The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease


{kind=link}
Abstract
1. Introduction
2. Identification of a Zn2+-Sensing Receptor, ZnR/GPR39
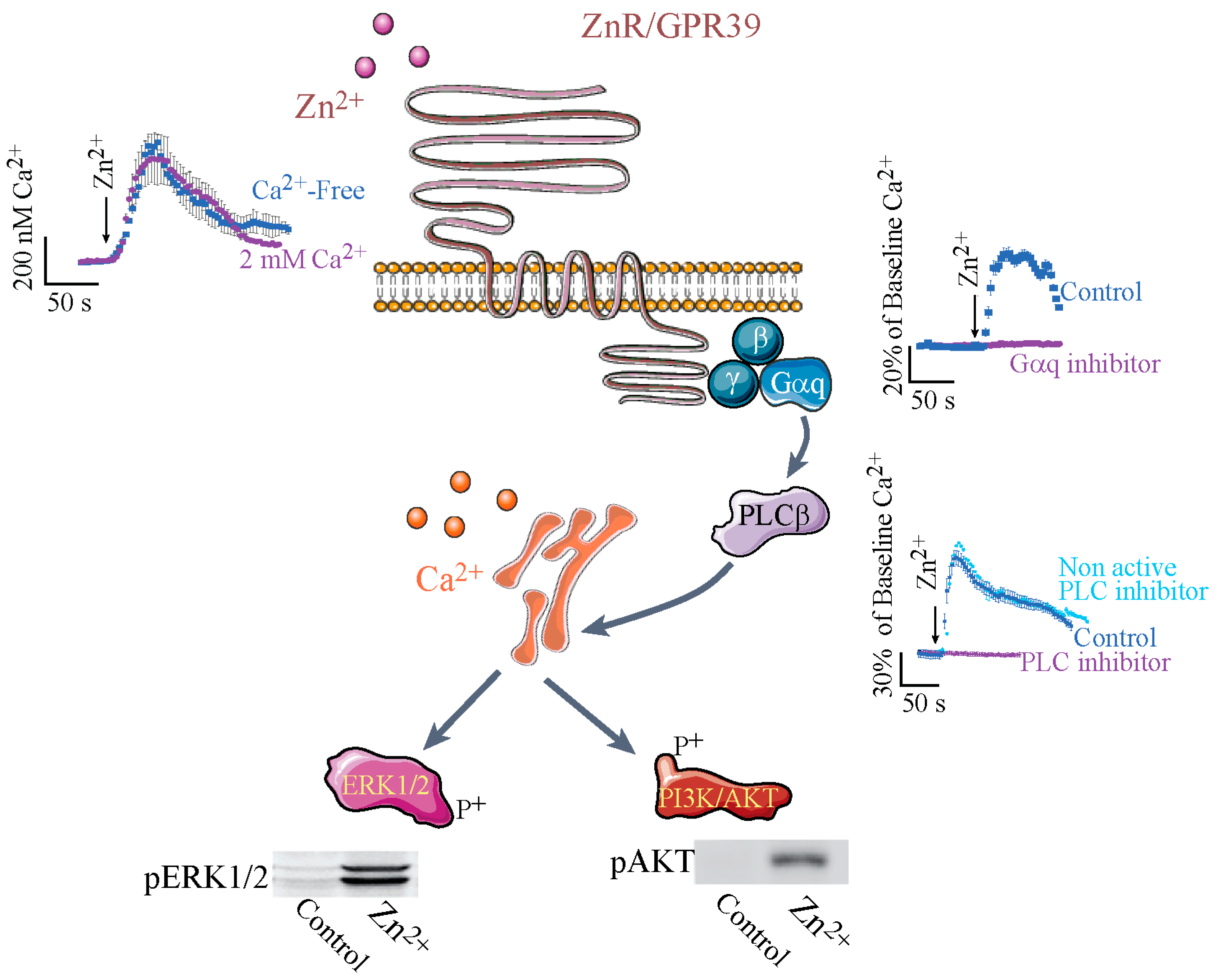
3. ZnR/GPR39-Dependent Signaling
4. ZnR/GPR39 Regulation of Physiological Functions
4.1. ZnR/GPR39 Regulates Ion Transport Mechanisms
4.2. ZnR/GPR39 Regulates Tight Junction Formation
5. A Role for ZnR/GPR39 in Disease
5.1. ZnR/GPR39 in Wound Healing
5.2. Diarrhea and Inflammatory Bowel Diseases
5.3. Epilepsy
5.4. Depression
5.5. Insulin Secretion
5.6. Defects in Bone Composition
5.7. ZnR/GPR39 in Cancer
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Roohani, N.; Hurrell, R.; Kelishadi, R.; Schulin, R. Zinc and its importance for human health: An integrative review. J. Res. Med. Sci. 2013, 18, 144–157. [Google Scholar] [PubMed]
- Prasad, A.S. Zinc in human health: Effect of zinc on immune cells. Mol. Med. 2008, 14, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Sandstead, H.H.; Frederickson, C.J.; Penland, J.G. History of zinc as related to brain function. J. Nutr. 2000, 130, 496S–502S. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, S.L.; McCormick, N.H.; Velasquez, V.; Lopez, V. Zinc in specialized secretory tissues: Roles in the pancreas, prostate, and mammary gland. Adv. Nutr. 2011, 2, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Weaver, B.P.; Andrews, G.K. The genetics of essential metal homeostasis during development. Genesis 2008, 46, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L. Zinc and carbonic anhydrase content of red cells in normals and in pernicious anemia. J. Clin. Investig. 1948, 27, 559. [Google Scholar] [PubMed]
- Vallee, B.L.; Altschule, M.D. Zinc in the mammalian organism, with particular reference to carbonic anhydrase. Physiol. Rev. 1949, 29, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc biochemistry, physiology, and homeostasis—Recent insights and current trends. BioMetals 2001, 14, 187–190. [Google Scholar] [CrossRef]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc biochemistry: From a single zinc enzyme to a key element of life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in cellular regulation: The nature and significance of “zinc signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.; Massarotti, A.; Hogstrand, C.; Maret, W. Zinc ions modulate protein tyrosine phosphatase 1B activity. Metallomics 2014, 6, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef] [PubMed]
- Sekler, I.; Sensi, S.L.; Hershfinkel, M.; Silverman, W.F. Mechanism and regulation of cellular zinc transport. Mol. Med. 2007, 13, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Cousins, R.J. Mammalian zinc transporters. Annu. Rev. Nutr. 2004, 24, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Eide, D.J. Zinc transporters and the cellular trafficking of zinc. Biochim. Biophys. Acta 2006, 1763, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 2006, 11, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Aizenman, E.; Mastroberardino, P.G. Metals and neurodegeneration. Neurobiol. Dis. 2015, 81, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Hasegawa, A.; Hojyo, S.; Ohashi, W.; Fukada, T.; Nishida, K.; Hirano, T. A novel role of the l-type calcium channel α1D subunit as a gatekeeper for intracellular zinc signaling: Zinc wave. PLoS ONE 2012, 7, e39654. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K.; et al. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Fukada, T.; Shimoda, S.; Ohashi, W.; Bin, B.H.; Koseki, H.; Hirano, T. The zinc transporter SLC39A14/ZIP14 controls G-protein coupled receptor-mediated signaling required for systemic growth. PLoS ONE 2011, 6, e18059. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Sitren, H.S.; Cousins, R.J. The zinc transporter Zip14 influences c-Met phosphorylation and hepatocyte proliferation during liver regeneration in mice. Gastroenterology 2012, 142, 1536–1546.e5. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Liuzzi, J.P.; McClellan, S.; Cousins, R.J. Zinc transporter ZIP8 (SLC39A8) and zinc influence IFN-γ expression in activated human T cells. J. Leukoc. Biol. 2009, 86, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Nimmanon, T.; Ziliotto, S.; Morris, S.; Flanagan, L.; Taylor, K.M. Phosphorylation of zinc channel ZIP7 drives MAPK, PI3K and mTOR growth and proliferation signalling. Metallomics 2017, 9, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci. Signal 2012, 5, ra11. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Vichova, P.; Jordan, N.; Hiscox, S.; Hendley, R.; Nicholson, R.I. ZIP7-mediated intracellular zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Hessels, A.M.; Taylor, K.M.; Merkx, M. Monitoring cytosolic and ER Zn2+ in stimulated breast cancer cells using genetically encoded FRET sensors. Metallomics 2016, 8, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takeda, T.A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of zinc transporters: Molecular and genetic importance in zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Sullivan, P.G.; Sensi, S.L.; Steward, O.; Weiss, J.H. Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem. 2001, 276, 47524–47529. [Google Scholar] [CrossRef] [PubMed]
- Reilly-O’Donnell, B.; Robertson, G.B.; Karumbi, A.; McIntyre, C.; Bal, W.; Nishi, M.; Takeshima, H.; Stewart, A.J.; Pitt, S.J. Dysregulated Zn2+ homeostasis impairs cardiac type-2 ryanodine receptor and mitsugumin 23 functions, leading to sarcoplasmic reticulum Ca2+ leakage. J. Biol. Chem. 2017, 292, 13361–13373. [Google Scholar] [CrossRef] [PubMed]
- Woodier, J.; Rainbow, R.D.; Stewart, A.J.; Pitt, S.J. Intracellular zinc modulates cardiac ryanodine receptor-mediated calcium release. J. Biol. Chem. 2015, 290, 17599–17610. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in the biosciences. Metallomics 2014, 6, 1174. [Google Scholar] [CrossRef] [PubMed]
- Kocyla, A.; Adamczyk, J.; Krezel, A. Interdependence of free zinc changes and protein complex assembly—Insights into zinc signal regulation. Metallomics 2017, 10, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Hershfinkel, M. Zinc, a dynamic signaling molecule. In Molecular Biology of Metal Homeostasis and Detoxification; Tamas, M., Martinoia, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 14, pp. 131–153. [Google Scholar]
- Frederickson, C.J.; Rampy, B.A.; Reamy Rampy, S.; Howell, G.A. Distribution of histochemically reactive zinc in the forebrain of the rat. J. Chem. Neuroanat. 1992, 5, 521–530. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Perez-Clausell, J.; Danscher, G. Zinc-containing 7S-NGF complex. Evidence from zinc histochemistry for localization in salivary secretory granules. J. Histochem. Cytochem. 1987, 35, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Danscher, G. Zinc-containing neurons in hippocampus and related CNS structures. Prog. Brain Res. 1990, 83, 71–84. [Google Scholar] [PubMed]
- Danscher, G.; Stoltenberg, M. Zinc-enriched neurons. J. Neurochem. 2003, 85, 10. [Google Scholar] [CrossRef]
- Ishii, K.; Sato, M.; Akita, M.; Tomita, H. Localization of zinc in the rat submandibular gland and the effect of its deficiency on salivary secretion. Ann. Otol. Rhinol. Laryngol. 1999, 108, 300–308. [Google Scholar] [CrossRef] [PubMed]
- McCormick, N.; Velasquez, V.; Finney, L.; Vogt, S.; Kelleher, S.L. X-ray fluorescence microscopy reveals accumulation and secretion of discrete intracellular zinc pools in the lactating mouse mammary gland. PLoS ONE 2010, 5, e11078. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Zinger, A.; Nevo, A.; Sekler, I.; Hershfinkel, M. Zinc released from injured cells is acting via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J. Biol. Chem. 2010, 285, 26097–26106. [Google Scholar] [CrossRef] [PubMed]
- Hosie, A.M.; Dunne, E.L.; Harvey, R.J.; Smart, T.G. Zinc-mediated inhibition of GABAA receptors: Discrete binding sites underlie subtype specificity. Nat. Neurosci. 2003, 6, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wu, S.M. Modulation of glycine receptors in retinal ganglion cells by zinc. Proc. Natl. Acad. Sci. USA 1999, 96, 3234–3238. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.W.; Jacques, P.; Pierce, K.D.; Schofield, P.R. Zinc potentiation of the glycine receptor chloride channel is mediated by allosteric pathways. J. Neurochem. 1998, 71, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of nmda NR1-NR2A receptors. J. Neurosci. 1997, 17, 5711–5725. [Google Scholar] [PubMed]
- Herin, G.A.; Aizenman, E. Amino terminal domain regulation of NMDA receptor function. Eur. J. Pharmacol. 2004, 500, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Moran, A.; Hershfinkel, M.; Sekler, I. Inhibitory mechanism of store-operated Ca2+ channels by zinc. J. Biol. Chem. 2004, 279, 11106–11111. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S.S.; King, B.F.; Burnstock, G. Modulatory activity of extracellular h+ and Zn2+ on ATP-responses at rP2X1 and rP2X3 receptors. Br. J. Pharmacol. 1999, 128, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castillo, C.; Morales, B.; Huidobro-Toro, J.P. Zinc and copper modulate differentially the P2X4 receptor. J. Neurochem. 2000, 74, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jung, Y.; Kim, D.; Koh, H.; Chung, J. Extracellular zinc activates p70 S6 kinase through the phosphatidylinositol 3-kinase signaling pathway. J. Biol. Chem. 2000, 275, 25979–25984. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Park, K.S.; Kim, J.A.; Choi, K.Y. Differential modulation of zinc-stimulated p21(Cip/WAF1) and cyclin D1 induction by inhibition of PI3 kinase in HT-29 colorectal cancer cells. Exp. Mol. Med. 2002, 34, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Hershfinkel, M.; Moran, A.; Grossman, N.; Sekler, I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc. Natl. Acad. Sci. USA 2001, 98, 11749–11754. [Google Scholar] [CrossRef] [PubMed]
- Sunuwar, L.; Gilad, D.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, in health and disease. Front. Biosci. 2017, 22, 1469–1492. [Google Scholar]
- Hershfinkel, M. The zinc-sensing receptor, ZnR/GPR39: Signaling and significance. In Zinc Signals in Cellular Functions and Disorders; Fukada, T., Kambe, T., Eds.; Springer: Tokyo, Japan, 2014; pp. 11–134. [Google Scholar]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Azriel-Tamir, H.; Sharir, H.; Schwartz, B.; Hershfinkel, M. Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J. Biol. Chem. 2004, 279, 51804–51816. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Hershfinkel, M. The extracellular zinc-sensing receptor mediates intercellular communication by inducing ATP release. Biochem. Biophys. Res. Commun. 2005, 332, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Dubi, N.; Gheber, L.; Fishman, D.; Sekler, I.; Hershfinkel, M. Extracellular zinc and zinc-citrate, acting through a putative zinc-sensing receptor, regulate growth and survival of prostate cancer cells. Carcinogenesis 2008, 29, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Egerod, K.L.; Jin, C.; Petersen, P.S.; Ostergaard, M.V.; Hald, J.; Sprinkel, A.M.; Storling, J.; Mandrup-Poulsen, T.; Holst, J.J.; et al. G protein-coupled receptor 39 deficiency is associated with pancreatic islet dysfunction. Endocrinology 2009, 150, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Schmidt, F.; Guterman-Ram, G.; Khayyeri, H.; Jähn, K.; Hiram-Bab, S.; Orenbuch, A.; Katchkovsky, S.; Aflalo, A.; Isaksson, H.; et al. Perturbed bone composition and integrity with disorganized osteoblast function in zinc receptor/GPR39 deficient mice. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chorin, E.; Vinograd, O.; Fleidervish, I.; Gilad, D.; Herrmann, S.; Sekler, I.; Aizenman, E.; Hershfinkel, M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 2011, 31, 12916–12926. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rosello, T.; Anderson, C.T.; Schopfer, F.J.; Zhao, Y.; Gilad, D.; Salvatore, S.R.; Freeman, B.A.; Hershfinkel, M.; Aizenman, E.; Tzounopoulos, T. Synaptic Zn2+ inhibits neurotransmitter release by promoting endocannabinoid synthesis. J. Neurosci. 2013, 33, 9259–9272. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Hildebrand, M.S.; Mullen, S.A.; Hildebrand, J.M.; Berkovic, S.F.; Petrou, S. Synaptic Zn2+ and febrile seizure susceptibility. Br. J. Pharmacol. 2017, 174, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Iida, M.; Ando, M.; Nakamura, M.; Tamano, H.; Oku, N. Enhanced susceptibility to spontaneous seizures of noda epileptic rats by loss of synaptic Zn2+. PLoS ONE 2013, 8, e71372. [Google Scholar] [CrossRef] [PubMed]
- Nasehi, M.M.; Sakhaei, R.; Moosazadeh, M.; Aliramzany, M. Comparison of serum zinc levels among children with simple febrile seizure and control group: A systematic review. Iran. J. Child. Neurol. 2015, 9, 17–24. [Google Scholar] [PubMed]
- Ganesh, R.; Janakiraman, L. Serum zinc levels in children with simple febrile seizure. Clin. Pediatr. (Phila) 2008, 47, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Elsas, S.M.; Hazany, S.; Gregory, W.L.; Mody, I. Hippocampal zinc infusion delays the development of afterdischarges and seizures in a kindling model of epilepsy. Epilepsia 2009, 50, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Azriel-Tamir, H.; Arotsker, N.; Sekler, I.; Hershfinkel, M. Zinc sensing receptor signaling, mediated by GPR39, reduces butyrate-induced cell death in HT29 colonocytes via upregulation of clusterin. PLoS ONE 2012, 7, e35482. [Google Scholar]
- Moechars, D.; Depoortere, I.; Moreaux, B.; de Smet, B.; Goris, I.; Hoskens, L.; Daneels, G.; Kass, S.; Ver Donck, L.; Peeters, T.; et al. Altered gastrointestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology 2006, 131, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Depoortere, I. Gi functions of GPR39: Novel biology. Curr. Opin. Pharmacol. 2012, 12, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Zhao, H.; Zhang, Y.; Peng, H.; Chen, Q.; Zhang, H.; Zheng, X.; Jin, Y.; Ni, H.; Duan, E.; et al. GPR39 is region-specifically expressed in mouse oviduct correlating with the Zn2+ distribution. Theriogenology 2017, 88, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tang, S.; Zhang, W.; Gao, W.; Chen, Y. GPR39 activates proliferation and differentiation of porcine intramuscular preadipocytes through targeting the PI3K/AKT cell signaling pathway. J. Recept. Signal Transduct. Res. 2016, 36, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, Y.; Zhang, X.; Chen, D. Zac1/GPR39 phosphorylating CaMK-II contributes to the distinct roles of Pax3 and Pax7 in myogenic progression. Biochim. Biophys. Acta 2018, 1864, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, G.L.; Alpers, D.H.; Binder, H.J.; Tran, C.D.; Ramakrishna, B.S.; Brown, I.; Manary, M.; Mortimer, E.; Young, G.P. The relevance of the colon to zinc nutrition. Nutrients 2015, 7, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Kirschke, C.P.; Huang, L. Immunohistochemical analysis of ZnT1, 4, 5, 6, and 7 in the mouse gastrointestinal tract. J. Histochem. Cytochem. 2007, 55, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Hershfinkel, M.; Silverman, W.F.; Sekler, I. The zinc sensing receptor, a link between zinc and cell signaling. Mol. Med. 2007, 13, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, J.; Saito, T.; Taniguchi, M.; Kawasaki, T.; Moritani, Y.; Hayashi, K.; Kobori, M. A novel Gαq/11-selective inhibitor. J. Biol. Chem. 2004, 279, 47438–47445. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Suzumura, K.; Nagai, K.; Kawasaki, T.; Takasaki, J.; Sekiguchi, M.; Moritani, Y.; Saito, T.; Hayashi, K.; Fujita, S.; et al. YM-254890 analogues, novel cyclic depsipeptides with Gαq/11 inhibitory activity from Chromobacterium sp. QS3666. Bioorg. Med. Chem. 2004, 12, 3125–3133. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.H.; Brown, E.M. Disorders of calcium ion sensing. J. Clin. Endocrinol. Metab. 1996, 81, 2030–2035. [Google Scholar] [PubMed]
- Yasuda, S.; Miyazaki, T.; Munechika, K.; Yamashita, M.; Ikeda, Y.; Kamizono, A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J. Recept. Signal Transduct. Res. 2007, 27, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Popovics, P.; Stewart, A.J. GPR39: A Zn2+-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell. Mol. Life Sci. 2011, 68, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Asraf, H.; Salomon, S.; Nevo, A.; Sekler, I.; Mayer, D.; Hershfinkel, M. The ZnR/GPR39 interacts with the CaSR to enhance signaling in prostate and salivary epithelia. J. Cell. Physiol. 2013, 229, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.V.; Ren, P.G.; Avsian-Kretchmer, O.; Luo, C.W.; Rauch, R.; Klein, C.; Hsueh, A.J. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 2005, 310, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, B. Consideration in estimates of requirements and critical intake of zinc. Adaption, availability and interactions. Analyst 1995, 120, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.; Jensen, M.; Solgaard, P.; Sorensen, S.S.; Sandstrom, B. Zinc absorption estimated by fecal monitoring of zinc stable isotopes validated by comparison with whole-body retention of zinc radioisotopes in humans. J. Nutr. 1995, 125, 1274–1282. [Google Scholar] [PubMed]
- Sandstrom, B.; Cederblad, A.; Kivisto, B.; Stenquist, B.; Andersson, H. Retention of zinc and calcium from the human colon. Am. J. Clin. Nutr. 1986, 44, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Sekler, I.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, controls proliferation and differentiation of colonocytes and thereby tight junction formation in the colon. Cell Death Dis. 2014, 5, e1307. [Google Scholar] [CrossRef] [PubMed]
- Sunuwar, L.; Medini, M.; Cohen, L.; Sekler, I.; Hershfinkel, M. The zinc sensing receptor, ZnR/GPR39, triggers metabotropic calcium signalling in colonocytes and regulates occludin recovery in experimental colitis. Philos. Trans. R Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef]
- Sunuwar, L.; Asraf, H.; Donowitz, M.; Sekler, I.; Hershfinkel, M. The Zn2+-sensing receptor, ZnR/GPR39, upregulates colonocytic Cl− absorption, via basolateral KCC1, and reduces fluid loss. Biochim. Biophys. Acta 2017, 1863, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.; Chorin, E.; Sekler, I.; Silverman, W.F.; Atkin, S.; Russell, J.T.; Hershfinkel, M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 2009, 29, 2890–2901. [Google Scholar] [CrossRef] [PubMed]
- Albizu, L.; Balestre, M.N.; Breton, C.; Pin, J.P.; Manning, M.; Mouillac, B.; Barberis, C.; Durroux, T. Probing the existence of g protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol. Pharmacol. 2006, 70, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Frimurer, T.M.; Mende, F.; Graae, A.S.; Engelstoft, M.S.; Egerod, K.L.; Nygaard, R.; Gerlach, L.O.; Hansen, J.B.; Schwartz, T.W.; Holst, B. Model-based discovery of synthetic agonists for the Zn2+-sensing G-protein-coupled receptor 39 (GPR39) reveals novel biological functions. J. Med. Chem. 2017, 60, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/raf/MEK/ERK and PI3K/PTEN/AKT/MTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.P.; Rubinek, T.; Ryvo, L.; Wolf, I. Endocrine resistance in breast cancer: Focus on the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling pathway. Breast Care 2013, 8, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Fassnacht, M.; Weismann, D.; Ebert, S.; Adam, P.; Zink, M.; Beuschlein, F.; Hahner, S.; Allolio, B. AKT is highly phosphorylated in pheochromocytomas but not in benign adrenocortical tumors. J. Clin. Endocrinol. Metab. 2005, 90, 4366–4370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Huang, Y.X.; Sun, Y.; Zheng, L.H.; Li, Y.X. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem. Biol. Interact. 2010, 185, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.C.; Waby, J.S.; Chirakkal, H.; Staton, C.A.; Corfe, B.M. Butyrate suppresses expression of neuropilin I in colorectal cell lines through inhibition of Sp1 transactivation. Mol. Cancer 2010, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Scharlau, D.; Borowicki, A.; Habermann, N.; Hofmann, T.; Klenow, S.; Miene, C.; Munjal, U.; Stein, K.; Glei, M. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 2009, 682, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. Butyrate and wnt signaling: A possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle 2008, 7, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Opoka, W.; Adamek, D.; Plonka, M.; Reczynski, W.; Bas, B.; Drozdowicz, D.; Jagielski, P.; Sliwowski, Z.; Adamski, P.; Brzozowski, T. Importance of luminal and mucosal zinc in the mechanism of experimental gastric ulcer healing. J. Physiol. Pharmacol. 2010, 61, 581–591. [Google Scholar] [PubMed]
- Barceloux, D.G. Zinc. J. Toxicol. Clin. Toxicol. 1999, 37, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Lansdown, A.B.; Mirastschijski, U.; Stubbs, N.; Scanlon, E.; Agren, M.S. Zinc in wound healing: Theoretical, experimental, and clinical aspects. Wound Repair Regen. 2007, 15, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Lansdown, A.B. Zinc in the healing wound. Lancet 1996, 347, 706–707. [Google Scholar] [CrossRef]
- Schwartz, J.R.; Marsh, R.G.; Draelos, Z.D. Zinc and skin health: Overview of physiology and pharmacology. Dermatol. Surg. 2005, 31, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Dong, X.; Zhang, W. Obestatin changes proliferation, differentiation and apoptosis of porcine preadipocytes. Ann. Endocrinol. 2014, 75, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ganay, T.; Asraf, H.; Aizenman, E.; Bogdanovic, M.; Sekler, I.; Hershfinkel, M. Regulation of neuronal pH by the metabotropic zinc receptor mZnR/GPR39. J. Neurochem. 2015, 135, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Volk, L.J.; Daly, C.A.; Huber, K.M. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J. Neurophysiol. 2006, 95, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Fibuch, E.E.; Mao, L. Regulation of mitogen-activated protein kinases by glutamate receptors. J. Neurochem. 2007, 100, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.L.; Vasudevan, N.T.; Gupta, M.K.; Martelli, E.E.; Naga Prasad, S.V. G-protein coupled receptor resensitization-appreciating the balancing act of receptor function. Curr. Mol. Pharmacol. 2012, 5, 350–361. [Google Scholar] [CrossRef]
- Shimizu, Y.; Koyama, R.; Kawamoto, T. Rho kinase-dependent desensitization of GPR39; a unique mechanism of gpcr downregulation. Biochem. Pharmacol. 2017, 140, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Storjohann, L.; Holst, B.; Schwartz, T.W. Molecular mechanism of Zn2+ agonism in the extracellular domain of GPR39. FEBS Lett. 2008, 582, 2583–2588. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Asraf, H.; Sekler, I.; Hershfinkel, M. Extracellular ph regulates zinc signaling via an ASP residue of the zinc-sensing receptor (ZnR/GPR39). J. Biol. Chem. 2012, 287, 33339–33350. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, R.S. The role of zinc in growth and cell proliferation. J. Nutr. 2000, 130, 1500S–1508S. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.G.; Kumar, D.; Rampton, D.S.; Evans, D.F. Intestinal luminal ph in inflammatory bowel disease: Possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 2001, 48, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, J.; Grinstein, S. Na+/H+ exchangers. Compr. Physiol. 2011, 1, 2083–2100. [Google Scholar] [PubMed]
- Vaneckova, I.; Vylitova-Pletichova, M.; Beskid, S.; Zicha, J.; Pacha, J. Intracellular pH regulation in colonocytes of rat proximal colon. Biochim. Biophys. Acta 2001, 1536, 103–115. [Google Scholar] [CrossRef]
- Hachem, J.P.; Behne, M.; Aronchik, I.; Demerjian, M.; Feingold, K.R.; Elias, P.M.; Mauro, T.M. Extracellular ph controls NHE1 expression in epidermis and keratinocytes: Implications for barrier repair. J. Investig. Dermatol. 2005, 125, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Cardone, R.A.; Busco, G.; Krahling, H.; Schwab, A.; Reshkin, S.J. Protons extruded by NHE1: Digestive or glue? Eur. J. Cell. Biol. 2008, 87, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Panula, P.; Karhunen, T.; Heinonen, E. Fall in intracellular pH mediated by gabaa receptors in cultured rat astrocytes. Neurosci. Lett. 1991, 126, 9–12. [Google Scholar] [CrossRef]
- Manhas, N.; Shi, Y.; Taunton, J.; Sun, D. P90RSK activation contributes to cerebral ischemic damage via phosphorylation of Na+/H+ exchanger isoform 1. J. Neurochem. 2010, 114, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Aspects Med. 2013, 34, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Jurd, R.; Moss, S.J. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 2010, 45, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Watanabe, M.; Moorhouse, A.J.; Kanematsu, T.; Horibe, S.; Matsukawa, N.; Asai, K.; Ojika, K.; Hirata, M.; Nabekura, J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 2007, 27, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Kaila, K. The cellular, molecular and ionic basis of GABAA receptor signalling. Prog. Brain Res. 2007, 160, 59–87. [Google Scholar] [PubMed]
- Viitanen, T.; Ruusuvuori, E.; Kaila, K.; Voipio, J. The K+-Cl− cotransporter KCC2 promotes gabaergic excitation in the mature rat hippocampus. J. Physiol. 2010, 588, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chen, C.X.; Liu, Y.J.; Aizenman, E.; Kandler, K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of gaba responses. Eur J. Neurosci. 2005, 21, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Karadsheh, M.; Delpire, E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J. Neurobiol. 1999, 39, 558–568. [Google Scholar] [CrossRef]
- Saadi, R.A.; He, K.; Hartnett, K.A.; Kandler, K.; Hershfinkel, M.; Aizenman, E. Snare-dependent upregulation of potassium chloride co-transporter 2 activity after metabotropic zinc receptor activation in rat cortical neurons in vitro. Neuroscience 2012, 210, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Donowitz, M.; Verkman, A.S. Secretory diarrhoea: Mechanisms and emerging therapies. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Valenzano, M.C.; Mercado, J.M.; Zurbach, E.P.; Mullin, J.M. Zinc supplementation modifies tight junctions and alters barrier function of CACO-2 human intestinal epithelial layers. Dig. Dis. Sci. 2013, 58, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Finamore, A.; Massimi, M.; Conti Devirgiliis, L.; Mengheri, E. Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J. Nutr. 2008, 138, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.X.; Lei, Z.; Wolf, P.G.; Gao, Y.; Guo, Y.M.; Zhang, B.K. Zinc supplementation, via GPR39, upregulates PKCζ to protect intestinal barrier integrity in Caco-2 cells challenged by salmonella enterica serovar typhimurium. J. Nutr. 2017, 147, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, Y.B.; Sekler, I.; Silverman, W.F. Histochemical and histofluorescence tracing of chelatable zinc in the developing mouse. J. Histochem. Cytochem. 2004, 52, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Takagishi, T.; Hara, T.; Fukada, T. Recent advances in the role of SLC39A/ZIP zinc transporters in vivo. Int. J. Mol. Sci. 2017, 18, 2708. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Lin, P.; Zhu, H.; Ko, J.K.; Hwang, M.; Tan, T.; Pan, Z.; Korichneva, I.; Ma, J. Zinc binding to MG53 protein facilitates repair of injury to cell membranes. J. Biol. Chem. 2015, 290, 13830–13839. [Google Scholar] [CrossRef] [PubMed]
- Krasovec, M.; Frenk, E. Acrodermatitis enteropathica secondary to crohn’s disease. Dermatology 1996, 193, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Sturniolo, G.C.; Fries, W.; Mazzon, E.; Di Leo, V.; Barollo, M.; D’Inca, R. Effect of zinc supplementation on intestinal permeability in experimental colitis. J. Lab. Clin. Med. 2002, 139, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Luk, H.H.; Ko, J.K.; Fung, H.S.; Cho, C.H. Delineation of the protective action of zinc sulfate on ulcerative colitis in rats. Eur. J. Pharmacol. 2002, 443, 197–204. [Google Scholar] [CrossRef]
- Walker, C.L.; Black, R.E. Zinc for the treatment of diarrhoea: Effect on diarrhoea morbidity, mortality and incidence of future episodes. Int. J. Epidemiol. 2010, 39, i63–i69. [Google Scholar] [CrossRef] [PubMed]
- Alam, D.S.; Yunus, M.; El Arifeen, S.; Chowdury, H.R.; Larson, C.P.; Sack, D.A.; Baqui, A.H.; Black, R.E. Zinc treatment for 5 or 10 days is equally efficacious in preventing diarrhea in the subsequent 3 months among bangladeshi children. J. Nutr. 2011, 141, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Yang, J.; Chen, T.E.; Zachos, N.C.; Kovbasnjuk, O.; Verkman, A.S.; Donowitz, M. Translating molecular physiology of intestinal transport into pharmacologic treatment of diarrhea: Stimulation of Na+ absorption. Clin. Gastroenterol. Hepatol. 2014, 12, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Di Sole, F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am. J. Physiol. Cell Physiol. 2012, 302, C1569–C1587. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Ko, E.A.; Tradtrantip, L.; Donowitz, M.; Verkman, A.S. Discovery and development of antisecretory drugs for treating diarrheal diseases. Clin. Gastroenterol. Hepatol. 2014, 12, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Passariello, A.; Terrin, G.; De Marco, G.; Cecere, G.; Ruotolo, S.; Marino, A.; Cosenza, L.; Tardi, M.; Nocerino, R.; Berni Canani, R. Efficacy of a new hypotonic oral rehydration solution containing zinc and prebiotics in the treatment of childhood acute diarrhea: A randomized controlled trial. J. Pediatr. 2011, 158, 288–292.e281. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.S.; Phillips, A.M.; Mullen, S.A.; Adlard, P.A.; Hardies, K.; Damiano, J.A.; Wimmer, V.; Bellows, S.T.; McMahon, J.M.; Burgess, R.; et al. Loss of synaptic Zn2+ transporter function increases risk of febrile seizures. Sci. Rep. 2015, 5, 17816. [Google Scholar] [CrossRef] [PubMed]
- Farahani, H.N.; Ashthiani, A.R.; Masihi, M.S. Study on serum zinc and selenium levels in epileptic patients. Neurosciences 2013, 18, 138–142. [Google Scholar] [PubMed]
- Mitsuya, K.; Nitta, N.; Suzuki, F. Persistent zinc depletion in the mossy fiber terminals in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. Epilepsia 2009, 50, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Xu, K.; Yoo, J.; Chen, T.T.; Andrews, G.; Noebels, J.L. Knockout of Zn transporters ZIP-1 and ZIP-3 attenuates seizure-induced ca1 neurodegeneration. J. Neurosci. 2011, 31, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Robbins, C.A.; Wenzel, H.J.; Schwartzkroin, P.A.; Palmiter, R.D. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000, 39, 153–169. [Google Scholar] [CrossRef]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lovinger, D.; Delpire, E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J. Neurophysiol. 2005, 93, 1557–1568. [Google Scholar] [CrossRef] [PubMed]
- Woo, N.S.; Lu, J.; England, R.; McClellan, R.; Dufour, S.; Mount, D.B.; Deutch, A.Y.; Lovinger, D.M.; Delpire, E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 2002, 12, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Gilad, D.; Shorer, S.; Ketzef, M.; Friedman, A.; Sekler, I.; Aizenman, E.; Hershfinkel, M. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol. Dis. 2015, 81, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Q.; Yao, H.; Haddad, G.G. Increased neuronal excitability and seizures in the Na+/H+ exchanger null mutant mouse. Am. J. Physiol. Cell Physiol. 2001, 281, C496–C503. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhao, P.; Xue, J.; Gu, X.Q.; Sun, X.; Yao, H.; Haddad, G.G. Na+ channel expression and neuronal function in the Na+/H+ exchanger 1 null mutant mouse. J. Neurophysiol. 2003, 89, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.P.; Tanzi, R.E.; Bush, A.I. Zinc-induced alzheimer’s Aβ1-40 aggregation is mediated by conformational factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Tempaku, M.; Sasaki, M.; Uematsu, C.; Sato, S.; Kanazawa, H.; Datki, Z.L.; Adlard, P.A.; Bush, A.I. Extracellular Zn2+ is essential for amyloid β1-42-induced cognitive decline in the normal brain and its rescue. J. Neurosci. 2017, 37, 7253–7262. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch-Dahan, C.; Asraf, H.; Bogdanovic, M.; Sekler, I.; Bush, A.I.; Hershfinkel, M. Amyloid β attenuates metabotropic zinc sensing receptor, mZnR/GPR39, dependent Ca2+, ERK1/2 and clusterin signaling in neurons. J. Neurochem. 2016, 139, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, M.A.; Kranz, T.M.; Kleinhaus, K.; Joe, P.; Getz, M.; Johnson, P.; Chao, M.V.; Malaspina, D. The emerging role for zinc in depression and psychosis. Front. Pharmacol. 2017, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K.; Doboszewska, U.; Szewczyk, B.; Sowa-Kucma, M.; Misztak, P.; Piekoszewski, W.; Trela, F.; Ostachowicz, B.; Nowak, G. The involvement of the GPR39-Zn2+-sensing receptor in the pathophysiology of depression. Studies in rodent models and suicide victims. Neuropharmacology 2014, 79, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K.; Nowak, G. GPR39 up-regulation after selective antidepressants. Neurochem. Int. 2013, 62, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Mlyniec, K.; Gawel, M.; Librowski, T.; Reczynski, W.; Bystrowska, B.; Holst, B. Investigation of the GPR39 zinc receptor following inhibition of monoaminergic neurotransmission and potentialization of glutamatergic neurotransmission. Brain Res. Bull. 2015, 115, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Cichy, A.; Sowa-Kucma, M.; Legutko, B.; Pomierny-Chamiolo, L.; Siwek, A.; Piotrowska, A.; Szewczyk, B.; Poleszak, E.; Pilc, A.; Nowak, G. Zinc-induced adaptive changes in NMDA/glutamatergic and serotonergic receptors. Pharmacol. Rep. 2009, 61, 1184–1191. [Google Scholar] [CrossRef]
- Mlyniec, K.; Nowak, G. Up-regulation of the GPR39 Zn2+-sensing receptor and CREB/BDNF/TrkB pathway after chronic but not acute antidepressant treatment in the frontal cortex of zinc-deficient mice. Pharmacol. Rep. 2015, 67, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.J.; Gee, K.R.; Kennedy, R.T. Imaging of Zn2+ release from pancreatic β-cells at the level of single exocytotic events. Anal. Chem. 2003, 75, 3468–3475. [Google Scholar] [CrossRef] [PubMed]
- Bloc, A.; Cens, T.; Cruz, H.; Dunant, Y. Zinc-induced changes in ionic currents of clonal rat pancreatic β-cells: Activation of ATP-sensitive K+ channels. J. Physiol. (Lond.) 2000, 529, 723–734. [Google Scholar] [CrossRef]
- Ferrer, R.; Soria, B.; Dawson, C.M.; Atwater, I.; Rojas, E. Effects of Zn2+ on glucose-induced electrical activity and insulin release from mouse pancreatic islets. Am. J. Physiol. 1984, 246, C520–C527. [Google Scholar] [CrossRef] [PubMed]
- Franklin, I.; Gromada, J.; Gjinovci, A.; Theander, S.; Wollheim, C.B. Beta-cell secretory products activate α-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005, 54, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in pancreatic islet biology, insulin sensitivity, and diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Richard, A.M.; Will, S.; Syed, J.; Stedman, N.; Perreault, M.; Gimeno, R.E. Disruption of G protein-coupled receptor 39 impairs insulin secretion in vivo. Endocrinology 2009, 150, 2586–2595. [Google Scholar] [CrossRef] [PubMed]
- Verhulst, P.J.; Lintermans, A.; Janssen, S.; Loeckx, D.; Himmelreich, U.; Buyse, J.; Tack, J.; Depoortere, I. GPR39, a receptor of the ghrelin receptor family, plays a role in the regulation of glucose homeostasis in a mouse model of early onset diet-induced obesity. J. Neuroendocrinol. 2011, 23, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Egerod, K.L.; Jin, C.; Petersen, P.S.; Wierup, N.; Sundler, F.; Holst, B.; Schwartz, T.W. Beta-cell specific overexpression of GPR39 protects against streptozotocin-induced hyperglycemia. Int. J. Endocrinol. 2011, 2011, 401258. [Google Scholar] [CrossRef] [PubMed]
- Moran, B.M.; Abdel-Wahab, Y.H.; Vasu, S.; Flatt, P.R.; McKillop, A.M. GPR39 receptors and actions of trace metals on pancreatic β cell function and glucose homoeostasis. Acta Diabetol. 2016, 53, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: A small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, H.; Zhu, Y.H.; Qin, Y.R.; Dai, Y.; Zeng, T.; Chen, L.; Nie, C.; Tang, H.; Li, Y.; et al. Overexpression of GPR39 contributes to malignant development of human esophageal squamous cell carcinoma. BMC Cancer 2011, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Alen, B.O.; Leal-Lopez, S.; Alen, M.O.; Viano, P.; Garcia-Castro, V.; Mosteiro, C.S.; Beiras, A.; Casanueva, F.F.; Gallego, R.; Garcia-Caballero, T.; et al. The role of the obestatin/GPR39 system in human gastric adenocarcinomas. Oncotarget 2016, 7, 5957–5971. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Egerod, K.L.; Schild, E.; Vickers, S.P.; Cheetham, S.; Gerlach, L.O.; Storjohann, L.; Stidsen, C.E.; Jones, R.; Beck-Sickinger, A.G.; et al. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 2007, 148, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M. Liv-1 breast cancer protein belongs to new family of histidine-rich membrane proteins with potential to control intracellular Zn2+ homeostasis. IUBMB Life 2000, 49, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M. A distinct role in breast cancer for two liv-1 family zinc transporters. Biochem. Soc. Trans. 2008, 36, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Li, P.; Wang, W.; Wang, J.; Gerry, E.; Wang, T.L.; Shih, I.M.; Nephew, K.P.; Xu, Y. The novel ZIP4 regulation and its role in ovarian cancer. Oncotarget 2017, 8, 90090–90107. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Custodi, C.; Nuti, R.; Oprea, T.I.; Macchiarulo, A. Fitting the complexity of GPCRs modulation into simple hypotheses of ligand design. J. Mol. Graph. Model. 2012, 38, 70–81. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hershfinkel, M. The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease. Int. J. Mol. Sci. 2018, 19, 439. https://doi.org/10.3390/ijms19020439
Hershfinkel M. The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease. International Journal of Molecular Sciences. 2018; 19(2):439. https://doi.org/10.3390/ijms19020439
Chicago/Turabian StyleHershfinkel, Michal. 2018. "The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease" International Journal of Molecular Sciences 19, no. 2: 439. https://doi.org/10.3390/ijms19020439
APA StyleHershfinkel, M. (2018). The Zinc Sensing Receptor, ZnR/GPR39, in Health and Disease. International Journal of Molecular Sciences, 19(2), 439. https://doi.org/10.3390/ijms19020439