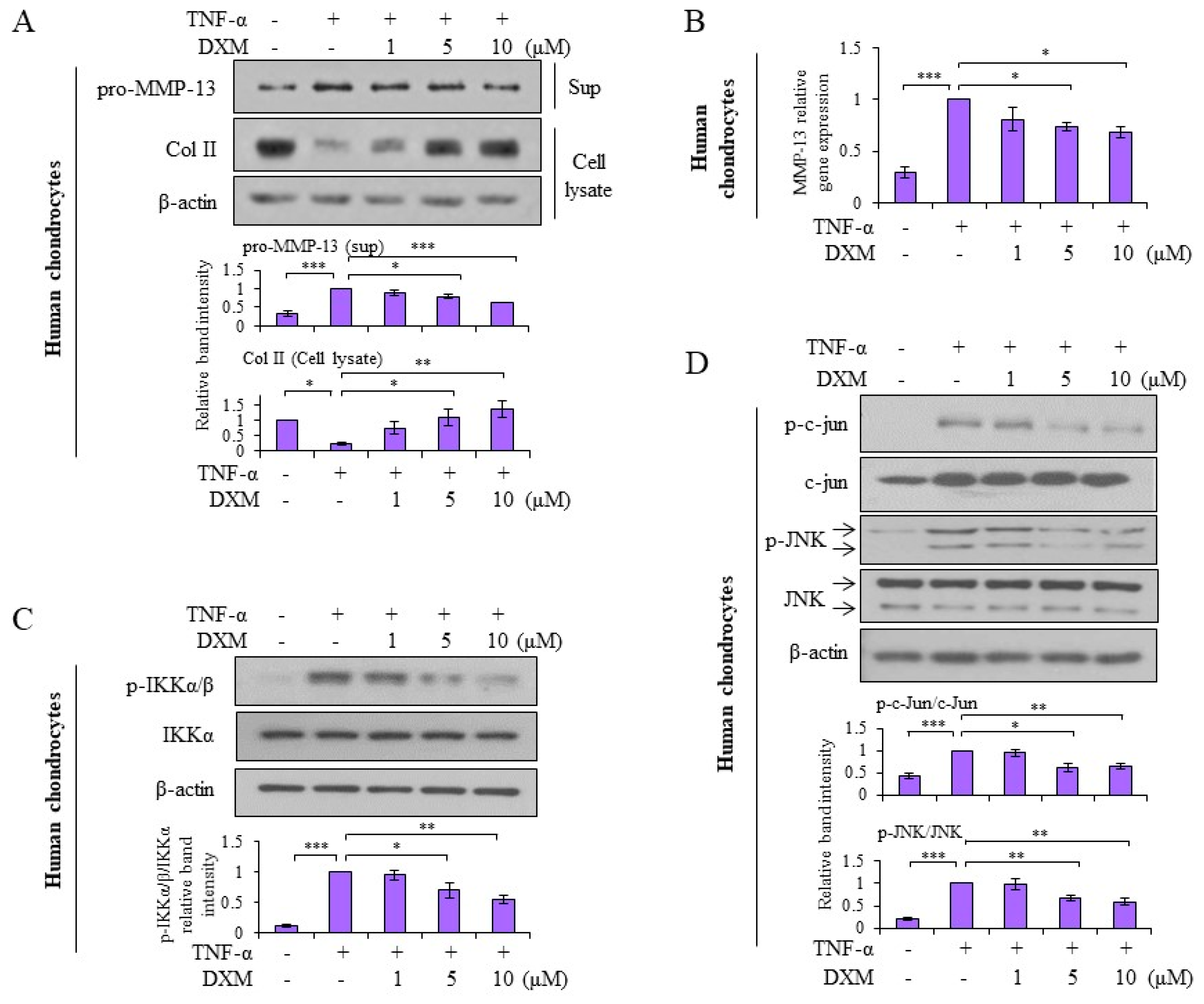
2.4. Effects and Mechanisms of DXM on TNF-α-Stimulated Human OA Chondrocytes
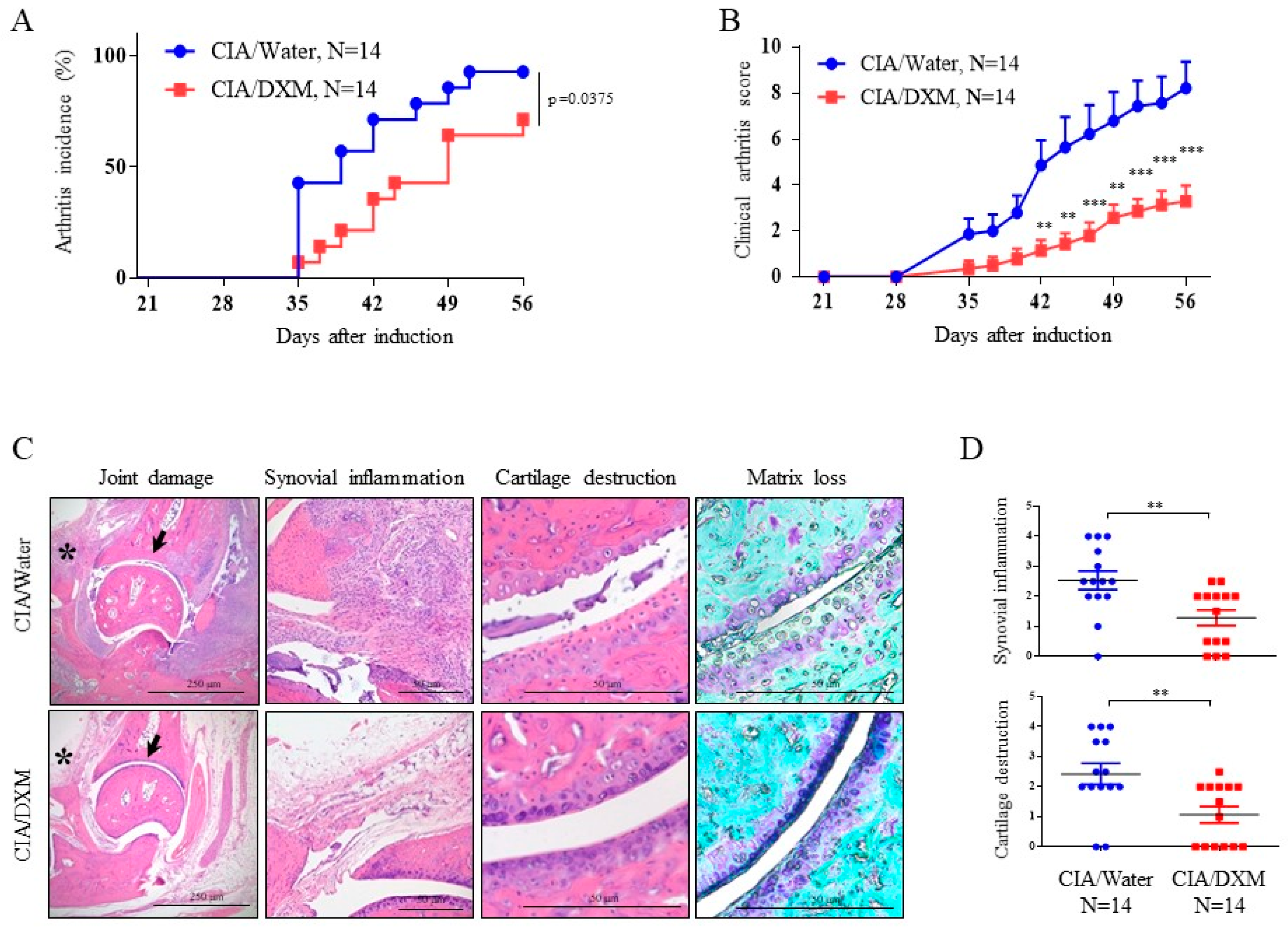
2.5. DXM Successfully Inhibited Progression and Severity in CIA Mice
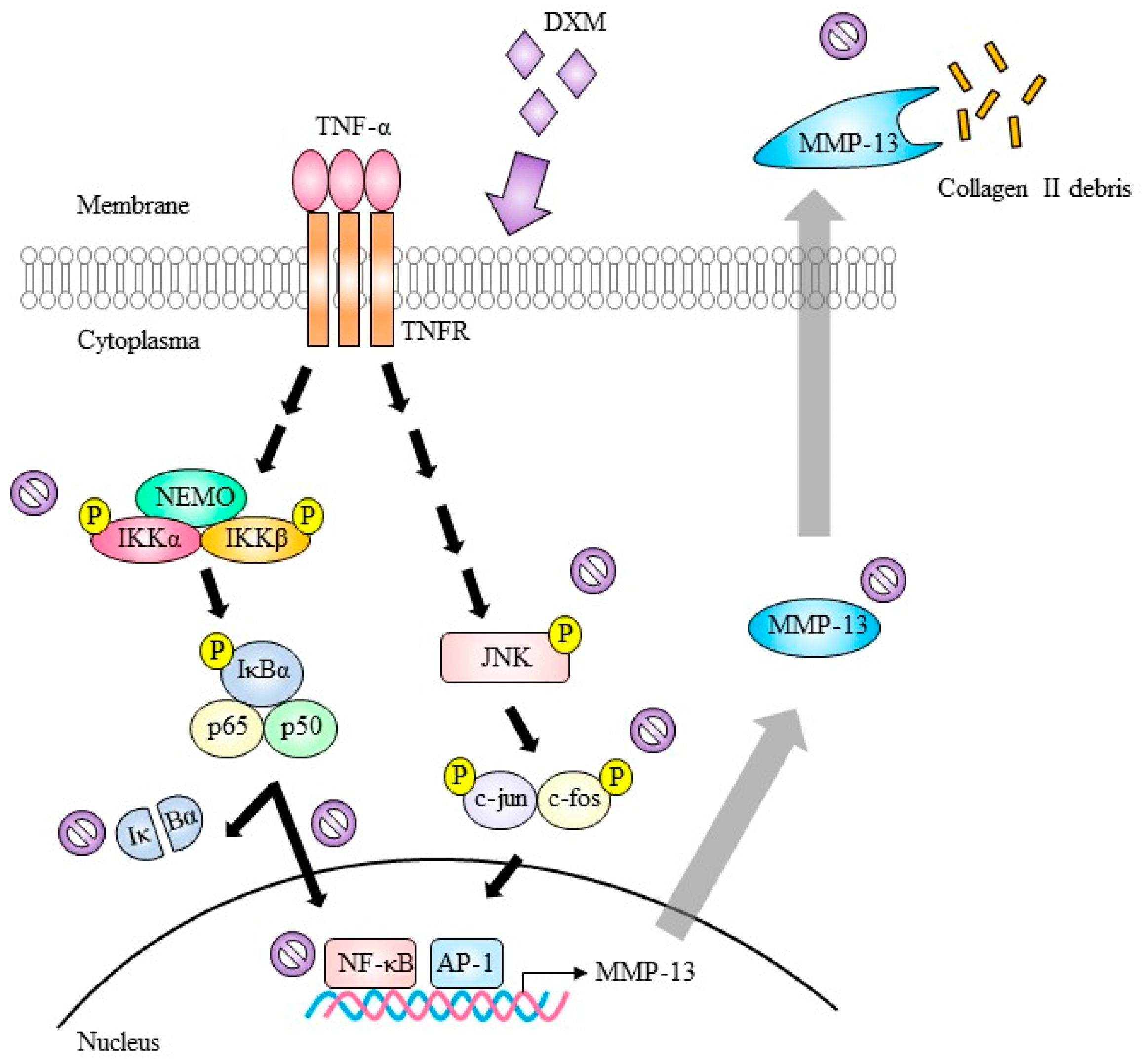
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Isolation and Culture of Human and Porcine Chondrocytes
4.3. Preparation of Porcine Synoviocytes
4.4. Preparation of Cartilage Explants
4.5. Analysis by a Real-Time Polymerase Chain Reaction with Reverse Transcription
4.6. Western Blotting
4.7. Nuclear Extract Preparation
4.8. Electrophoretic Mobility Shift Assay (EMSA)
4.9. 3-D Alginate Bead Experiments
4.10. Immunostaining of Porcine Cartilage
4.11. Collagen-Induced Arthritis in Mice
4.12. Histological Analysis in CIA
4.13. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AP-1 | activator protein-1 |
| CIA | collagen-induced arthritis |
| Col II | collagen II |
| DXM | dextromethorphan |
| ECM | extracellular matrix |
| ERK | extracellular-signal-regulated kinase |
| IKK | I kappa B kinase |
| IP-10 | interferon-gamma-inducible protein 10 |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinases |
| NF-κB | nuclear factor-kappaB |
| NMDA | N-methyl-d-aspartate |
| OA | osteoarthritis |
| OSM | oncostatin M |
| RA | rheumatoid arthritis |
| TNF-α | tumor necrosis factor-alpha |
References
- Parkinson, L.; Waters, D.L.; Franck, L. Systematic review of the impact of osteoarthritis on health outcomes for comorbid disease in older people. Osteoarthr. Cartil. 2017, 25, 1751–1770. [Google Scholar] [CrossRef] [PubMed]
- Pap, T.; Korb-Pap, A. Cartilage damage in osteoarthritis and rheumatoid arthritis—Two unequal siblings. Nat. Rev. Rheumatol. 2015, 11, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Saxby, D.J.; Lloyd, D.G. Osteoarthritis year in review 2016: Mechanics. Osteoarthr. Cartil. 2017, 25, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Tiku, M.L.; Madhan, B. Preserving the longevity of long-lived type II collagen and its implication for cartilage therapeutics. Ageing Res. Rev. 2016, 28, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.; Englund, M.; Struglics, A.; Lohmander, L.S. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthr. Cartil. 2015, 23, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Thomas, K.L.; Lucke-Wold, B.P.; Cavendish, J.Z.; Crowe, M.S.; Matsumoto, R.R. Dextromethorphan: An update on its utility for neurological and neuropsychiatric disorders. Pharmacol. Ther. 2016, 159, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Chechneva, O.V.; Mayrhofer, F.; Daugherty, D.J.; Pleasure, D.E.; Hong, J.S.; Deng, W. Low dose dextromethorphan attenuates moderate experimental autoimmune encephalomyelitis by inhibiting NOX2 and reducing peripheral immune cells infiltration in the spinal cord. Neurobiol. Dis. 2011, 44, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Li, Y.H.; Shi, G.Y.; Tang, S.H.; Jiang, S.J.; Huang, C.W.; Liu, P.Y.; Hong, J.S.; Wu, H.L. Dextromethorphan reduces oxidative stress and inhibits atherosclerosis and neointima formation in mice. Cardiovasc. Res. 2009, 82, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Marquard, J.; Otter, S.; Welters, A.; Stirban, A.; Fischer, A.; Eglinger, J.; Herebian, D.; Kletke, O.; Klemen, M.S.; Stozer, A.; et al. Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat. Med. 2015, 21, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Michaelis, M.; Ladel, C.; Siebuhr, A.S.; Bihlet, A.R.; Andersen, J.R.; Guehring, H.; Christiansen, C.; Bay-Jensen, A.C.; Kraus, V.B. Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: Lessons learned from failures and opportunities for the future. Osteoarthr. Cartil. 2016, 24, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, B.C.; Schultz, N.; Madsen, S.H.; Bay-Jensen, A.C.; Kassem, M.; Karsdal, M.A. MAPKs are essential upstream signaling pathways in proteolytic cartilage degradation—Divergence in pathways leading to aggrecanase and MMP-mediated articular cartilage degradation. Osteoarthr. Cartil. 2010, 18, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Billinghurst, R.C.; Dahlberg, L.; Ionescu, M.; Reiner, A.; Bourne, R.; Rorabeck, C.; Mitchell, P.; Hambor, J.; Diekmann, O.; Tschesche, H.; et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Investig. 1997, 99, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.H.; Luo, S.F.; Hung, L.F.; Huang, C.Y.; Lien, S.B.; Lin, L.C.; Liu, F.C.; Yen, B.L.; Ho, L.J. Physiological concentrations of soluble uric acid are chondroprotective and anti-inflammatory. Sci. Rep. 2017, 7, 2359. [Google Scholar] [CrossRef] [PubMed]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Huang, W.; Dehnade, F.; Ahmad, M.; Zafarullah, M. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp. Cell Res. 2003, 288, 208–217. [Google Scholar] [CrossRef]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Cantini, F.; Niccoli, L.; Nannini, C.; Cassara, E.; Kaloudi, O.; Giulio Favalli, E.; Becciolini, A.; Benucci, M.; Gobbi, F.L.; Guiducci, S.; et al. Second-line biologic therapy optimization in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Semin. Arthritis Rheum. 2017, 47, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B. TNF and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Lin, C.C.; Chen, Y.M.; Chao, Y.H.; Yang, D.H. Dextromethorphan Exhibits Anti-inflammatory and Immunomodulatory Effects in a Murine Model of Collagen-Induced Arthritis and in Human Rheumatoid Arthritis. Sci. Rep. 2017, 7, 11353. [Google Scholar] [CrossRef] [PubMed]
- Roman-Blas, J.A.; Jimenez, S.A. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr. Cartil. 2006, 14, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Park, S.H.; Kim, S.K.; Kim, J.H.; Ha, C.W.; Chun, C.H.; Chun, J.S. Inhibition of BATF/JUN transcriptional activity protects against osteoarthritic cartilage destruction. Ann. Rheum. Dis. 2017, 76, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.; Matthys, P. Collagen-induced arthritis and related animal models: How much of their pathogenesis is auto-immune, how much is auto-inflammatory? Cytokine Growth Factor Rev. 2011, 22, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hunter, D.J.; Jin, X.; Ding, C. The importance of synovial inflammation in osteoarthritis: Current evidence from imaging assessments and clinical trials. Osteoarthr. Cartil. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.B.P.; Filiberti, A.; Husain, S.A.; Humphrey, M.B. Immune Contributions to Osteoarthritis. Curr. Osteoporos. Rep. 2017, 15, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Mobasheri, A.; Mozafari, M. Inflammatory mediators in osteoarthritis: A critical review of the state-of-the-art, current prospects, and future challenges. Bone 2016, 85, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Biologic basis of osteoarthritis: State of the evidence. Curr. Opin. Rheumatol. 2015, 27, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Ramage, L.; Martel, M.A.; Hardingham, G.E.; Salter, D.M. NMDA receptor expression and activity in osteoarthritic human articular chondrocytes. Osteoarthr. Cartil. 2008, 16, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Salter, D.M.; Wright, M.O.; Millward-Sadler, S.J. NMDA receptor expression and roles in human articular chondrocyte mechanotransduction. Biorheology 2004, 41, 273–281. [Google Scholar] [PubMed]
- Piepoli, T.; Mennuni, L.; Zerbi, S.; Lanza, M.; Rovati, L.C.; Caselli, G. Glutamate signaling in chondrocytes and the potential involvement of NMDA receptors in cell proliferation and inflammatory gene expression. Osteoarthr. Cartil. 2009, 17, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yue, J.; Yang, C. Unraveling the role of Mg(++) in osteoarthritis. Life Sci. 2016, 147, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wen, Z.H.; Chang, Y.C.; Huang, S.Y.; Tang, C.C.; Chen, W.F.; Hsieh, S.P.; Hsieh, C.S.; Jean, Y.H. Intra-articular magnesium sulfate (MgSO4) reduces experimental osteoarthritis and nociception: Association with attenuation of N-methyl-d-aspartate (NMDA) receptor subunit 1 phosphorylation and apoptosis in rat chondrocytes. Osteoarthr. Cartil. 2009, 17, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.; Muschter, D. Do Neuroendocrine Peptides and Their Receptors Qualify as Novel Therapeutic Targets in Osteoarthritis? Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Hunter, D.J. Pharmacologic therapy for osteoarthritis—The era of disease modification. Nat. Rev. Rheumatol. 2011, 7, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Vinatier, C.; Merceron, C.; Guicheux, J. Osteoarthritis: From pathogenic mechanisms and recent clinical developments to novel prospective therapeutic options. Drug Discov. Today 2016, 21, 1932–1937. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.C.; Hung, L.F.; Wu, W.L.; Chang, D.M.; Huang, C.Y.; Lai, J.H.; Ho, L.J. Chondroprotective effects and mechanisms of resveratrol in advanced glycation end products-stimulated chondrocytes. Arthritis Res. Ther. 2010, 12, R167. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.J.; Lin, L.C.; Hung, L.F.; Wang, S.J.; Lee, C.H.; Chang, D.M.; Lai, J.H.; Tai, T.Y. Retinoic acid blocks pro-inflammatory cytokine-induced matrix metalloproteinase production by down-regulating JNK-AP-1 signaling in human chondrocytes. Biochem. Pharmacol. 2005, 70, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Von der Mark, K.; Gauss, V.; von der Mark, H.; Muller, P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature 1977, 267, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Benya, P.D.; Padilla, S.R.; Nimni, M.E. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell 1978, 15, 1313–1321. [Google Scholar] [CrossRef]
- Orosa, B.; González, A.; Mera, A.; Gómez-Reino, J.J.; Conde, C. Lysophosphatidic acid receptor 1 suppression sensitizes rheumatoid fibroblast-like synoviocytes to tumor necrosis factor-induced apoptosis. Arthritis Rheum. 2012, 64, 2460–2470. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Cheng, W.T.; Kuo, T.F.; Sun, J.S.; Lin, F.H.; Tsai, J.C. Fibrin glue mixed with gelatin/hyaluronic acid/chondroitin-6-sulfate tri-copolymer for articular cartilage tissue engineering: The results of real-time polymerase chain reaction. J. Biomed. Mater. Res. A 2007, 82, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Upton, M.L.; Chen, J.; Setton, L.A. Region-specific constitutive gene expression in the adult porcine meniscus. J. Orthop. Res. 2006, 24, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.J.; Hung, L.F.; Liu, F.C.; Hou, T.Y.; Lin, L.C.; Huang, C.Y.; Lai, J.H. Ginkgo biloba extract individually inhibits JNK activation and induces c-Jun degradation in human chondrocytes: Potential therapeutics for osteoarthritis. PLoS ONE 2013, 8, e82033. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Françon, B.; et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chen, R.N.; Jhan, H.J.; Liu, D.Z.; Ho, H.O.; Mao, Y.; Kohn, J.; Sheu, M.T. Development and Characterization of Acellular Extracellular Matrix Scaffolds from Porcine Menisci for Use in Cartilage Tissue Engineering. Tissue Eng. Part C Methods 2015, 21, 971–986. [Google Scholar] [CrossRef] [PubMed]

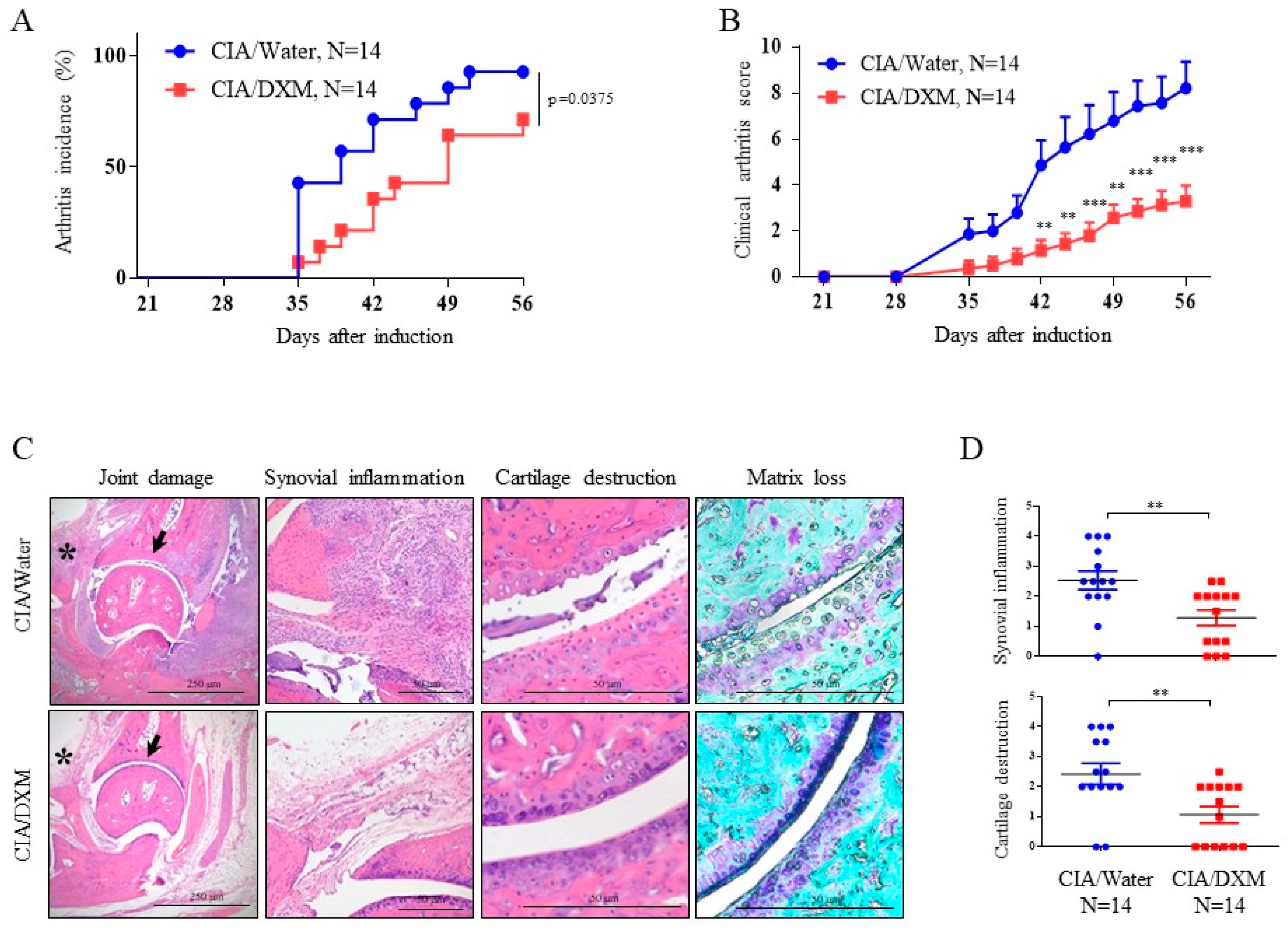
 indicate inhibition.
indicate inhibition.
indicate inhibition.
indicate inhibition.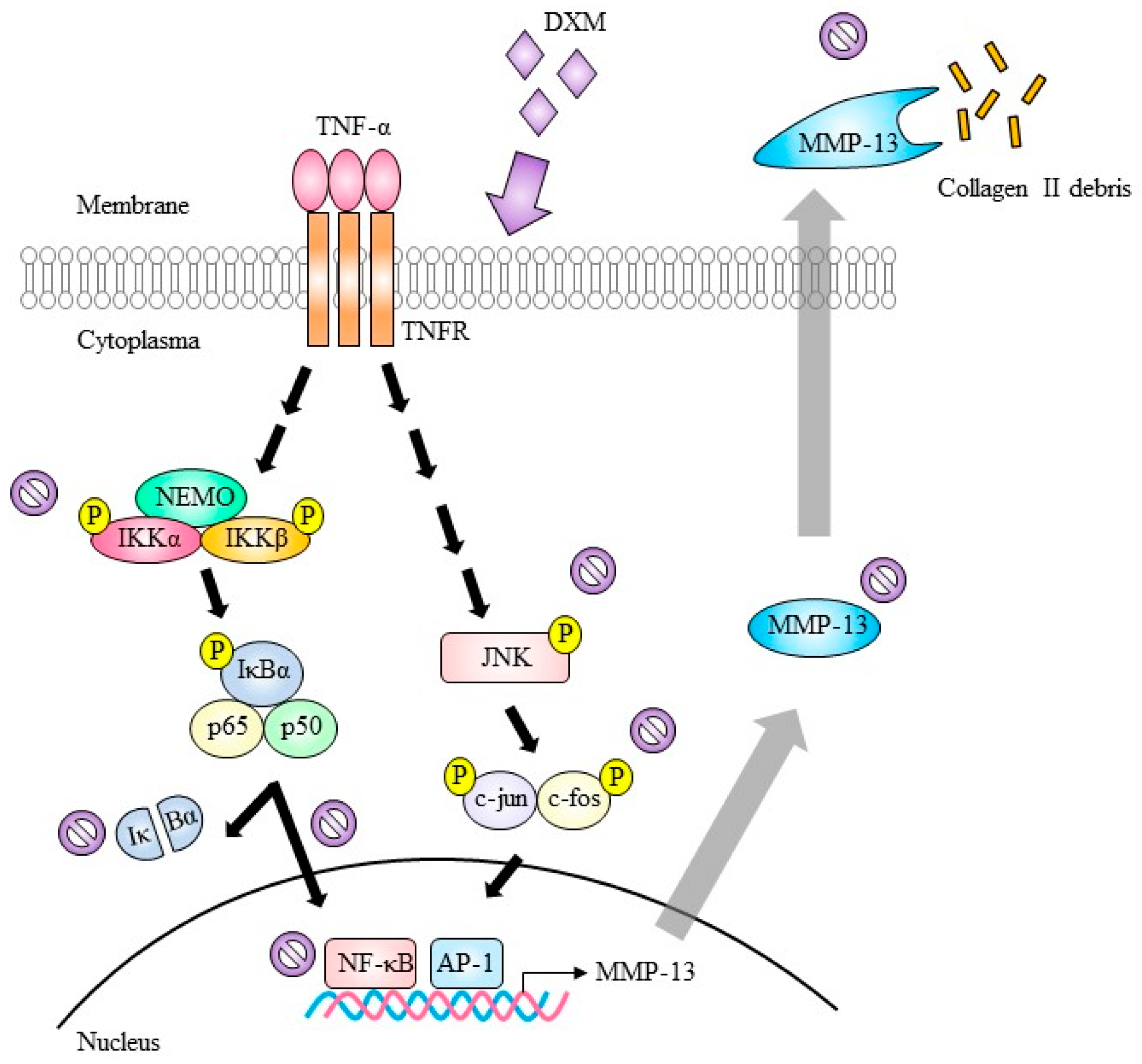
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.W.; Liu, F.-C.; Hung, L.-F.; Huang, C.-Y.; Lien, S.-B.; Lin, L.-C.; Lai, J.-H.; Ho, L.-J. Chondroprotective Effects and Mechanisms of Dextromethorphan: Repurposing Antitussive Medication for Osteoarthritis Treatment. Int. J. Mol. Sci. 2018, 19, 825. https://doi.org/10.3390/ijms19030825
Chen LW, Liu F-C, Hung L-F, Huang C-Y, Lien S-B, Lin L-C, Lai J-H, Ho L-J. Chondroprotective Effects and Mechanisms of Dextromethorphan: Repurposing Antitussive Medication for Osteoarthritis Treatment. International Journal of Molecular Sciences. 2018; 19(3):825. https://doi.org/10.3390/ijms19030825
Chicago/Turabian StyleChen, Liv Weichien, Feng-Cheng Liu, Li-Feng Hung, Chuan-Yueh Huang, Shiu-Bii Lien, Leou-Chyr Lin, Jenn-Haung Lai, and Ling-Jun Ho. 2018. "Chondroprotective Effects and Mechanisms of Dextromethorphan: Repurposing Antitussive Medication for Osteoarthritis Treatment" International Journal of Molecular Sciences 19, no. 3: 825. https://doi.org/10.3390/ijms19030825
APA StyleChen, L. W., Liu, F.-C., Hung, L.-F., Huang, C.-Y., Lien, S.-B., Lin, L.-C., Lai, J.-H., & Ho, L.-J. (2018). Chondroprotective Effects and Mechanisms of Dextromethorphan: Repurposing Antitussive Medication for Osteoarthritis Treatment. International Journal of Molecular Sciences, 19(3), 825. https://doi.org/10.3390/ijms19030825