Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy



Abstract
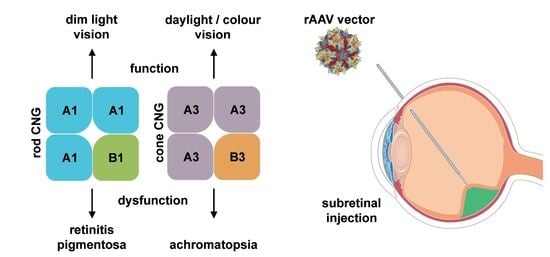
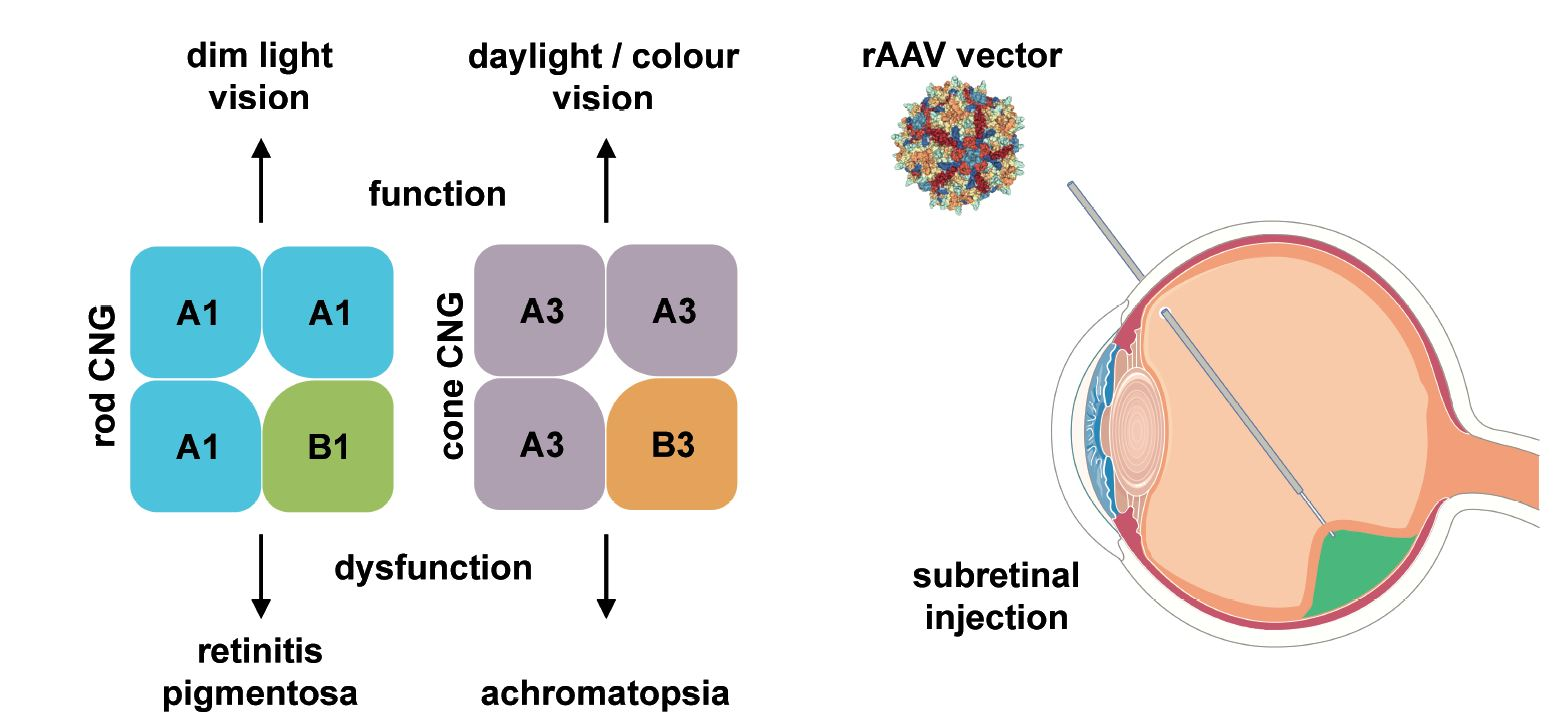
1. Introduction
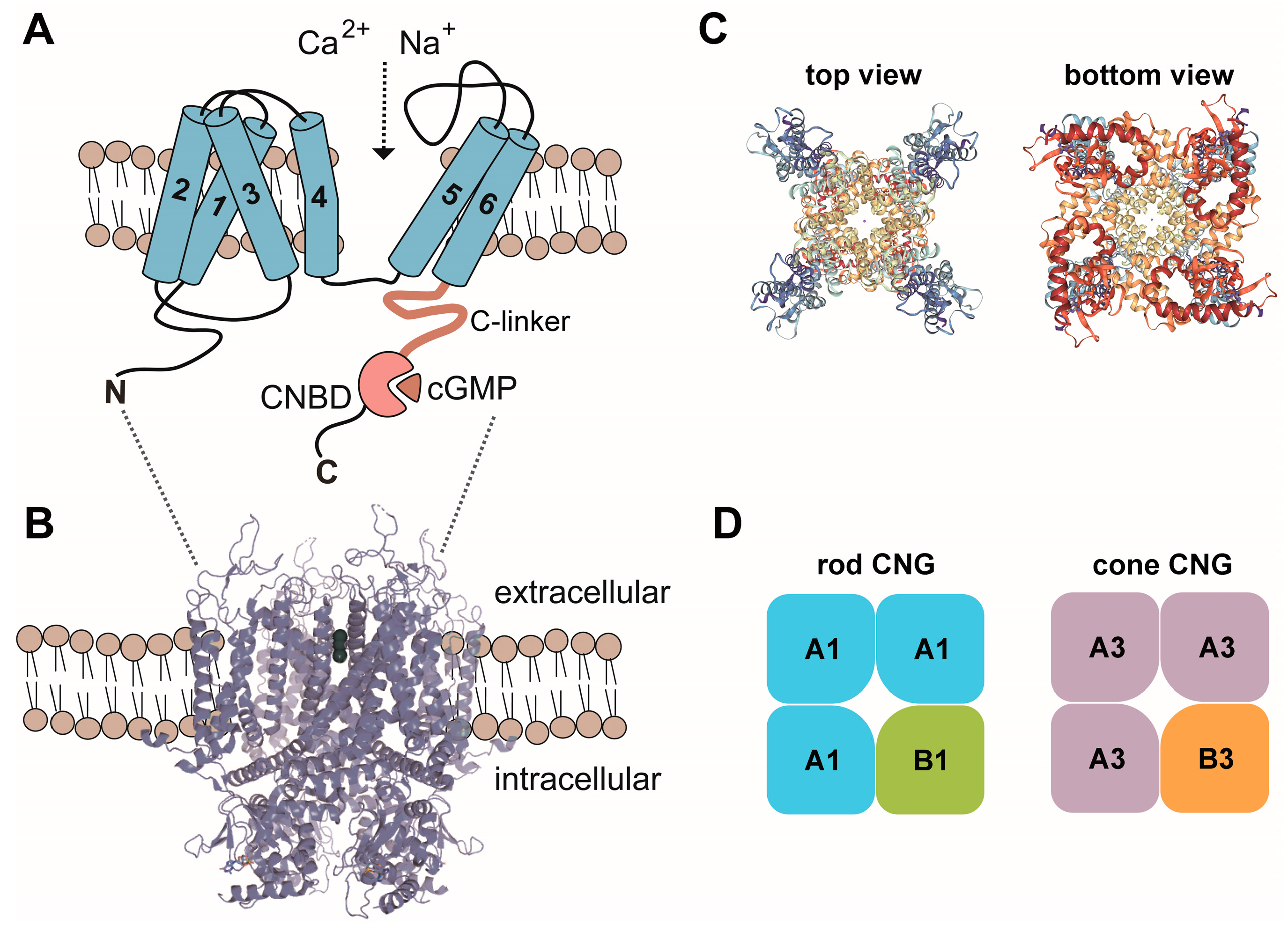
2. Structure, Basic Properties and Activation of CNG Channels
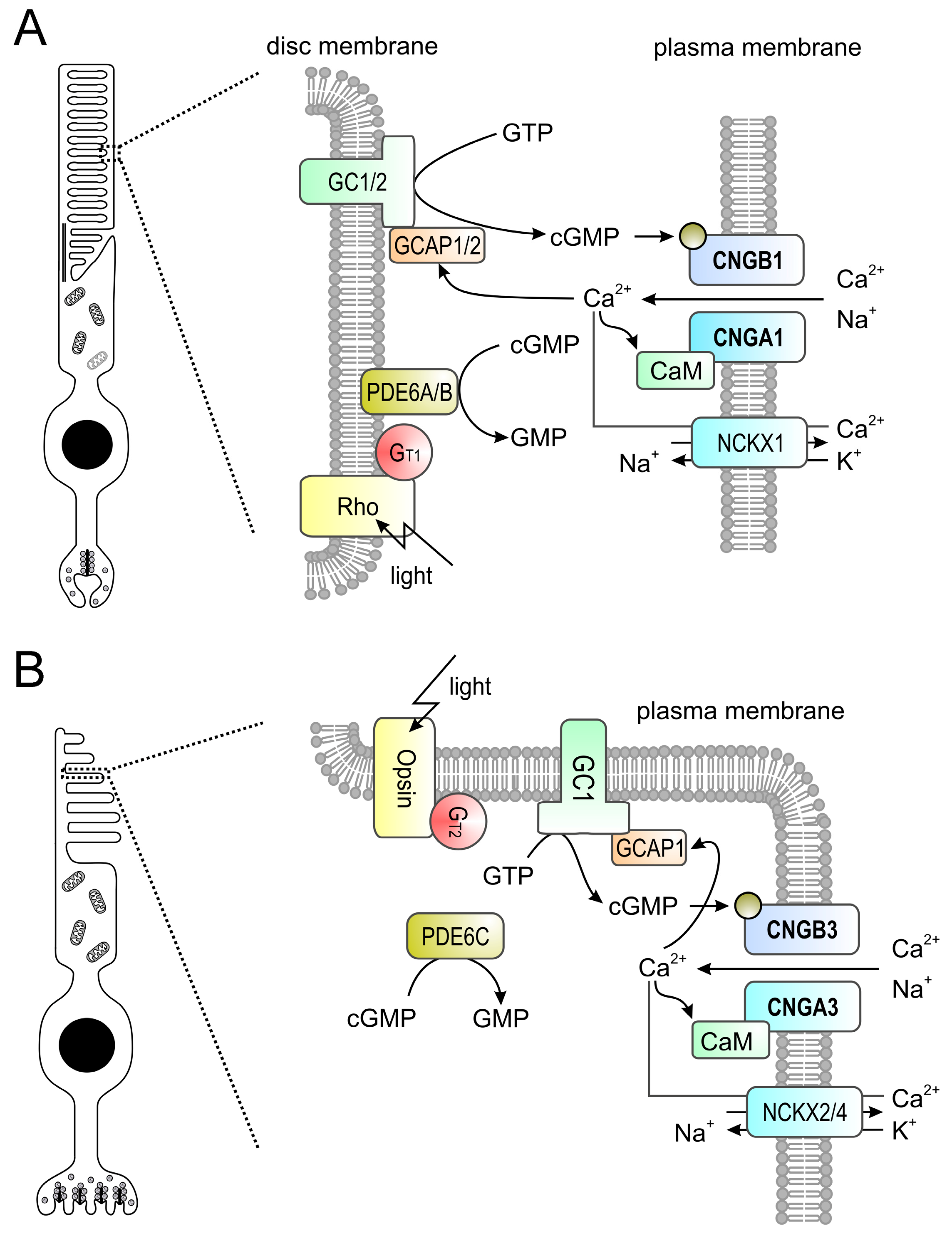
3. Signal Transduction in Photoreceptors
4. In Vivo Analysis of CNG Channel Function: Analysis of Genetic Mouse Models and Human Channelopathies
4.1. CNGA1 and CNGB1
4.2. CNGA3 and CNGB3
5. Gene Therapy for the Treatment of CNG Channelopathies
6. Gene Therapy for CNG Channel-Linked RP
7. Gene Therapy for CNG Channel-Linked ACHM
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [PubMed]
- James, Z.M.; Zagotta, W.N. Structural insights into the mechanisms of cnbd channel function. J. Gen. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 111–136. [Google Scholar] [CrossRef]
- Hofmann, F.; Biel, M.; Kaupp, U.B. International union of pharmacology. Li. Nomenclature and structure-function relationships of cyclic nucleotide-regulated channels. Pharmacol. Rev. 2005, 57, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Weitz, D.; Ficek, N.; Kremmer, E.; Bauer, P.J.; Kaupp, U.B. Subunit stoichiometry of the cng channel of rod photoreceptors. Neuron 2002, 36, 881–889. [Google Scholar] [CrossRef]
- Zheng, J.; Trudeau, M.C.; Zagotta, W.N. Rod cyclic nucleotide-gated channels have a stoichiometry of three cnga1 subunits and one cngb1 subunit. Neuron 2002, 36, 891–896. [Google Scholar] [CrossRef]
- Zhong, H.; Molday, L.L.; Molday, R.S.; Yau, K.W. The heteromeric cyclic nucleotide-gated channel adopts a 3a:1b stoichiometry. Nature 2002, 420, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Shuart, N.G.; Haitin, Y.; Camp, S.S.; Black, K.D.; Zagotta, W.N. Molecular mechanism for 3:1 subunit stoichiometry of rod cyclic nucleotide-gated ion channels. Nat. Commun. 2011, 2, 457. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, X.; Wang, S.; Michailidis, I.; Gong, Y.; Su, D.; Li, H.; Li, X.; Yang, J. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature 2017, 542, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Zagotta, W.N.; Olivier, N.B.; Black, K.D.; Young, E.C.; Olson, R.; Gouaux, E. Structural basis for modulation and agonist specificity of hcn pacemaker channels. Nature 2003, 425, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Siegelbaum, S.A. Gating of hcn channels by cyclic nucleotides: Residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 2007, 15, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Taraska, J.W.; Zagotta, W.N. Cyclic nucleotide-regulated ion channels: Spotlight on symmetry. Structure 2007, 15, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.E.; Black, K.D.; Islas, L.D.; Sankaran, B.; Zagotta, W.N. Structure and rearrangements in the carboxy-terminal region of spih channels. Structure 2007, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, M.; Arcangeletti, M.; Marchesi, A.; Napolitano, L.M.R.; Grosa, D.; Maity, S.; Anselmi, C.; Torre, V. The gating mechanism in cyclic nucleotide-gated ion channels. Sci. Rep. 2018, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002, 417, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Nair, A.V.; Codega, P.; Torre, V.; Carloni, P. Structural basis of gating of cng channels. FEBS Lett. 2005, 579, 1968–1972. [Google Scholar] [CrossRef] [PubMed]
- Matulef, K.; Zagotta, W.N. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell Dev. Biol. 2003, 19, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Frings, S.; Seifert, R.; Godde, M.; Kaupp, U.B. Profoundly different calcium permeation and blockage determine the specific function of distinct cyclic nucleotide-gated channels. Neuron 1995, 15, 169–179. [Google Scholar] [CrossRef]
- Dzeja, C.; Hagen, V.; Kaupp, U.B.; Frings, S. Ca2+ permeation in cyclic nucleotide-gated channels. EMBO J. 1999, 18, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Reisert, J.; Frings, S. Regulation of cyclic nucleotide-gated channels. Curr. Opin. Neurobiol. 2005, 15, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, S.; Boccaccio, A.; Menini, A. Cyclic nucleotide-gated ion channels in sensory transduction. FEBS Lett. 2006, 580, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Weitz, D.; Zoche, M.; Muller, F.; Beyermann, M.; Korschen, H.G.; Kaupp, U.B.; Koch, K.W. Calmodulin controls the rod photoreceptor cng channel through an unconventional binding site in the n-terminus of the beta-subunit. EMBO J. 1998, 17, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Bonigk, W.; Yau, K.W.; Frings, S. Calmodulin permanently associates with rat olfactory cng channels under native conditions. Nat. Neurosci. 2004, 7, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Rebrik, T.I.; Botchkina, I.; Arshavsky, V.Y.; Craft, C.M.; Korenbrot, J.I. Cng-modulin: A novel ca-dependent modulator of ligand sensitivity in cone photoreceptor cgmp-gated ion channels. J. Neurosci. 2012, 32, 3142–3153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahnelt, P.K.; Kolb, H. The mammalian photoreceptor mosaic-adaptive design. Prog. Retin. Eye Res. 2000, 19, 711–777. [Google Scholar] [CrossRef]
- Yokoyama, S. Molecular evolution of vertebrate visual pigments. Prog. Retin. Eye Res. 2000, 19, 385–419. [Google Scholar] [CrossRef]
- Yang, R.B.; Foster, D.C.; Garbers, D.L.; Fulle, H.J. Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. USA 1995, 92, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Pugh, E.N., Jr.; Duda, T.; Sitaramayya, A.; Sharma, R.K. Photoreceptor guanylate cyclases: A review. Biosci. Rep. 1997, 17, 429–473. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Nakatani, K. Cation selectivity of light-sensitive conductance in retinal rods. Nature 1984, 309, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; McNaughton, P.A.; Nunn, B.J. The ionic selectivity and calcium dependence of the light-sensitive pathway in toad rods. J. Physiol. 1985, 358, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Nakatani, K. Light-induced reduction of cytoplasmic free calcium in retinal rod outer segment. Nature 1985, 313, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Schnetkamp, P.P. The slc24 Na+/Ca2+-K+ exchanger family: Vision and beyond. Pflugers Arch. 2004, 447, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, L.; Lagnado, L.; Perry, R.J.; Robinson, D.W.; McNaughton, P.A. Extrusion of calcium from rod outer segments is driven by both sodium and potassium gradients. Nature 1989, 337, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Vinberg, F.; Wang, T.; De Maria, A.; Zhao, H.; Bassnett, S.; Chen, J.; Kefalov, V.J. The Na+/Ca2+, K+ exchanger nckx4 is required for efficient cone-mediated vision. eLife 2017, 6, e24550. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Sokal, I.; Baehr, W. Guanylate cyclase-activating proteins: Structure, function, and diversity. Biochem. Biophys. Res. Commun. 2004, 322, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Korenbrot, J.I. Speed, sensitivity, and stability of the light response in rod and cone photoreceptors: Facts and models. Prog. Retin. Eye Res. 2012, 31, 442–466. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, T.; Hackos, D.H.; Frings, S.; Hagen, V.; Kaupp, U.B.; Korenbrot, J.I. Fraction of the dark current carried by Ca2+ through cgmp-gated ion channels of intact rod and cone photoreceptors. J. Gen. Physiol. 2000, 116, 735–754. [Google Scholar] [CrossRef] [PubMed]
- Bareil, C.; Hamel, C.P.; Delague, V.; Arnaud, B.; Demaille, J.; Claustres, M. Segregation of a mutation in cngb1 encoding the beta-subunit of the rod cgmp-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum. Genet. 2001, 108, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; Finn, J.T.; Peng, Y.W.; McGee, T.L.; Berson, E.L.; Yau, K.W. Mutations in the gene encoding the alpha subunit of the rod cgmp-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 10177–10181. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Kennan, A.; Aherne, A.; Humphries, P. Light in retinitis pigmentosa. Trends Genet. 2005, 21, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kalloniatis, M.; Fletcher, E.L. Retinitis pigmentosa: Understanding the clinical presentation, mechanisms and treatment options. Clin. Exp. Optom. 2004, 87, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, K.; Sheng, X.; Li, Y.; Gao, X.; Zhang, X.; Kang, X.; Pan, X.; Liu, Y.; Jiang, C.; et al. Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Akahori, M.; Sergeev, Y.; Yoshitake, K.; Ikeo, K.; Furuno, M.; Hayashi, T.; Kondo, M.; Ueno, S.; Tsunoda, K.; et al. Whole exome analysis identifies frequent cnga1 mutations in japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE 2014, 9, e108721. [Google Scholar] [CrossRef] [PubMed]
- Paloma, E.; Martinez-Mir, A.; Garcia-Sandoval, B.; Ayuso, C.; Vilageliu, L.; Gonzalez-Duarte, R.; Balcells, S. Novel homozygous mutation in the alpha subunit of the rod cgmp gated channel (cnga1) in two spanish sibs affected with autosomal recessive retinitis pigmentosa. J. Med. Genet. 2002, 39, E66. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Function and dysfunction of cng channels: Insights from channelopathies and mouse models. Mol. Neurobiol. 2007, 35, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Mallouk, N.; Ildefonse, M.; Pages, F.; Ragno, M.; Bennett, N. Basis for intracellular retention of a human mutant of the retinal rod channel alpha subunit. J. Membr. Biol. 2002, 185, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Qin, M.; Mizota, A.; Kondo, M.; Hayashi, H.; Hayashi, K.; Oshima, K.; Tahira, T.; Hayashi, K. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4433–4439. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.A.; Clark, G.R.; Alexander, S.; Silvestri, G.; Willoughby, C.E. Molecular diagnosis for heterogeneous genetic diseases with targeted high-throughput DNA sequencing applied to retinitis pigmentosa. J. Med. Genet. 2011, 48, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Becirovic, E.; Nakova, K.; Hammelmann, V.; Hennel, R.; Biel, M.; Michalakis, S. The retinitis pigmentosa mutation c.3444+1g>a in cngb1 results in skipping of exon 32. PLoS ONE 2010, 5, e8969. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Zong, X.; Becirovic, E.; Hammelmann, V.; Wein, T.; Wanner, K.T.; Biel, M. The glutamic acid-rich protein is a gating inhibitor of cyclic nucleotide-gated channels. J. Neurosci. 2011, 31, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Leconte, L.; Barnstable, C.J. Impairment of rod cgmp-gated channel alpha-subunit expression leads to photoreceptor and bipolar cell degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 917–926. [Google Scholar]
- Wiik, A.C.; Ropstad, E.O.; Ekesten, B.; Karlstam, L.; Wade, C.M.; Lingaas, F. Progressive retinal atrophy in shetland sheepdog is associated with a mutation in the cnga1 gene. Anim. Genet. 2015, 46, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Hüttl, S.; Michalakis, S.; Seeliger, M.; Luo, D.G.; Acar, N.; Geiger, H.; Hudl, K.; Mader, R.; Haverkamp, S.; Moser, M.; et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit cngb1. J. Neurosci. 2005, 25, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Molday, L.L.; Molday, R.S.; Sarfare, S.S.; Woodruff, M.L.; Fain, G.L.; Kraft, T.W.; Pittler, S.J. Knockout of garps and the beta-subunit of the rod cgmp-gated channel disrupts disk morphogenesis and rod outer segment structural integrity. J. Cell Sci. 2009, 122, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Blank, T.; Goldmann, T.; Koch, M.; Amann, L.; Schon, C.; Bonin, M.; Pang, S.; Prinz, M.; Burnet, M.; Wagner, J.E.; et al. Early microglia activation precedes photoreceptor degeneration in a mouse model of cngb1-linked retinitis pigmentosa. Front. Immunol. 2017, 8, 1930. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Jones, S.M.; Occelli, L.M.; Winkler, P.A.; Lee, W.; Sparrow, J.R.; Tsukikawa, M.; Boye, S.L.; Chiodo, V.; Capasso, J.E.; Becirovic, E.; et al. Patients and animal models of cngbeta1-deficient retinitis pigmentosa support gene augmentation approach. J. Clin. Investig. 2018, 128, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Jones, S.M.; Winkler, P.A.; Bartoe, J.T.; Venta, P.J.; Ekenstedt, K. Large animal model of autosomal recessive rp due to a cngb1 gene mutation. Investig. Ophthalmol. Vis. Sci. 2013, 2013, 684. [Google Scholar]
- Kohl, S.; Marx, T.; Giddings, I.; Jagle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cgmp-gated cation channel. Nat. Genet. 1998, 19, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Baumann, B.; Broghammer, M.; Jagle, H.; Sieving, P.; Kellner, U.; Spegal, R.; Anastasi, M.; Zrenner, E.; Sharpe, L.T.; et al. Mutations in the cngb3 gene encoding the beta-subunit of the cone photoreceptor cgmp-gated channel are responsible for achromatopsia (achm3) linked to chromosome 8q21. Hum. Mol. Genet. 2000, 9, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H.; Yang, J.M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Silva, E.D.; Maumenee, I.H. Genetic basis of total colourblindness among the pingelapese islanders. Nat. Genet. 2000, 25, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S. The molecular basis of variation in human color vision. Clin. Genet. 2005, 67, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Jagla, W.M.; Jagle, H.; Hayashi, T.; Sharpe, L.T.; Deeb, S.S. The molecular basis of dichromatic color vision in males with multiple red and green visual pigment genes. Hum. Mol. Genet. 2002, 11, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Eksandh, L.; Kohl, S.; Wissinger, B. Clinical features of achromatopsia in swedish patients with defined genotypes. Ophthalmic Genet. 2002, 23, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J.; Smith, V.C.; Pinckers, A.J.; Cozijnsen, M. Classification of complete and incomplete autosomal recessive achromatopsia. Graefes Arch. Clin. Exp. Ophthalmol. 1982, 219, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Schön, C.; Becirovic, E.; Biel, M. Gene therapy for achromatopsia. J. Gene Med. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Aboshiha, J.; Dubis, A.M.; Carroll, J.; Hardcastle, A.J.; Michaelides, M. The cone dysfunction syndromes. Br. J. Ophthalmol. 2016, 100, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Poloschek, C.M.; Kohl, S. [achromatopsia]. Ophthalmologe 2010, 107, 571–580; quiz 581–572. [Google Scholar] [CrossRef] [PubMed]
- Zobor, D.; Zobor, G.; Kohl, S. Achromatopsia: On the doorstep of a possible therapy. Ophthalmic Res. 2015, 54, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Remmer, M.H.; Rastogi, N.; Ranka, M.P.; Ceisler, E.J. Achromatopsia: A review. Curr. Opin. Ophthalmol. 2015, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Varsanyi, B.; Antunes, G.A.; Baumann, B.; Hoyng, C.B.; Jagle, H.; Rosenberg, T.; Kellner, U.; Lorenz, B.; Salati, R.; et al. Cngb3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur. J. Hum. Genet. 2005, 13, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Sacks, O.W. The Island of the Colourblind; Alfred A. Knopf: New York, NY, USA, 1997. [Google Scholar]
- Biel, M.; Seeliger, M.; Pfeifer, A.; Kohler, K.; Gerstner, A.; Ludwig, A.; Jaissle, G.; Fauser, S.; Zrenner, E.; Hofmann, F. Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel cng3. Proc. Natl. Acad. Sci. USA 1999, 96, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Geiger, H.; Haverkamp, S.; Hofmann, F.; Gerstner, A.; Biel, M. Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel cnga3. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of cone vision in the cnga3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Xu, J.; Biel, M.; Ding, X.Q. Detection of cgmp in the degenerating retina. Methods Mol. Biol. 2013, 1020, 235–245. [Google Scholar] [PubMed]
- Pang, J.J.; Deng, W.T.; Dai, X.; Lei, B.; Everhart, D.; Umino, Y.; Li, J.; Zhang, K.; Mao, S.; Boye, S.L.; et al. Aav-mediated cone rescue in a naturally occurring mouse model of cnga3-achromatopsia. PLoS ONE 2012, 7, e35250. [Google Scholar] [CrossRef] [PubMed]
- Reicher, S.; Seroussi, E.; Gootwine, E. A mutation in gene cnga3 is associated with day blindness in sheep. Genomics 2010, 95, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Shamir, M.H.; Ofri, R.; Bor, A.; Brenner, O.; Reicher, S.; Obolensky, A.; Averbukh, E.; Banin, E.; Gootwine, E. A novel day blindness in sheep: Epidemiological, behavioural, electrophysiological and histopathological studies. Vet. J. 2010, 185, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Dutrow, E.V.; Miyadera, K.; Delemotte, L.; MacDermaid, C.M.; Reinstein, S.L.; Crumley, W.R.; Dixon, C.J.; Casal, M.L.; Klein, M.L.; et al. Canine cnga3 gene mutations provide novel insights into human achromatopsia-associated channelopathies and treatment. PLoS ONE 2015, 10, e0138943. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Harry, C.S.; Umino, Y.; Matveev, A.V.; Fliesler, S.J.; Barlow, R.B. Impaired cone function and cone degeneration resulting from cngb3 deficiency: Down-regulation of cnga3 biosynthesis as a potential mechanism. Hum. Mol. Genet. 2009, 18, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Morris, L.; Fliesler, S.J.; Sherry, D.M.; Ding, X.Q. Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in cng channel subunit cngb3 deficiency. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3557–3566. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sidjanin, D.J.; Lowe, J.K.; McElwee, J.L.; Milne, B.S.; Phippen, T.M.; Sargan, D.R.; Aguirre, G.D.; Acland, G.M.; Ostrander, E.A. Canine cngb3 mutations establish cone degeneration as orthologous to the human achromatopsia locus achm3. Hum. Mol. Genet. 2002, 11, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, G.D.; Rubin, L.F. Pathology of hemeralopia in the alaskan malamute dog. Investig. Ophthalmol. 1974, 13, 231–235. [Google Scholar]
- Aguirre, G.D.; Rubin, L.F. The electroretinogram in dogs with inherited cone degeneration. Investig. Ophthalmol. 1975, 14, 840–847. [Google Scholar]
- Rubin, L.F. Hemeralopia in alaskan malamute pups. J. Am. Vet. Med. Assoc. 1971, 158, 1699–1701. [Google Scholar] [PubMed]
- Rubin, L.F. Clinical features of hemeralopia in the adult alaskan malamute. J. Am. Vet. Med. Assoc. 1971, 158, 1696–1698. [Google Scholar] [PubMed]
- Winkler, P.A.; Ekenstedt, K.J.; Occelli, L.M.; Frattaroli, A.V.; Bartoe, J.T.; Venta, P.J.; Petersen-Jones, S.M. A large animal model for cngb1 autosomal recessive retinitis pigmentosa. PLoS ONE 2013, 8, e72229. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Sothilingam, V.; Garcia Garrido, M.; Tanimoto, N.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; Seeliger, M.W.; Biel, M.; et al. Gene therapy restores vision and delays degeneration in the cngb1(−/−) mouse model of retinitis pigmentosa. Hum. Mol. Genet. 2012, 21, 4486–4496. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.J.; Alexander, J.; Lei, B.; Deng, W.; Zhang, K.; Li, Q.; Chang, B.; Hauswirth, W.W. Achromatopsia as a potential candidate for gene therapy. Adv. Exp. Med. Biol. 2010, 664, 639–646. [Google Scholar] [PubMed]
- Banin, E.; Gootwine, E.; Obolensky, A.; Ezra-Elia, R.; Ejzenberg, A.; Zelinger, L.; Honig, H.; Rosov, A.; Yamin, E.; Sharon, D.; et al. Gene augmentation therapy restores retinal function and visual behavior in a sheep model of cnga3 achromatopsia. Mol. Ther. 2015, 23, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Mühlfriedel, R.; Tanimoto, N.; Schon, C.; Sothilingam, V.; Garcia Garrido, M.; Beck, S.C.; Huber, G.; Biel, M.; Seeliger, M.W.; Michalakis, S. Aav-mediated gene supplementation therapy in achromatopsia type 2: Preclinical data on therapeutic time window and long-term effects. Front. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Tao, Y.; Deng, W.T.; Zhu, P.; Li, J.; Dai, X.; Zhang, Y.; Shi, W.; Liu, X.; Chiodo, V.A.; et al. Vitreal delivery of aav vectored cnga3 restores cone function in cnga3−/−/nrl−/− mice, an all-cone model of cnga3 achromatopsia. Hum. Mol. Genet. 2015, 24, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; He, Y.; Zhang, H.; Zhang, Y.; Liu, Y.; Wang, M.; Chen, H.; Pang, J.J. Long-term retinal cone rescue using a capsid mutant aav8 vector in a mouse model of cnga3-achromatopsia. PLoS ONE 2017, 12, e0188032. [Google Scholar] [CrossRef] [PubMed]
- Gootwine, E.; Abu-Siam, M.; Obolensky, A.; Rosov, A.; Honig, H.; Nitzan, T.; Shirak, A.; Ezra-Elia, R.; Yamin, E.; Banin, E.; et al. Gene augmentation therapy for a missense substitution in the cgmp-binding domain of ovine cnga3 gene restores vision in day-blind sheep. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. Aav8 can induce innate and adaptive immune response in the primate eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef] [PubMed]
- Gootwine, E.; Ofri, R.; Banin, E.; Obolensky, A.; Averbukh, E.; Ezra-Elia, R.; Ross, M.; Honig, H.; Rosov, A.; Yamin, E.; et al. Safety and efficacy evaluation of raav2tyf-pr1.7-hcnga3 vector delivered by subretinal injection in cnga3 mutant achromatopsia sheep. Hum. Gene Ther. Clin. Dev. 2017, 28, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Ochakovski, G.A.; Peters, T.; Michalakis, S.; Wilhelm, B.; Wissinger, B.; Biel, M.; Bartz-Schmidt, K.U.; Fischer, M.D.; Consortium, R.-C. Subretinal injection for gene therapy does not cause clinically significant outer nuclear layer thinning in normal primate foveae. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Seitz, I.P.; Michalakis, S.; Wilhelm, B.; Reichel, F.F.; Ochakovski, G.A.; Zrenner, E.; Ueffing, M.; Biel, M.; Wissinger, B.; Bartz-Schmidt, K.U.; et al. Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of aav8 in nonhuman primates. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5792–5801. [Google Scholar] [CrossRef] [PubMed]
- Komaromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Tanaka, J.C.; Acland, G.M.; Hauswirth, W.W.; Aguirre, G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Komaromy, A.M.; Rowlan, J.S.; Corr, A.T.; Reinstein, S.L.; Boye, S.L.; Cooper, A.E.; Gonzalez, A.; Levy, B.; Wen, R.; Hauswirth, W.W.; et al. Transient photoreceptor deconstruction by cntf enhances raav-mediated cone functional rescue in late stage cngb3-achromatopsia. Mol. Ther. 2013, 21, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Xu, J.; Pearson, R.A.; Smith, A.J.; Bainbridge, J.W.; Morris, L.M.; Fliesler, S.J.; Ding, X.Q.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of cngb3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Komaromy, A.M.; Zeiss, C.; Calcedo, R.; Harman, C.D.; Koehl, K.L.; Stewart, G.A.; Iwabe, S.; Chiodo, V.A.; Hauswirth, W.W.; et al. Safety and efficacy of aav5 vectors expressing human or canine cngb3 in cngb3-mutant dogs. Hum. Gene Ther. Clin. Dev. 2017, 28, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; Sharma, A.K.; Ver Hoeve, J.N.; Smith, L.; Arndt, T.; Calcedo, R.; et al. Safety and biodistribution evaluation in cynomolgus macaques of raav2tyf-pr1.7-hcngb3, a recombinant aav vector for treatment of achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; McPherson, L.; Ver Hoeve, J.N.; Smith, L.; Arndt, T.; Mandapati, S.; et al. Safety and biodistribution evaluation in cngb3-deficient mice of raav2tyf-pr1.7-hcngb3, a recombinant aav vector for treatment of achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Hamel, C. Clinical utility gene card for: Achromatopsia—Update 2013. Eur. J. Hum. Genet. 2013, 21. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Schön, C.; Becirovic, E.; Biel, M.; Michalakis, S. Design and development of aav-based gene supplementation therapies for achromatopsia and retinitis pigmentosa. Methods Mol. Biol. 2018, 1715, 33–46. [Google Scholar] [PubMed]
- Schön, C.; Biel, M.; Michalakis, S. Retinal gene delivery by adeno-associated virus (aav) vectors: Strategies and applications. Eur. J. Pharm. Biopharm. 2015, 95, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Tay, S.S.; Alexander, I.E. Adeno-associated virus serotypes for gene therapeutics. Curr. Opin. Pharmacol. 2015, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [PubMed]
- Willett, K.; Bennett, J. Immunology of aav-mediated gene transfer in the eye. Front. Immunol. 2013, 4, 261. [Google Scholar] [CrossRef] [PubMed]
- Dismuke, D.J.; Tenenbaum, L.; Samulski, R.J. Biosafety of recombinant adeno-associated virus vectors. Curr. Gene Ther. 2013, 13, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using aav: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Kaeppel, C.; Beattie, S.G.; Fronza, R.; van Logtenstein, R.; Salmon, F.; Schmidt, S.; Wolf, S.; Nowrouzi, A.; Glimm, H.; von Kalle, C.; et al. A largely random aav integration profile after lpld gene therapy. Nat. Med. 2013, 19, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Mehta, A.; Wang, G.; Hauswirth, W.W.; Chiodo, V.; Boye, S.L.; Guy, J. Next-generation sequencing of mitochondrial targeted aav transfer of human nd4 in mice. Mol. Vis. 2013, 19, 1482–1491. [Google Scholar] [PubMed]
- Fischer, M.D.; Wilhelm, B.; Michalakis, S.; Zobor, D.; Kohl, S.; Seeliger, M.; Zrenner, E.; Ueffing, M.; Wissinger, B.; Biel, M.; et al. Successful delivery of raav8.Cnga3 in a patient with cnga3 achromatopsia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5207. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Gene (Cell Type) | Associated Human Disease | Animal Models | POC Studies | Preclinical Safety Studies | Clinical Trials (ID) |
|---|---|---|---|---|---|
| CNGA1 (rods) | Retinitis pigmentosa, RP49 | Cnga1 antisense expressing mice: retinal degeneration [53] Canine model (missense mutation): Progressive retinal atrophy [54] | - | - | - |
| CNGB1 (rods) | Retinitis pigmentosa, RP45 | Cngb1-deficient mice: impaired rod function and retinal degeneration [55,56] Canine model (missense mutation): impaired rod function and retinal degeneration [89] | [58,90] | - | - |
| CNGA3 (cones) | Achromatopsia, ACHM2 | Cnga3-deficient mice: loss of cone function and cone degeneration [74] cpfl5 mouse: loss of cone function and cone cell degeneration [91] Sheep model (missense mutation): loss of cone function and day blindness [80] | [76,78,92,93,94,95,96] | [97,98,99,100] | NCT02610582 NCT02935517 |
| CNGB3 (cones) | Achromatopsia, ACHM1 | Canine models (null-deletion or missense mutation): cone degeneration [84] Cngb3-deficient mice: Impaired cone function and cone degeneration [82] | [101,102,103] | [98,104,105,106] | NCT02599922 NCT03001310 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. https://doi.org/10.3390/ijms19030749
Michalakis S, Becirovic E, Biel M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. International Journal of Molecular Sciences. 2018; 19(3):749. https://doi.org/10.3390/ijms19030749
Chicago/Turabian StyleMichalakis, Stylianos, Elvir Becirovic, and Martin Biel. 2018. "Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy" International Journal of Molecular Sciences 19, no. 3: 749. https://doi.org/10.3390/ijms19030749
APA StyleMichalakis, S., Becirovic, E., & Biel, M. (2018). Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. International Journal of Molecular Sciences, 19(3), 749. https://doi.org/10.3390/ijms19030749