Temporospatial Analysis and New Players in the Immunology of Amyotrophic Lateral Sclerosis



Abstract
1. Amyotrophic Lateral Sclerosis (ALS)
2. New Immune Players in ALS
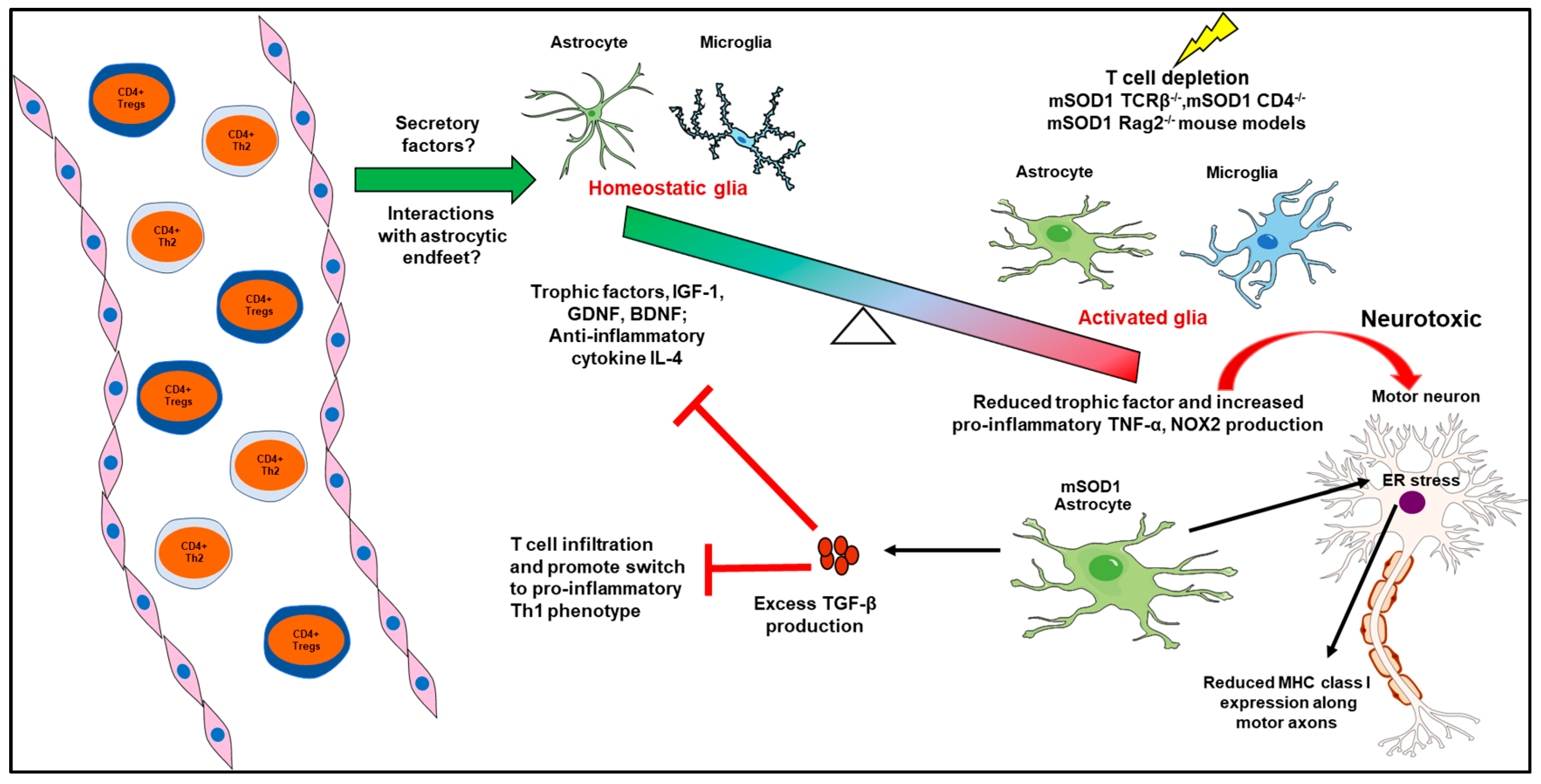
2.1. Peripheral Immune System—CNS Glia Interactions
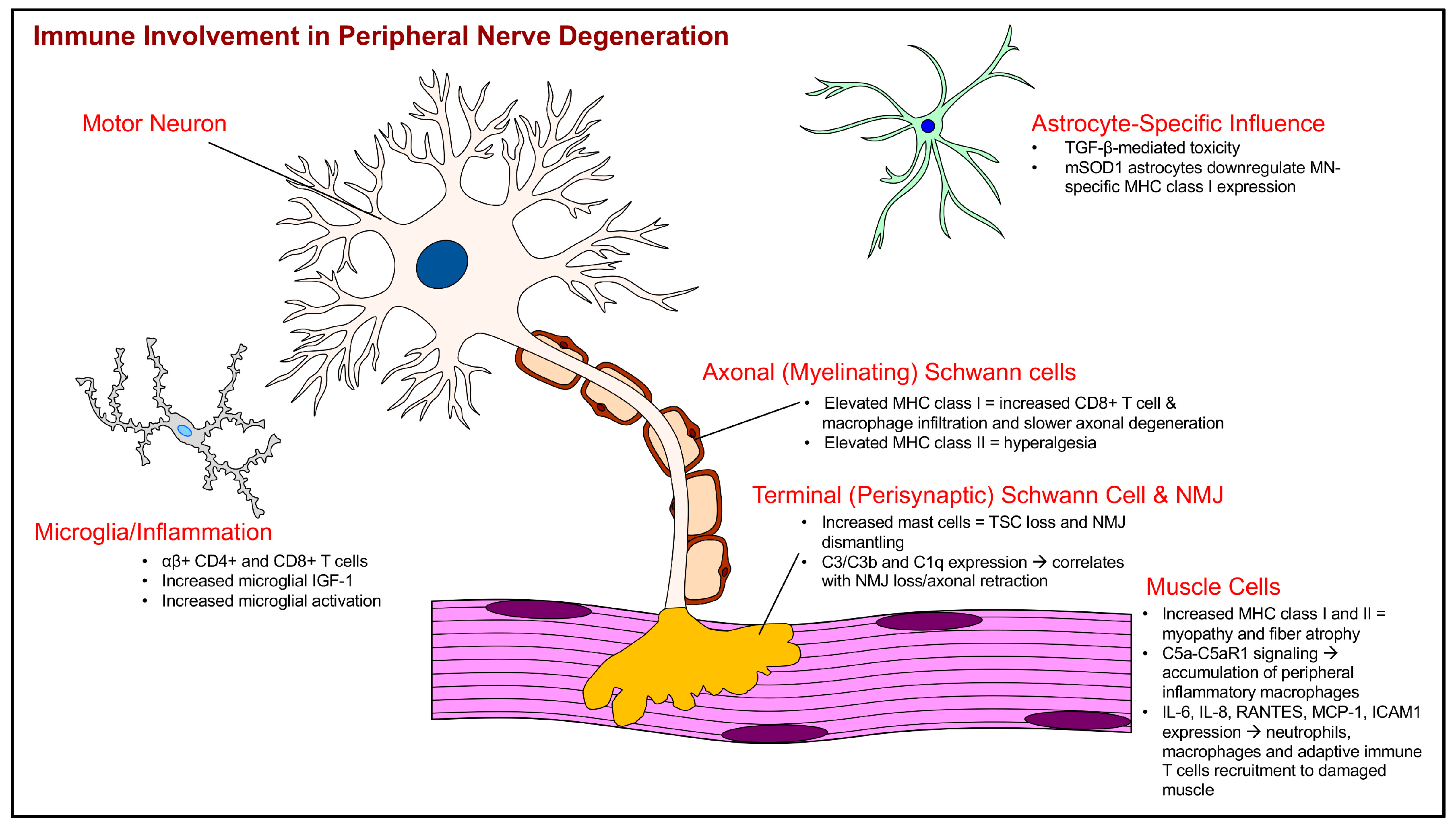
2.2. Immune System Interactions with the Tripartite Neuromuscular Junction
2.3. Schwann Cell (SC) Abnormalities and Immune Interactions
2.4. Skeletal Muscle Immune System Cross-Talk
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Talbot, K. Motor neuron disease: The bare essentials. Pract. Neurol. 2009, 9, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 2015. [Google Scholar] [CrossRef]
- Kraemer, M.; Buerger, M.; Berlit, P. Diagnostic problems and delay of diagnosis in amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg. 2010, 112, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Puentes, F.; Topping, J.; Kuhle, J.; van der Star, B.J.; Douiri, A.; Giovannoni, G.; Baker, D.; Amor, S.; Malaspina, A. Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, A.; Puentes, F.; Amor, S. Disease origin and progression in amyotrophic lateral sclerosis: An immunology perspective. Int. Immunol. 2015, 27, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in pu.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Cozzolino, M.; Carri, M.T. Old versus new mechanisms in the pathogenesis of ALS. Brain Pathol. 2016, 26, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H., Jr.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, D.M.; Camirand, G. New insights into the mechanisms of Treg function. Curr. Opin. Organ Transplant. 2015, 20, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Deboy, C.A.; Xin, J.; Byram, S.C.; Serpe, C.J.; Sanders, V.M.; Jones, K.J. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4+ T cells. Exp. Neurol. 2006, 201, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Setter, D.O.; Runge, E.M.; Schartz, N.D.; Kennedy, F.M.; Brown, B.L.; McMillan, K.P.; Miller, W.M.; Shah, K.M.; Haulcomb, M.M.; Sanders, V.M.; et al. Impact of peripheral immune status on central molecular responses to facial nerve axotomy. Brain Behav. Immun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Miranda, C.J.; Braun, L.; Meyer, K.; Frakes, A.E.; Ferraiuolo, L.; Likhite, S.; Bevan, A.K.; Foust, K.D.; McConnell, M.J.; et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat. Med. 2016, 22, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular junction formation, aging, and disorders. Annu. Rev. Physiol. 2017, 80, 159–188. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.L.; Thams, S.; Lidman, O.; Piehl, F.; Hokfelt, T.; Karre, K.; Linda, H.; Cullheim, S. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. USA 2004, 101, 17843–17848. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Thams, S.; Oliveira, A.; Cullheim, S. MHC class I expression and synaptic plasticity after nerve lesion. Brain Res. Rev. 2008, 57, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Maehlen, J.; Nennesmo, I.; Olsson, A.B.; Olsson, T.; Schroder, H.D.; Kristensson, K. Peripheral nerve injury causes transient expression of MHC class I antigens in rat motor neurons and skeletal muscles. Brain Res. 1989, 481, 368–372. [Google Scholar] [CrossRef]
- Thams, S.; Brodin, P.; Plantman, S.; Saxelin, R.; Karre, K.; Cullheim, S. Classical major histocompatibility complex class I molecules in motoneurons: New actors at the neuromuscular junction. J. Neurosci. 2009, 29, 13503–13515. [Google Scholar] [CrossRef] [PubMed]
- Staats, K.A.; Schonefeldt, S.; Van Rillaer, M.; Van Hoecke, A.; Van Damme, P.; Robberecht, W.; Liston, A.; Van Den Bosch, L. Β-2 microglobulin is important for disease progression in a murine model for amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2013, 7, 249. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Trolese, M.C.; de Vito, G.; Cecchi, R.; Riva, N.; Dina, G.; Heath, P.R.; Quattrini, A.; Shaw, P.J.; Piazza, V.; et al. Immune response in peripheral axons delays disease progression in SOD1 G93A mice. J. Neuroinflamm. 2016, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Horste, G.; Hu, W.; Hartung, H.P.; Lehmann, H.C.; Kieseier, B.C. The immunocompetence of schwann cells. Muscle Nerve 2008, 37, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Dalakas, M.C. Expression of the co-stimulatory molecule BB-1, the ligands CTLA-4 and CD28 and their mRNAs in chronic inflammatory demyelinating polyneuropathy. Brain 2000, 123, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Painter, M.W. Aging Schwann cells: Mechanisms, implications, future directions. Curr. Opin. Neurobiol. 2017, 47, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Toews, A.D.; Barrett, C.; Morell, P. Monocyte chemoattractant protein 1 is responsible for macrophage recruitment following injury to sciatic nerve. J. Neurosci. Res. 1998, 53, 260–267. [Google Scholar] [CrossRef]
- Turner, B.J.; Ackerley, S.; Davies, K.E.; Talbot, K. Dismutase-competent SOD1 mutant accumulation in myelinating Schwann cells is not detrimental to normal or transgenic ALS model mice. Hum. Mol. Genet. 2010, 19, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Lobsiger, C.S.; Boillee, S.; McAlonis-Downes, M.; Khan, A.M.; Feltri, M.L.; Yamanaka, K.; Cleveland, D.W. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. USA 2009, 106, 4465–4470. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pytel, P.; Feltri, M.L.; Wrabetz, L.; Roos, R.P. Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice. Neurobiol. Dis. 2012, 48, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Arbour, D.; Vande Velde, C.; Robitaille, R. New perspectives on amyotrophic lateral sclerosis: The role of glial cells at the neuromuscular junction. J. Physiol. 2017, 595, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.I.; Seburn, K.L.; Pinter, M.J. Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp. Neurol. 2016, 275, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.I.; Bahr, B.A.; Seburn, K.L.; Pinter, M.J. Abnormal response of distal Schwann cells to denervation in a mouse model of motor neuron disease. Exp. Neurol. 2016, 278, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Nunez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2017, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Nunez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Diaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef] [PubMed]
- Hartlehnert, M.; Derksen, A.; Hagenacker, T.; Kindermann, D.; Schafers, M.; Pawlak, M.; Kieseier, B.C.; Meyer Zu Horste, G. Schwann cells promote post-traumatic nerve inflammation and neuropathic pain through MHC class Ii. Sci. Rep. 2017, 7, 12518. [Google Scholar] [CrossRef] [PubMed]
- Burden, S.J.; Yumoto, N.; Zhang, W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a009167. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzo, S.; Zucca, I.; Mastropietro, A.; de Rosbo, N.K.; Cavalcante, P.; Tartari, S.; Bonanno, S.; Preite, L.; Mantegazza, R.; Bernasconi, P. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: A longitudinal MRI study. Exp. Neurol. 2011, 231, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, J.; Putman, C.T.; Gordon, T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007, 28, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Dengler, R.; Konstanzer, A.; Kuther, G.; Hesse, S.; Wolf, W.; Struppler, A. Amyotrophic lateral sclerosis: Macro-EMG and twitch forces of single motor units. Muscle Nerve 1990, 13, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Sanjak, M.; Brinkmann, J.; Belden, D.S.; Roelke, K.; Waclawik, A.; Neville, H.E.; Ringel, S.P.; Murphy, J.R.; Brooks, B.R. Quantitative assessment of motor fatigue in amyotrophic lateral sclerosis. J. Neurol. Sci. 2001, 191, 55–59. [Google Scholar] [CrossRef]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Garlepp, M.J.; Chen, W.; Tabarias, H.; Baines, M.; Brooks, A.; McCluskey, J. Antigen processing and presentation by a murine myoblast cell line. Clin. Exp. Immunol. 1995, 102, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Mitsdoerffer, M.; Schneider, D.; Melms, A.; Lochmuller, H.; Hohlfeld, R.; Weller, M. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible co-stimulatory molecule, ICOSL: Implications for the pathogenesis of inflammatory myopathies. Brain 2003, 126, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Blumbergs, P.C.; Manavis, J.; Limaye, V.S. Major histocompatibility complex class I and II expression in idiopathic inflammatory myopathy. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Englund, P.; Lindroos, E.; Nennesmo, I.; Klareskog, L.; Lundberg, I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am. J. Pathol. 2001, 159, 1263–1273. [Google Scholar] [CrossRef]
- Wiendl, H.; Behrens, L.; Maier, S.; Johnson, M.A.; Weiss, E.H.; Hohlfeld, R. Muscle fibers in inflammatory myopathies and cultured myoblasts express the nonclassical major histocompatibility antigen HLA-G. Ann. Neurol. 2000, 48, 679–684. [Google Scholar] [CrossRef]
- Wiendl, H.; Mitsdoerffer, M.; Weller, M. Express and protect yourself: The potential role of HLA-G on muscle cells and in inflammatory myopathies. Hum. Immunol. 2003, 64, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Rouas-Freiss, N.; Goncalves, R.M.; Menier, C.; Dausset, J.; Carosella, E.D. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 11520–11525. [Google Scholar] [CrossRef] [PubMed]
- Riteau, B.; Rouas-Freiss, N.; Menier, C.; Paul, P.; Dausset, J.; Carosella, E.D. HLA-G2, -G3, and -G4 isoforms expressed as nonmature cell surface glycoproteins inhibit NK and antigen-specific CTl cytolysis. J. Immunol. 2001, 166, 5018–5026. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Mitsdoerffer, M.; Schneider, D.; Chen, L.; Lochmuller, H.; Melms, A.; Weller, M. Human muscle cells express a B7-related molecule, B7-H1, with strong negative immune regulatory potential: A novel mechanism of counterbalancing the immune attack in idiopathic inflammatory myopathies. FASEB J. 2003, 17, 1892–1894. [Google Scholar] [CrossRef] [PubMed]
- Waschbisch, A.; Wintterle, S.; Lochmuller, H.; Walter, M.C.; Wischhusen, J.; Kieseier, B.C.; Wiendl, H. Human muscle cells express the costimulatory molecule B7-H3, which modulates muscle-immune interactions. Arthritis Rheum. 2008, 58, 3600–3608. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; D’Aigle, T.; Gowing, G.; Julien, J.P.; Rivest, S. Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2004, 24, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Rivest, S. MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. J. Cell Biol. 2007, 179, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Hulderman, T.; Liston, A.; Simeonova, P.P. Toll-like and adenosine receptor expression in injured skeletal muscle. Muscle Nerve 2011, 44, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Voss, J.; Wischhusen, J.; Dombrowski, Y.; Steinle, A.; Lochmuller, H.; Dalakas, M.; Melms, A.; Wiendl, H. Expression of toll-like receptors by human muscle cells in vitro and in vivo: TLR3 is highly expressed in inflammatory and HIV myopathies, mediates IL-8 release and up-regulation of NKG2D-ligands. FASEB J. 2006, 20, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Krook, A. Innate immune receptors in skeletal muscle metabolism. Exp. Cell Res. 2017, 360, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Henriques-Pons, A.; Yu, Q.; Rayavarapu, S.; Cohen, T.V.; Ampong, B.; Cha, H.J.; Jahnke, V.; Van der Meulen, J.; Wang, D.; Jiang, W.; et al. Role of toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet. 2014, 23, 2604–2617. [Google Scholar] [CrossRef] [PubMed]
- Fellner, A.; Barhum, Y.; Angel, A.; Perets, N.; Steiner, I.; Offen, D.; Lev, N. Toll-like receptor-4 inhibitor TAK-242 attenuates motor dysfunction and spinal cord pathology in an amyotrophic lateral sclerosis mouse model. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Letiembre, M.; Liu, Y.; Walter, S.; Hao, W.; Pfander, T.; Wrede, A.; Schulz-Schaeffer, W.; Fassbender, K. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol. Aging 2009, 30, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Sta, M.; Sylva-Steenland, R.M.; Casula, M.; de Jong, J.M.; Troost, D.; Aronica, E.; Baas, F. Innate and adaptive immunity in amyotrophic lateral sclerosis: Evidence of complement activation. Neurobiol. Dis. 2011, 42, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Heath, P.R.; Holden, H.; Kasher, P.; Kirby, J.; Shaw, P.J. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 2007, 27, 9201–9219. [Google Scholar] [CrossRef] [PubMed]
- Lobsiger, C.S.; Boillee, S.; Cleveland, D.W. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7319–7326. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.M.; Costantini, K.J.; Crane, J.W.; Atkin, J.D.; Monk, P.N.; Taylor, S.M.; Noakes, P.G. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J. Immunol. 2008, 181, 8727–8734. [Google Scholar] [CrossRef] [PubMed]
- Heurich, B.; El Idrissi, N.B.; Donev, R.M.; Petri, S.; Claus, P.; Neal, J.; Morgan, B.P.; Ramaglia, V. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J. Neuroimmunol. 2011, 235, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Bahia El Idrissi, N.; Bosch, S.; Ramaglia, V.; Aronica, E.; Baas, F.; Troost, D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Presumey, J.; Bialas, A.R.; Carroll, M.C. Complement system in neural synapse elimination in development and disease. Adv. Immunol. 2017, 135, 53–79. [Google Scholar] [PubMed]
- Wang, H.A.; Lee, J.D.; Lee, K.M.; Woodruff, T.M.; Noakes, P.G. Complement C5a-C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. Skelet. Muscle 2017, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Figarella-Branger, D.; Civatte, M.; Bartoli, C.; Pellissier, J.F. Cytokines, chemokines, and cell adhesion molecules in inflammatory myopathies. Muscle Nerve 2003, 28, 659–682. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Sharma, M.C.; Sarkar, C.; Bhatia, R.; Singh, S.; Handa, R. Increased expression of cell adhesion molecules in inflammatory myopathies: Diagnostic utility and pathogenetic insights. Folia Neuropathol. 2009, 47, 33–42. [Google Scholar] [PubMed]
- Peake, J.M.; Della Gatta, P.; Suzuki, K.; Nieman, D.C. Cytokine expression and secretion by skeletal muscle cells: Regulatory mechanisms and exercise effects. Exerc. Immunol. Rev. 2015, 21, 8–25. [Google Scholar] [PubMed]
- Tidball, J.G. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017, 17, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.T.; Hwang, C.S.; Hsieh, C.H.; Lu, C.H.; Chang, S.L.; Lee, J.C.; Huang, C.F.; Chang, H.T. Eosinophil-derived neurotoxin is elevated in patients with amyotrophic lateral sclerosis. Mediat. Inflamm. 2013, 2013, 421389. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Nikolaou, C.; Rombos, A.; Boufidou, F.; Zoga, M.; Dimitrakopoulos, A.; Tsoutsou, A.; Vassilopoulos, D. Rantes levels are elevated in serum and cerebrospinal fluid in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2007, 8, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Schroder, T.; Fuchss, J.; Schneider, I.; Stoltenburg-Didinger, G.; Hanisch, F. Eosinophils in hereditary and inflammatory myopathies. Acta Myol. 2013, 32, 148–153. [Google Scholar] [PubMed]
- Sunohara, N.; Furukawa, S.; Nishio, T.; Mukoyama, M.; Satoyoshi, E. Neurotoxicity of human eosinophils towards peripheral nerves. J. Neurol. Sci. 1989, 92, 1–7. [Google Scholar] [CrossRef]
- Kingham, P.J.; McLean, W.G.; Walsh, M.T.; Fryer, A.D.; Gleich, G.J.; Costello, R.W. Effects of eosinophils on nerve cell morphology and development: The role of reactive oxygen species and p38 MAP kinase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L915–L924. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.T.; Curran, D.R.; Kingham, P.J.; Morgan, R.K.; Durcan, N.; Gleich, G.J.; McLean, W.G.; Costello, R.W. Effect of eosinophil adhesion on intracellular signaling in cholinergic nerve cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Cantor, A.B.; Yang, H.; Browne, C.; Wells, R.A.; Fujiwara, Y.; Orkin, S.H. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J. Exp. Med. 2002, 195, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Pfluger, C.M.; Henderson, R.D.; McCombe, P.A. Reduced levels of interleukin 33 and increased levels of soluble ST2 in subjects with amyotrophic lateral sclerosis. J. Neuroimmunol. 2012, 249, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Walsh, J.T.; Smirnov, I.; Zheng, J.; Kipnis, J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 2015, 85, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, J.; Hot, D.; Hansmannel, F.; Kerdraon, O.; Ferreira, S.; Hubans, C.; Maurage, C.A.; Huot, L.; Bensemain, F.; Laumet, G.; et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for alzheimer’s disease. Mol. Psychiatry 2009, 14, 1004–1016. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.R.; Milovanovic, M.; Allan, D.; Niedbala, W.; Besnard, A.G.; Fukada, S.Y.; Alves-Filho, J.C.; Togbe, D.; Goodyear, C.S.; Linington, C.; et al. IL-33 attenuates eae by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur. J. Immunol. 2012, 42, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Hou, M.S.; Wang, S.W.; Chang, C.L.; Liou, Y.H.; Liao, N.S. Skeletal muscle interleukin 15 promotes CD8+ T-cell function and autoimmune myositis. Skelet. Muscle 2015, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.T.; Quinn, L.S. Sarcopenia, obesity, and natural killer cell immune senescence in aging: Altered cytokine levels as a common mechanism. Aging 2012, 4, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Rombos, A.; Nikolaou, C.; Zoga, M.; Zouvelou, V.; Dimitrakopoulos, A.; Alexakis, T.; Tsoutsou, A.; Samakovli, A.; Michalopoulou, M.; et al. Interleukin-15 and interleukin-12 are elevated in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur. Neurol. 2010, 63, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Staff, N.P.; Bornschlegl, S.; Butler, G.W.; Maas, M.L.; Kazamel, M.; Zubair, A.; Gastineau, D.A.; Windebank, A.J.; Dietz, A.B. Comprehensive immune profiling reveals substantial immune system alterations in a subset of patients with amyotrophic lateral sclerosis. PLoS ONE 2017, 12, e0182002. [Google Scholar] [CrossRef] [PubMed]
- Valdez, G.; Tapia, J.C.; Lichtman, J.W.; Fox, M.A.; Sanes, J.R. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS ONE 2012, 7, e34640. [Google Scholar] [CrossRef] [PubMed]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Moller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Hammarberg, H.; Lidman, O.; Lundberg, C.; Eltayeb, S.Y.; Gielen, A.W.; Muhallab, S.; Svenningsson, A.; Linda, H.; van Der Meide, P.H.; Cullheim, S.; et al. Neuroprotection by encephalomyelitis: Rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J. Neurosci. 2000, 20, 5283–5291. [Google Scholar] [PubMed]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Wiendl, H.; Marcenaro, E.; Kerlero de Rosbo, N.; Uccelli, A.; Laroni, A. Regulatory functions of natural killer cells in multiple sclerosis. Front. Immunol. 2016, 7, 606. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Shegarfi, H.; Naddafi, F.; Mirshafiey, A. The role of natural killer cells in Alzheimer’s disease. Scand. J. Immunol. 2012, 76, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Lincecum, J.M.; Vieira, F.G.; Wang, M.Z.; Thompson, K.; De Zutter, G.S.; Kidd, J.; Moreno, A.; Sanchez, R.; Carrion, I.J.; Levine, B.A.; et al. From transcriptome analysis to therapeutic anti-CD40l treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat. Genet. 2010, 42, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Pfohl, S.R.; Halicek, M.T.; Mitchell, C.S. Characterization of the contribution of genetic background and gender to disease progression in the SOD1 G93A mouse model of amyotrophic lateral sclerosis: A meta-analysis. J. Neuromuscul. Dis. 2015, 2, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Murdock, B.J.; Bender, D.E.; Kashlan, S.R.; Figueroa-Romero, C.; Backus, C.; Callaghan, B.C.; Goutman, S.A.; Feldman, E.L. Increased ratio of circulating neutrophils to monocytes in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e242. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, M.; Gerardi, F.; Santus, W.; Lizio, A.; Sansone, V.A.; Lunetta, C.; Zanoni, I.; Granucci, F. Inflammatory role of dendritic cells in amyotrophic lateral sclerosis revealed by an analysis of patients’ peripheral blood. Sci. Rep. 2017, 7, 7853. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Shefner, J.M.; Schoenfeld, D.A.; Zhang, H.; Andreasson, K.I.; Rothstein, J.D.; Drachman, D.B. Trial of celecoxib in amyotrophic lateral sclerosis. Ann. Neurol. 2006, 60, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.; Moglia, C.; Balma, M.; Chio, A. Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: A therapeutic opportunity? CNS Neurol. Disord. Drug Targets 2010, 9, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Miller, R.G.; Murphy, J.R.; Ringel, S.P. Treatment of ALS with high dose pulse cyclophosphamide. J. Neurol. Sci. 1994, 124, 84–87. [Google Scholar] [CrossRef]
- Werdelin, L.; Boysen, G.; Jensen, T.S.; Mogensen, P. Immunosuppressive treatment of patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 1990, 82, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, S.G.; Brajkovic, S.; Cipolat Mis, M.S.; Parente, V.; Corti, S. Therapeutic strategies under development targeting inflammatory mechanisms in amyotrophic lateral sclerosis. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
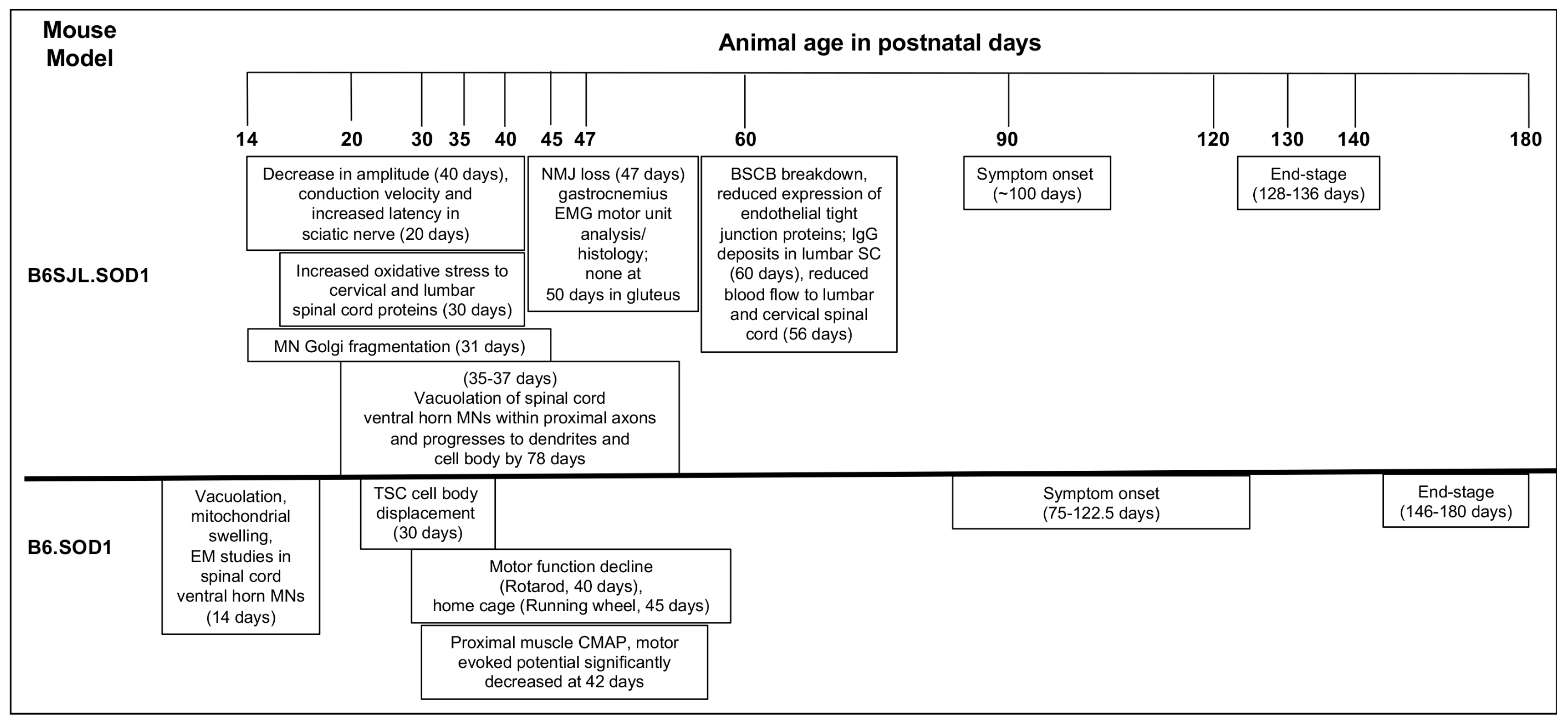
| Reported Immune Cellular/Marker Changes in ALS | ALS Model | Earliest Observed Immune System Changes in Postnatal Days of Age | Reference(s) |
|---|---|---|---|
| Increased lymphocyte cell numbers in spinal cord and altered microglial immune profile | B6SJL.SOD1G93A | 65 (presymptomatic) | [12] |
| Reduced growth factors and increased neurotoxic molecular expression in the absence of CD4+ T cells | B6.SOD1G93A | ~180 (end-stage) | [13] |
| Increase in percent CD4+CD25+, CD25+FoxP3 Tregs in SOD1 mice blood and reduced Tregs in ALS patients with rapidly progressing disease | B6.SOD1G93A and definite or probable sporadic ALS patients | 77 (symptom onset), peak at 112 (stable disease phase) in mice | [14] |
| Increased β2m in spinal cord of ALS mice | B6.SOD1G93A | 130 (mid-symptomatic) | [26] |
| Increase in MHC class I, β2m, LMP7, CCL2, C3, macrophages and CD8+ T cells along axons of peripheral nerve; MHC class I overlap with Schwann cells | B6.SOD1G93A | 135 (mid-symptomatic) | [27] |
| Endomysial infiltration of mast cells | SOD1G93A rats NTac:SD-Tg(SOD1G93A) L26H | Paralysis onset (187+/−15) | [38,39] |
| C3 activation products, C1q in motor end plates in mice; C1q and regulators CD55, CD59 on motor end plates in intercostal muscle | B6SJL.SOD1G93A; sporadic and familial ALS patients | 47 (presymptomatic); post-mortem | [72,73] |
| C1qB, C4, fB, C3, C5a, and C5aR1 in tibialis anterior (TA) muscles; C5a and its receptor C5aR1 in TA and soleus muscle with C5aR1 localization on macrophages | B6.SOD1G93A | Variable between 77 (symptom onset) and 130 (mid-symptomatic) for each listed molecule, however, most molecules are upregulated by 77; at 77 (symptom onset) | [75] |
| Elevated eosinophil-derived neurotoxin in serum and chemokine RANTES in serum and CSF | ALS patients | Mean ALSFRS-R = 17.8 (SD = 13.29) | [81,82] |
| Reduced IL-33 and elevated levels of soluble ST2 receptors in serum | ALS patients | met the modified El Escorial criteria for probable or definite ALS | [90] |
| Elevated serum IL-15 and NK cells | ALS patients | - | [96,97] |
| Increased macrophage accumulation in sciatic nerve, T cell co-stimulatory pathway upregulated in skeletal muscle, sciatic nerve, spinal cord and in blood samples from ALS patients | B6SJL.SOD1G93A and ALS patients | 50 (symptom onset) through 120 (end-stage) in mice | [103] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, A.K.; Jones, K.J.; Sanders, V.M.; Walker, C.L. Temporospatial Analysis and New Players in the Immunology of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 631. https://doi.org/10.3390/ijms19020631
Iyer AK, Jones KJ, Sanders VM, Walker CL. Temporospatial Analysis and New Players in the Immunology of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2018; 19(2):631. https://doi.org/10.3390/ijms19020631
Chicago/Turabian StyleIyer, Abhirami K., Kathryn J. Jones, Virginia M. Sanders, and Chandler L. Walker. 2018. "Temporospatial Analysis and New Players in the Immunology of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 19, no. 2: 631. https://doi.org/10.3390/ijms19020631
APA StyleIyer, A. K., Jones, K. J., Sanders, V. M., & Walker, C. L. (2018). Temporospatial Analysis and New Players in the Immunology of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 19(2), 631. https://doi.org/10.3390/ijms19020631