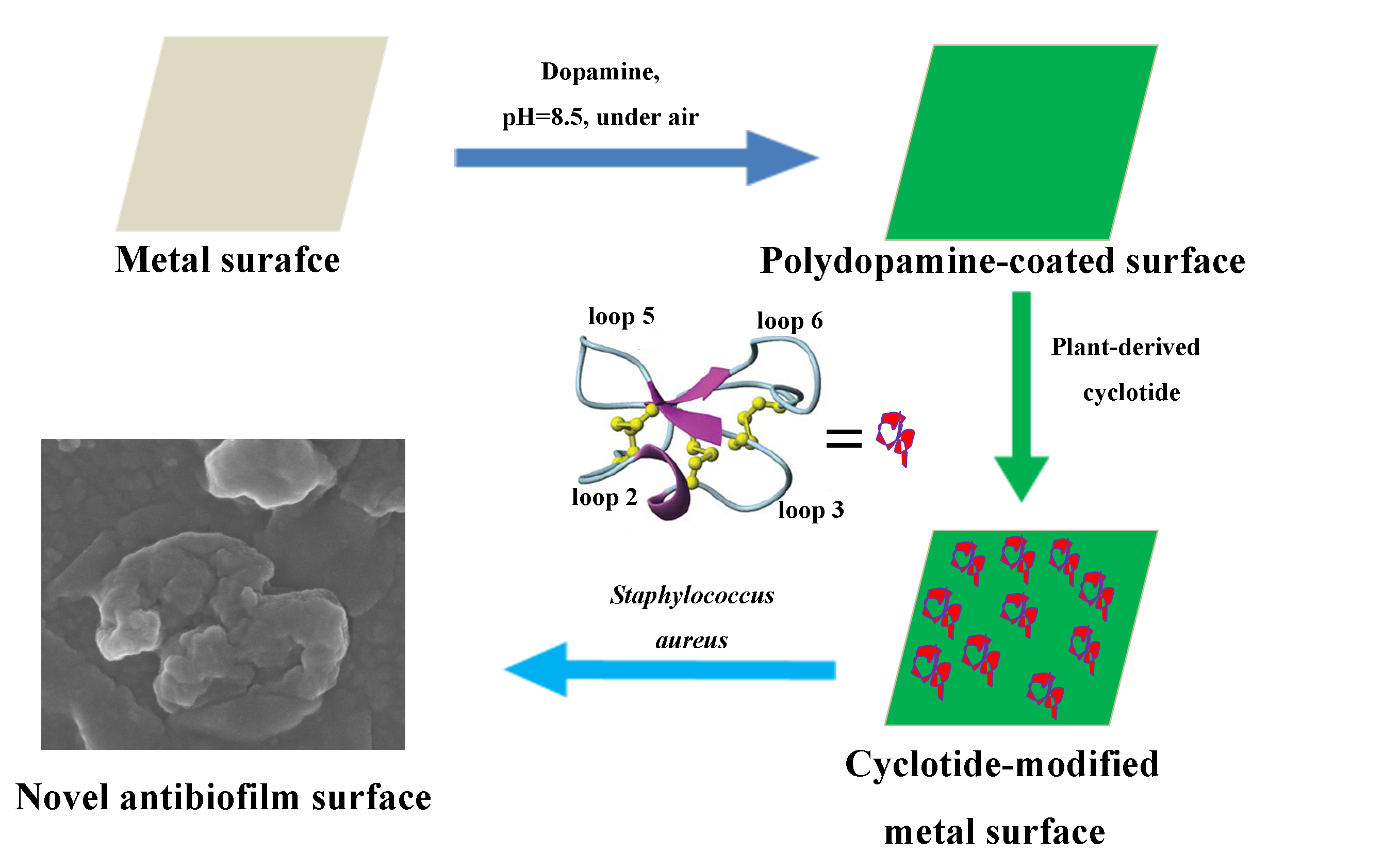
Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study


 , and
, and
Abstract

1. Introduction
2. Results
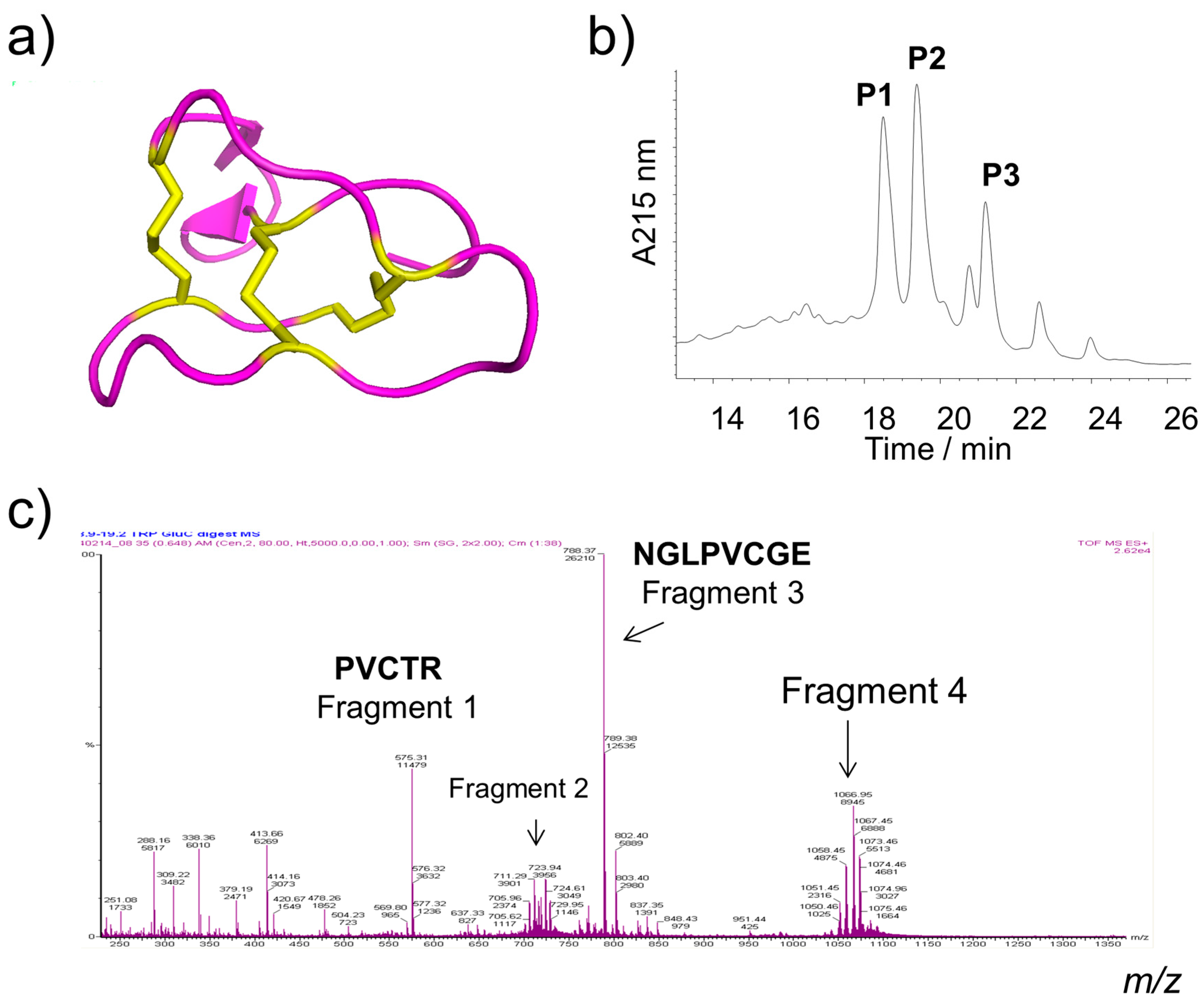
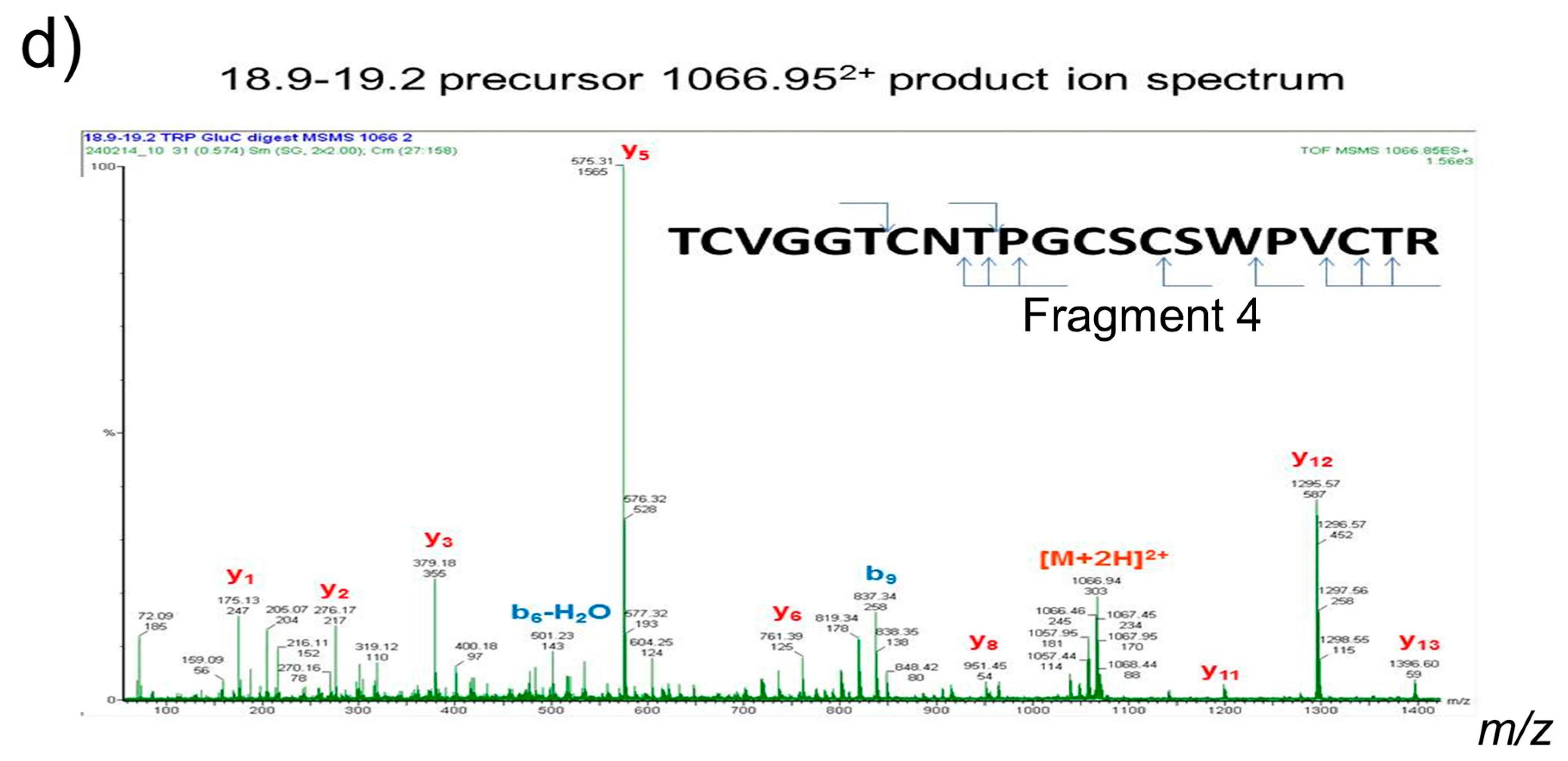
2.1. Isolation and Identification of Cyclotides
2.2. Characterization of Cyclotide-Modified Metal Surfaces
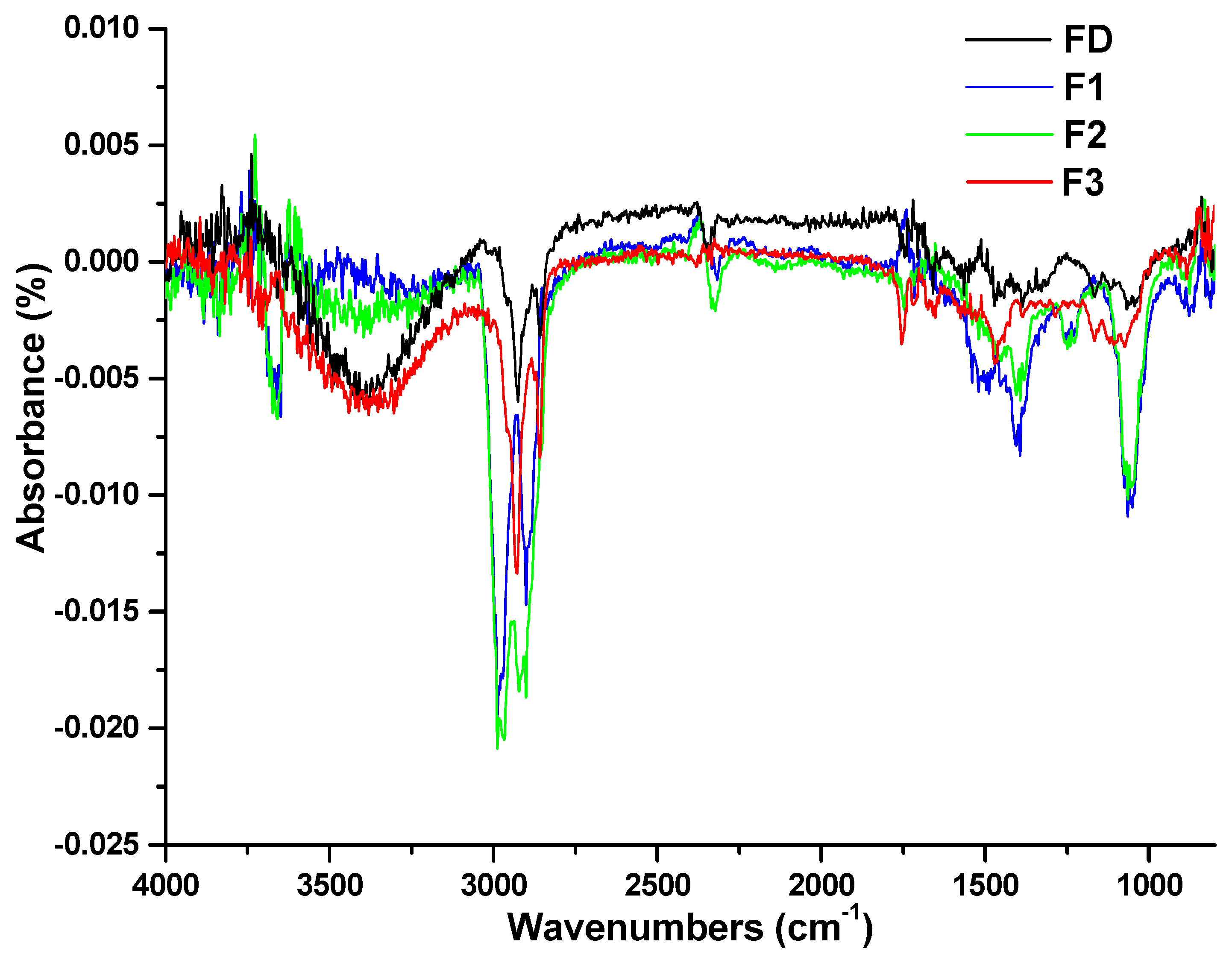
2.2.1. Fourier Transform Infrared (FTIR) Spectroscopy
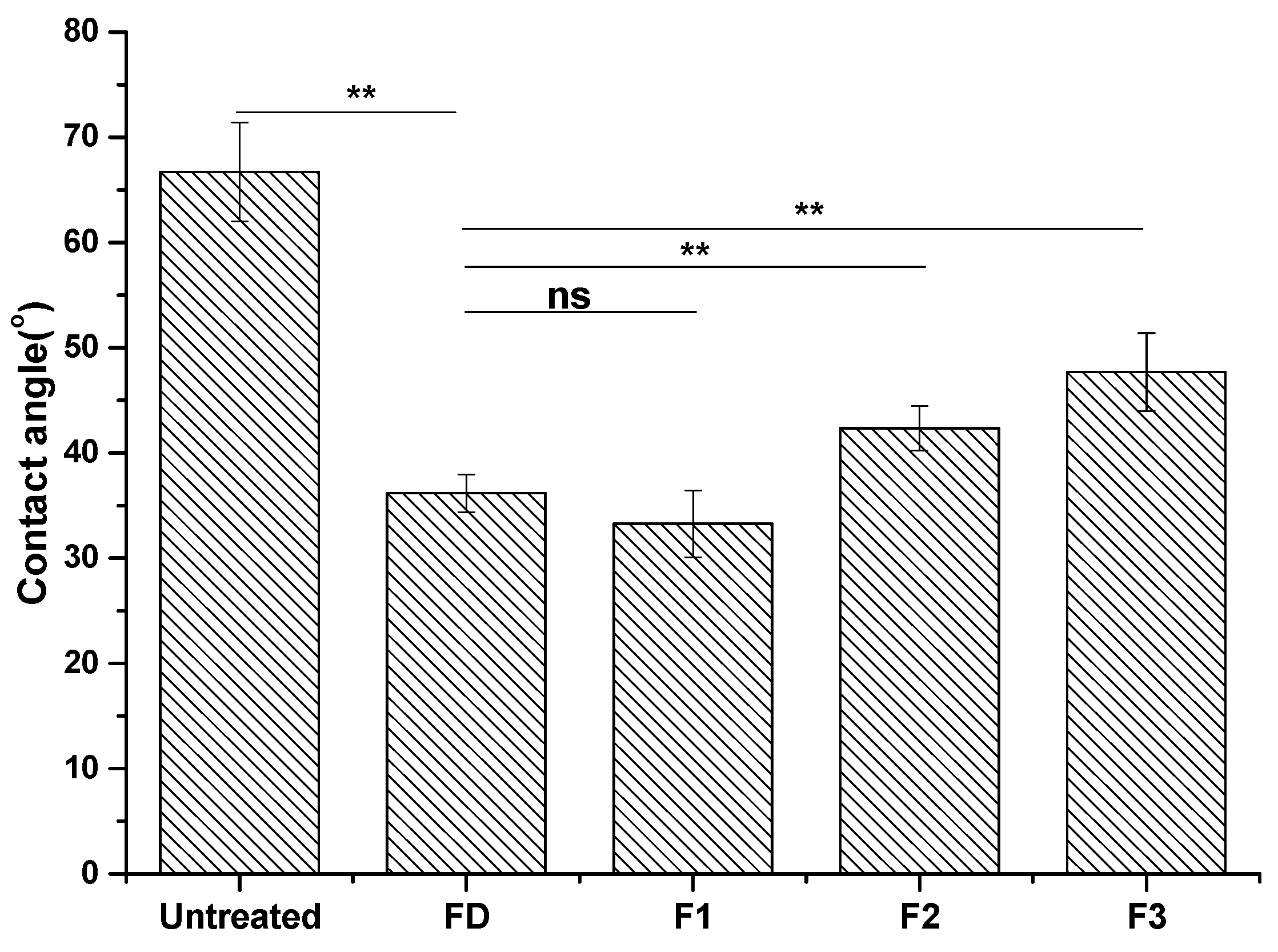
2.2.2. Contact Angle Analysis
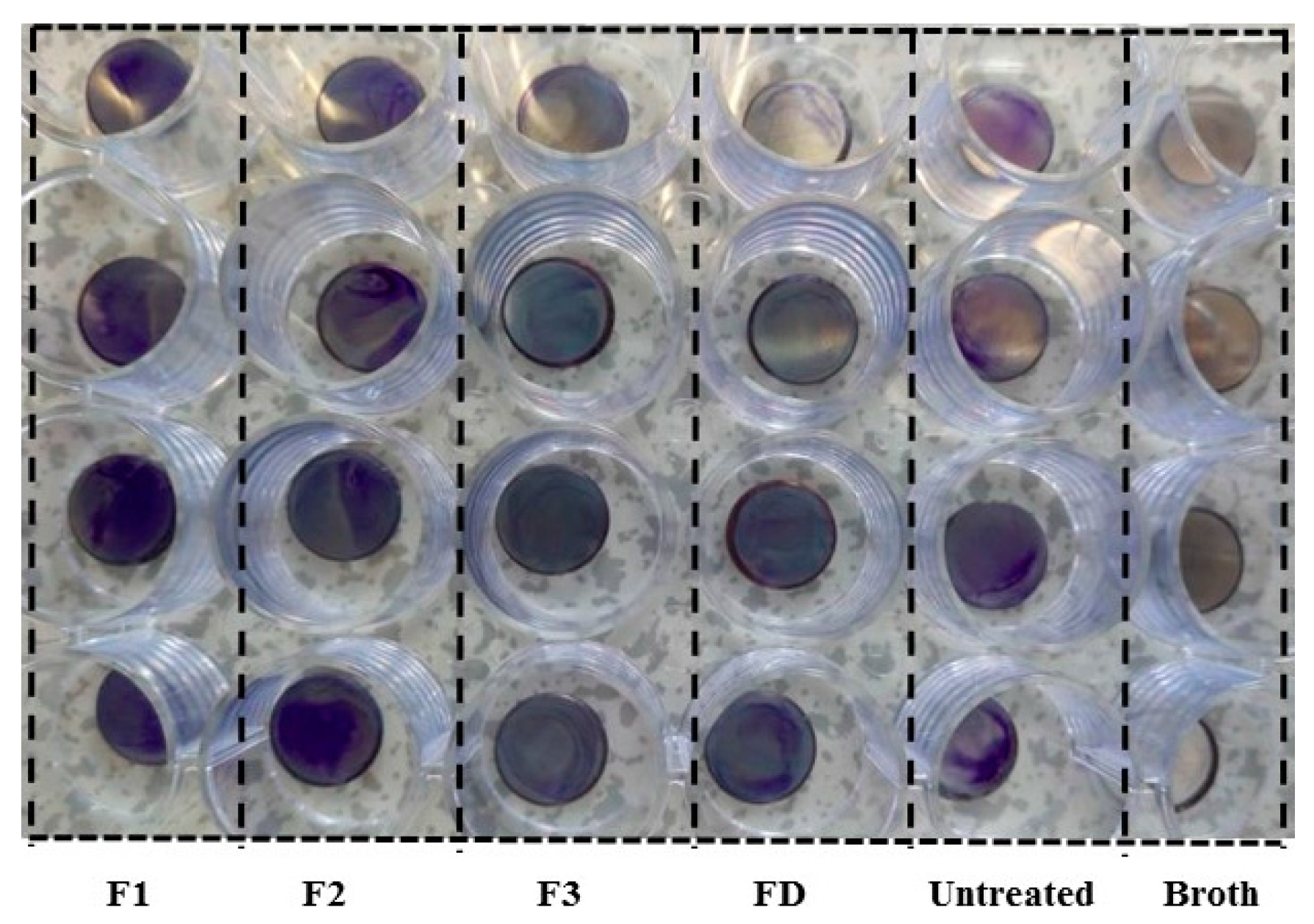
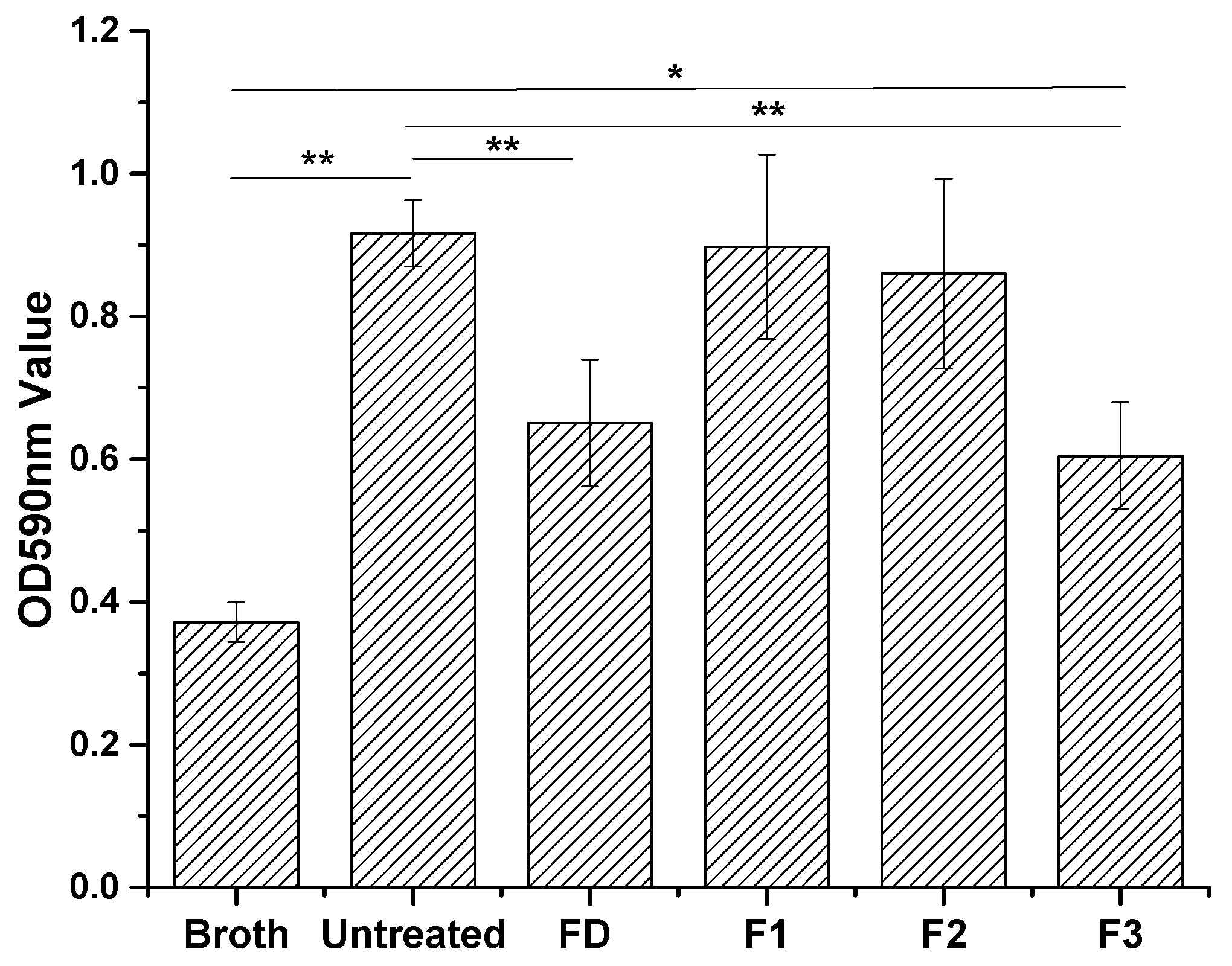
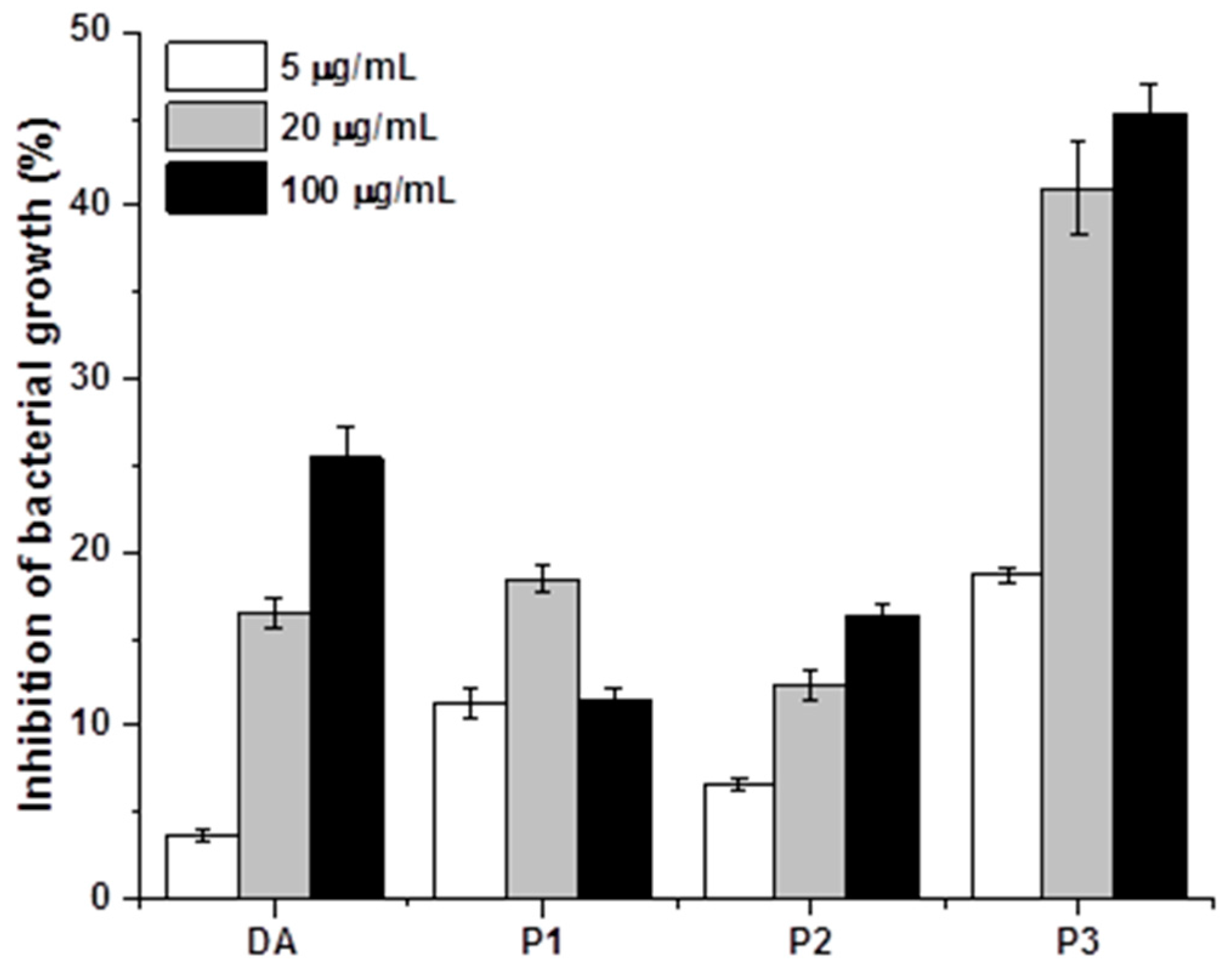
2.3. Antibacterial and Antibiofilm Activity
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Extraction and Isolation of Cyclotides
4.3. Mass Spectrometric Analysis of Cyclotides from V. Philippica
4.3.1. Methods for Cyclotides Reduction and Enzyme Digestion
4.3.2. Liquid Chromatography Electrospray Ionization/Mass Spectrometry (LC ESI/MS)
4.4. Immobilization of Cyclotides onto the Metal Surfaces
4.5. Surface Characterization
4.6. Antibiofilm Assay
4.7. Antibacterial (Alamar Blue) Assay
4.8. FESEM Analysis
4.9. Statistical Methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| ATR | Attenuated total reflection |
| CV | Crystal violet |
| DA | Dopamine |
| FT-IR | Fourier transform infrared |
| HPLC | High performance liquid chromatography |
| GRAVY | Grand average of hydropathicity |
| OD | Optical density |
| FESEM | Field emission Electron scanning microscopy |
| TCEP | Tris (2-caboxyethyyl) phosphine hydrochloride |
| TFA | Trifluoroacetic acid |
References
- Banerjee, I.; Pangule, R.C.; Kane, R.S. Antifouling coatings: Recent developments in the design of surfaces that prevent fouling by proteins, bacteria, and marine organisms. Adv. Mater. 2011, 23, 690–718. [Google Scholar] [CrossRef] [PubMed]
- Lejars, M.; Margaillan, A.; Bressy, C. Fouling Release Coatings: A Nontoxic Alternative to Biocidal Antifouling Coatings. Chem. Rev. 2012, 112, 4347–4390. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.P.; Bendick, J.A.; Holm, E.R.; Hertel, W.M. Economic impact of biofouling on a naval surface ship. Biofouling 2011, 27, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Callow, J.A.; Callow, M.E. Trends in the development of environmentally friendly fouling-resistant marine coatings. Nat. Commun. 2011, 2, 244. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wu, Z.Q.; Chen, H. Dual-function antibacterial surfaces for biomedical applications. Acta Biomater. 2015, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Seker, U.O.S.; Demir, H.V. Material Binding Peptides for Nanotechnology. Molecules 2011, 16, 1426–1451. [Google Scholar] [CrossRef] [PubMed]
- Glinel, K.; Thebault, P.; Humblot, V.; Pradier, C.M.; Jouenne, T. Antibacterial surfaces developed from bio-inspired approaches. Acta Biomater. 2012, 8, 1670–1684. [Google Scholar] [CrossRef] [PubMed]
- Alves, D.; Pereira, M.O. Mini-review: Antimicrobial peptides and enzymes as promising candidates to functionalize biomaterial surfaces. Biofouling 2014, 30, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.M.; Li, D.Y.; Irvin, R.T. A peptide-stainless steel reaction that yields a new bioorganic-metal state of matter. Biomaterials 2011, 32, 5311–5319. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.Y.; Bai, X.Q.; Yuan, C.Q.; Yang, Y.; Xie, H.; Cao, P.; Ma, C.Y.; Wang, X.J.; Yan, X.P. Protein engineering of a new recombinant peptide to increase the surface contact angle of stainless steel. RSC Adv. 2015, 5, 101309–101318. [Google Scholar] [CrossRef]
- Cao, P.; Yuan, C.Q.; Ma, C.Y.; Yang, Y.; Bai, X.Q.; Wang, X.J.; Ren, X.Y.; Xie, H.; Yan, X.P. Preparation and analysis of a new bioorganic metallic material. RSC Adv. 2015, 5, 78030–78037. [Google Scholar] [CrossRef]
- Liu, Y.; Ai, K.; Lu, L. Polydopamine and its derivative materials: Synthesis and promising applications in energy, environmental, and biomedical fields. Chem. Rev. 2014, 114, 5057–5115. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Barclay, T.G.; Hegab, H.M.; Clarke, S.R.; Ginic-Markovic, M. Versatile Surface Modification Using Polydopamine and Related Polycatecholamines: Chemistry, Structure, and Applications. Adv. Mater. Interfaces 2017, 4, 1601192. [Google Scholar] [CrossRef]
- Shalev, T.; Gopin, A.; Bauer, M.; Stark, R.W.; Rahimipour, S. Non-leaching antimicrobial surfaces through polydopamine bio-inspired coating of quaternary ammonium salts or an ultrashort antimicrobial lipopeptide. J. Mater. Chem. 2012, 22, 2026–2032. [Google Scholar] [CrossRef]
- Yeroslavsky, G.; Girshevitz, O.; Foster-Frey, J.; Donovan, D.M.; Rahimipour, S. Antibacterial and Antibiofilm Surfaces through Polydopamine-Assisted Immobilization of Lysostaphin as an Antibacterial Enzyme. Langmuir 2015, 31, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Li, W.W.; Morris, A.R.; Horrocks, P.; Yuan, C.Q.; Yang, Y. Investigation of the antibiofilm capacity of peptide-modified stainless steel. R. Soc. Open Sci. 2018, 5, 172165. [Google Scholar] [CrossRef]
- Craik, D.J.; Swedberg, J.E.; Mylne, J.S.; Cemazar, M. Cyclotides as a basis for drug design. Expert Opin. Drug Discov. 2012, 7, 179–194. [Google Scholar] [CrossRef] [PubMed]
- De Veer, S.J.; Weidmann, J.; Craik, D.J. Cyclotides as Tools in Chemical Biology. Acc. Chem. Res. 2017, 50, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Chan, L.Y.; Zeng, G.; Daly, N.L.; Craik, D.J.; Tan, N. Isolation and characterization of cytotoxic cyclotides from Viola philippica. Peptides 2011, 32, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Uche, F.I.; Li, W.W.; Richardson, A.; Greenhough, T.J. Anticancer activities of cyclotides from Viola yedeonsis Makino (Violaceae). Planta Medica 2014, 80. [Google Scholar] [CrossRef]
- Pranting, M.; Loov, C.; Burman, R.; Goransson, U.; Andersson, D.I. The cyclotide cycloviolacin O2 from Viola odorata has potent bactericidal activity against Gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Stromstedt, A.A.; Park, S.; Burman, R.; Goransson, U. Bactericidal activity of cyclotides where phosphatidylethanolamine-lipid selectivity determines antimicrobial spectra. Biochim. Biophys. Acta 2017, 1859, 1986–2000. [Google Scholar] [CrossRef] [PubMed]
- Goransson, U.; Sjogren, M.; Svangard, E.; Claeson, P.; Bohlin, L. Reversible antifouling effect of the cyclotide cycloviolacin O2 against barnacles. J. Nat. Prod. 2004, 67, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- ExPASy-Protparam Tool. Available online: https://web.expasy.org/protparam/ (accessed on 15 October 2017).
- O’Toole, G.A. Microtiter Dish Biofilm Formation Assay. J. Vis. Exp. 2011, 2437. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.A.; Franzblau, S.G. Microplate Alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [PubMed]
- Carbonaro, M.; Nucara, A. Secondary structure of food proteins by Fourier transform spectroscopy in the mid-infrared region. Amino Acids 2010, 38, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Coates, J. Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Kang, J.; Sakuragi, M.; Shibata, A.; Abe, H.; Kitajima, T.; Tada, S.; Mizutani, M.; Ohmori, H.; Ayame, H.; Son, T.I.; et al. Immobilization of epidermal growth factor on titanium and stainless steel surfaces via dopamine treatment. Mater. Sci. Eng. C 2012, 32, 2552–2561. [Google Scholar] [CrossRef]
- Su, L.; Yu, Y.; Zhao, Y.S.; Liang, F.; Zhang, X.J. Strong Antibacterial Polydopamine Coatings Prepared by a Shaking-assisted Method. Sci. Rep. 2016, 6, 24420. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Bi, T.; Camarero, J.A. Chemical and Biological Production of Cyclotides. Adv. Bot. Res. 2015, 76, 271–303. [Google Scholar] [PubMed]
- Goransson, U.; Svangard, E.; Claeson, P.; Bohlin, L. Novel strategies for isolation and characterization of cyclotides: The discovery of bioactive macrocyclic plant polypeptides in the Violaceae. Curr. Protein Pept. Sci. 2004, 5, 317–329. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Cyclotide | Sequence of Amino Acid Residues | Theoretical Monoisotopic Mass | Experimental Monoisotopic Mass | GRAVY for Their Linear Forms | Net Charges |
|---|---|---|---|---|---|---|
| P1 | Varv A Kalata b1 Viba 15 Viba 17 | Cyclo-(CGETCVGGTCNTPG---CSCSWPVCTRNGLPV) Cyclo-(CGETCVGGTCNTPG---CTCSWPVCTRNGLPV) Cyclo-(CGETCVGGTCNTPG---CACSWPVCTRNGLPV) Cyclo-(CGETCVGGTCNTPG---CGCSWPVCTRNGLPV) | 2876.17 2890.14 2860.18 2846.02 | 2876.06 2890.11 2860.12 2846.08 | 0.148 0.152 0.238 0.162 | 0 0 0 0 |
| P2 | Varv E | Cyclo-(CGETCVGGTCNTPG---CSCSWPVCTRNGLPI) | 2890.14 | 2890.00 | 0.159 | 0 |
| P3 | Viphi G | Cyclo-(CGESCVF I P C I SAIIGCSCSNKVCYKNGSIP) | 3170.43 | 3170.43 | 0.726 | +1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, P.; Yang, Y.; Uche, F.I.; Hart, S.R.; Li, W.-W.; Yuan, C. Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study. Int. J. Mol. Sci. 2018, 19, 793. https://doi.org/10.3390/ijms19030793
Cao P, Yang Y, Uche FI, Hart SR, Li W-W, Yuan C. Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study. International Journal of Molecular Sciences. 2018; 19(3):793. https://doi.org/10.3390/ijms19030793
Chicago/Turabian StyleCao, Pan, Ying Yang, Fidelia Ijeoma Uche, Sarah Ruth Hart, Wen-Wu Li, and Chengqing Yuan. 2018. "Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study" International Journal of Molecular Sciences 19, no. 3: 793. https://doi.org/10.3390/ijms19030793
APA StyleCao, P., Yang, Y., Uche, F. I., Hart, S. R., Li, W.-W., & Yuan, C. (2018). Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study. International Journal of Molecular Sciences, 19(3), 793. https://doi.org/10.3390/ijms19030793