Abstract
Zinc deficiency is a risk factor for obesity and diabetes. However, until recently, the underlying molecular mechanisms remained unclear. The breakthrough discovery that the common polymorphism in zinc transporter SLC30A8/ZnT8 may increase susceptibility to type 2 diabetes provided novel insights into the role of zinc in diabetes. Our group and others showed that altered ZnT8 function may be involved in the pathogenesis of type 2 diabetes, indicating that the precise control of zinc homeostasis is crucial for maintaining health and preventing various diseases, including lifestyle-associated diseases. Recently, the role of the zinc transporter ZIP13 in the regulation of beige adipocyte biogenesis was clarified, which indicated zinc homeostasis regulation as a possible therapeutic target for obesity and metabolic syndrome. Here we review advances in the role of zinc homeostasis in the pathophysiology of diabetes, and propose that inadequate zinc distribution may affect the onset of diabetes and metabolic diseases by regulating various critical biological events.
Keywords:
zinc; zinc transporters; diabetes; obesity; ZnT8; ZIP13; pancreatic β cell; beige adipocyte; therapeutic target 1. Introduction
Type 2 diabetes is now a crucial health problem in many parts of the world. Type 2 diabetes mellitus (T2DM) is characterized by peripheral insulin resistance and pancreatic beta (β) cell dysfunction. The disease is thought to be caused by defects in insulin signaling or secretion, the activation of various stress pathways, and dysregulation of the central nervous system (CNS). It is well accepted that the most accurate predictor for developing T2DM is obesity. Therefore, much attention has also been paid to the contribution of nutrients and nutrient-sensing pathways in situations of chronic caloric excess. Most of the interest in the role of nutrients in diabetes is centered on macronutrients, such as carbohydrate and fat, but micronutrients, such as iron and zinc, are also closely associated with diabetes [1,2]. Whole-body level dysregulation of zinc is known to occur in both type 1 and type 2 diabetes. However, it remains unclear as to whether zinc deficiency causes the disease or is merely a consequence of the disease. A possible causal link between changes in zinc homeostasis and pancreatic β cell function was suggested in 2007 with the identification of an association between the risk of T2DM and polymorphisms in the SLC30A8 gene, which encodes zinc transporter ZnT8 [3].
Several groups have been analyzing the roles of zinc homeostasis in the health and disease of endocrine organs, with particular focus on zinc transporter function. In this review, we will discuss the roles of zinc homeostasis in glucose metabolism, particularly in association with ZnT8. Furthermore, we will discuss the role of ZIP13 in beige adipocyte biogenesis and energy expenditure, which we have recently elucidated.
2. Zinc Homeostasis and Pancreatic β Cells
2.1. Insulin Biosynthesis in Pancreatic β Cells
Pancreatic β cells are known to contain very high concentrations of zinc compared with various other cells. In particular, insulin secretory granules have been shown to have the highest zinc content within β cells [4,5]. In vertebrates, three protein families have been shown to regulate cellular zinc homeostasis, namely, metallothioneins (MTs), zinc importers (ZIP, SLC39A), and zinc exporters (ZnT, SLC30A). MTs have been shown to bind zinc with low affinity, whereas zinc transporters mediate the compartmentalization of zinc into various organelles and vesicles for their storage, and to supply zinc to various proteins that require zinc for their function [6]. Nine ZnTs and 14 ZIP transporters have been identified to play important roles in whole body maintenance, as well as in zinc homeostasis at the cellular and subcellular levels. These transporters act either independently or coordinately, and in a cell-specific or tissue-specific manner [7,8]. ZnT8 plays a key role in the accumulation of zinc within insulin secretory granules [9]. Furthermore, zinc is essential for the appropriate synthesis of insulin, as well as its storage and structural stability [10].
Insulin comprises a hexamer of six insulin and two zinc molecules [11,12]. The mature insulin molecule comprises two polypeptide chains, namely, chains A and B. Initially, insulin mRNA is translated into an inactive preproinsulin molecule, which comprises two chains that are connected by a c-peptide, with the signal peptide at the N-terminus. Proinsulin is formed from preproinsulin by signal peptide cleavage in the endoplasmic reticulum (ER). Proinsulin then folds into the final three-dimensional structure, upon formation of the correct disulfide bonds. Subsequently, the proinsulin protein forms dimers via electrostatic interactions. Proinsulin hexamers are then formed by electrostatically-coupled proinsulin dimers and zinc binding to histidine residue 10 of the B chain (His B10) [13]. After entering the Golgi apparatus, insulin hexamer formation is completed upon the dissociation of c-peptide, mediated by prohormone convertase (PC) dissociation [14]. Insulin crystallization occurs under specific conditions in insulin secretory granules, in which both insulin and zinc exist in high concentrations and acidic pH is maintained [15,16]. This crystallized insulin can be observed as “dense core granules” by electron microscopy [17,18], and insulin crystals that are secreted from pancreatic β cells are believed to dissociate rapidly into monomers as they enter the bloodstream.
2.2. Zinc Supplementation in Diabetic Animals and Patients
Mice with zinc deficiency were found to have a decreased number of insulin granules in their pancreatic β cells [19], as well as impaired glucose-stimulated insulin secretion (GSIS) [20]. Since pancreatic β cells synthesize a large amount of ATP, this makes them prone to oxidative stress exposure, which can subsequently cause cellular damage [21]. As zinc is required for the actions of many antioxidative enzymes, including Cu-Zn-SOD (superoxide dismutase) [22] and catalase [23], a lack of zinc will lead to further damage of pancreatic β cells under oxidative stress, such as in T2DM.
Regarding humans, a prospective cohort study in the United States analyzed 82,000 women and demonstrated that low zinc intake results in a 17% increased risk of developing diabetes compared with those women taking sufficient amounts of zinc [24]. Recently, a study from China has reported a negative correlation between concentrations of plasma zinc and the onset of diabetes [25]. Interestingly, this report suggested that an interaction between SLC30A8 (ZnT8) dysfunction and decreased plasma zinc concentrations regulates glucose tolerance and diabetes. Furthermore, the authors suggested that a decrease in plasma zinc concentrations as well as ZnT8 function may coordinately increase the risk of diabetes. Although these data suggest that zinc supplementation prevents disruption of glucose homeostasis, particularly in people with zinc deficiency, prospective intervention studies should be performed to clarify the efficacy of zinc supplementation in preventing the onset of diabetes. On the other hand, excessive supplementation with zinc may have deleterious effects, as excessive zinc intake may cause an undesirable increase in HbA1c levels and high blood pressure [26].
A genome-wide association study (GWAS) demonstrated that a nonsynonymous single-nucleotide polymorphism, namely, rs13266634 in the SLC30A8 gene, results in the replacement of tryptophan-325 to arginine, which modestly increases the risk of T2DM [3]. Furthermore, recent studies on human SNPs demonstrated the association of 12 rare loss-of-function ZnT8 mutants with a 65% decreased risk of T2DM [27]. Taken together, the data indicate that polymorphisms in the SLC30A8 gene are associated with altered risk of T2DM.
3. ZnT8 Plays a Crucial Role in Glucose Homeostasis
3.1. Insulin Secretory Granule of ZnT8-KO Mice
ZnT8 is found in the plasma membrane of insulin secretory granules of pancreatic β cells, and is implicated in zinc transport into insulin secretory granules [9]. Several groups, including our own have aimed to clarify the role of ZnT8 in glucose homeostasis by establishing SLC30A8-deficient (ZnT8-KO) mice [18,28,29] (Supplemental Table S1). Each mouse model shows variation in certain phenotype traits, which are attributed to differences in deletion strategy, genetic background, and housing condition. Most of the ZnT8-KO mice have been reported to have mildly impaired glucose tolerance and there have been no reports of the improvement of glucose tolerance in the ZnT8-KO mice model [18,30,31,32,33,34,35] (Supplemental Table S1). Furthermore, hZnT8 transgenic mice showed mildly improved glucose tolerance [31], suggesting that the expression levels of ZnT8 determines the risk of T2DM in these mouse models. Electron microscopy analysis revealed that dense-core granules, which are a hallmark of crystallized insulin usually seen in normal β cells, were absent in the ZnT8-KO mouse β cells. In most of the ZnT8-KO mice, some of the granules appeared atypical granules possessing abnormal “rod-like” or empty cores, while some ZnT8-KO mice revealed the nearly complete loss of crystal containing granules [18,30].
3.2. Phenotypes of ZnT8-KO Mice Regarding Glucose Metabolism
As described above, glucose tolerance was mildly impaired in ZnT8-KO mice. This demonstrates that peripheral insulin levels in ZnT8-KO mice were reduced compared with control mice. Indeed, we and others have observed decreased insulin levels in ZnT8-KO mice, despite GSIS levels being unchanged or slightly increased in ZnT8-KO mouse islets [18,34] (Supplemental Table S1). To understand this discrepancy, we performed a pancreas perfusion experiment, and found that insulin secretion was still enhanced upon pancreas infusion in ZnT8-KO mice, further supporting that insulin secretion is increased in ZnT8-KO mice. However, pancreas-liver dual perfusion analysis demonstrated that in these mice, a large proportion of the secreted insulin is actually degraded during its passage through the liver, suggesting that ZnT8 regulates hepatic insulin clearance [18]. A set of in vivo and in vitro experiments demonstrated that ZnT8-mediated zinc inhibits hepatic insulin uptake by counteracting clathrin-mediated endocytosis of the insulin receptor. These results suggested that ZnT8 plays an important role in determining the amount of insulin that is delivered to the liver and other peripheral organs, and thus optimizes the effect of insulin on whole body glucose metabolism [18].
For the assessment of in vivo insulin clearance, the c-peptide/insulin ratio may be useful [36,37]. Consistently, although insulin secretion was increased in ZnT8-KO mice, peripheral insulin levels were lower and c-peptide/insulin ratios were increased [18]. Studies of c-peptide/insulin ratios and rates of insulin clearance in humans with the rs13266634 polymorphism also demonstrated results consistent with this idea [18]. Furthermore, the Eugene study showed that when human homozygous carriers of the SLC30A8 risk allele are subjected to the intravenous glucose tolerance test, they demonstrate low peripheral insulin levels in the early phases [38]. These results indicate that SLC30A8/ZnT8 regulates hepatic insulin clearance, and importantly, the same mechanism appears to be conserved in humans (Figure 1) [18].
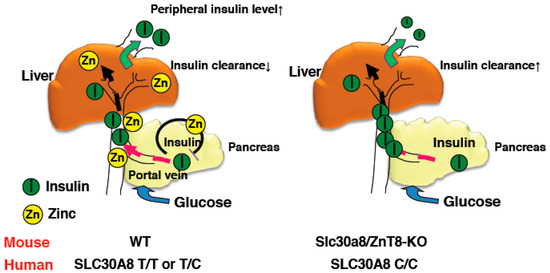
Figure 1.
Schematic representation of insulin clearance in WT and ZnT8-KO mice. Zinc co-secreted with insulin suppresses insulin secretion from pancreatic β cells and inhibits hepatic insulin clearance in WT mice (left). In contrast, reduced zinc secretion results in enhanced insulin secretion from β cells in ZnT8-KO mice and hepatic insulin clearance is not suppressed (right). Thus, peripheral insulin levels in ZnT8-KO mice are maintained at lower levels than in WT mice.
Clinically, the inhibition of hepatic insulin clearance seems likely to be a therapeutic target for diabetes. A previous study reported the therapeutic potential of small molecule inhibitors of insulin-degrading enzyme, which regulates insulin catabolism [39,40]. Our findings hence might provide novel insights into the molecular pathology of diabetes, which involves dysregulated insulin clearance from the liver, and hence may be a promising future therapeutic target for diabetes [18].
3.3. Involvement of Other ZnT Transporters
Although there is a substantial decrease in total zinc levels in the islets of ZnT8-KO mice compared with wild-type mice, the phenotypes of ZnT8-KO mice regarding glucose metabolism were fairly modest. Several other ZnT isoforms were expressed at low levels in the pancreatic islets. Thus, functional compensation by other ZnT isoforms might reduce the effect of the ZnT8-KO phenotype. ZnT3 is a candidate ZnT transporter for this compensation. ZnT3 is known to play a role in the uptake of zinc in the synaptic vesicles of glutaminergic hippocampal neurons [41,42]. Considering that β cells and neurons share some similar characteristics, ZnT3 might be involved in the transport of zinc into insulin secretory vesicles. However, it is unclear whether ZnT3 is expressed in the islets of mice [43], and ZnT3-KO mice appear to undergo normal glucose metabolism [44], suggesting that ZnT3 is not involved in this process.
As zinc is required for the hexamerization of insulin and its conversion from proinsulin to insulin in the Golgi compartment, a sufficient amount of import of zinc to this compartment is also required. ZnT5 and ZnT7 are also reported to be expressed in β cells and to co-localize with the Golgi apparatus and secretory vesicles [43,45,46]. Thus, ZnT5 and ZnT7 transporters might be involved in these processes. A recent study analyzed this possibility by crossing ZnT7-KO mice with ZnT8-KO mice. However, whether ZnT7 has a redundant role of ZnT8 remains to be clarified because global ZnT7-KO mice displayed several defects in insulin-sensitive tissues outside of β cells, as described below, and because the report did not include data on ZnT8 single-knockout mice [47]. Further analyses are needed to identify the zinc transporters involved in each step from insulin processing to storage.
3.4. Zinc Transport Activity of ZnT8 Variants
One of the important unresolved issue regarding ZnT8 is whether the ZnT8 variant 325Arg(R) increases or decreases zinc transport activity. In one study, the fluorescent dye FluoZin-3 was used to monitor cytosolic zinc and the fluorescent dye Zinquin was used to monitor vacuolar zinc accumulation in MIN6 cells transiently expressing the Arg(R) or Trp(W) variants of hZnT8. Cells expressing the variant (W) showed significantly greater fluorescence of both dyes, and the authors concluded that this variant was a more active transporter of zinc [34]. In another study, HEK293 cells inducibly expressing hZnT8 variants were established, and both the R and W variants of hZnT8 were purified in the native state, and a reconstitution system was developed to measure zinc transport activities [48]. The authors found the R variant to be more active than the W variant, suggesting that the common high-risk R variant is hyperactive and thus may be a therapeutic target to reduce the risk of T2DM in the general population [48]. An additional report described the possible association between the diabetes risk allele in hZnT8 and the higher zinc concentration in human islets [49]. These results suggest that β cells with lower zinc levels may be protection from T2DM, whereas higher zinc in β cells may be associated with T2DM. Interestingly, these new findings appear to be consistent with the finding that rare loss-of-function mutations in ZnT8 are associated with reduced T2DM risk in humans [27]. This suggests that the role of ZnT8 might be contradictory between humans and mice, as a loss-of-function of ZnT8 in humans decreases the risk for T2DM, whereas ZnT8-KO mice have impaired glucose tolerance [50]. In the course of evolution, the role of ZnT8 in glucose homeostasis has been altered. There are some factors to explain this discrepancy. Since synaptic ZnT3-mediated zinc contributes predominantly to amyloid deposition in human amyloid precursor protein (hAPP) mice [51,52], human islet amyloid polypeptide (hIAPP) might be able to explain this discrepancy. Compared to mouse IAPP, hIAPP can form toxic oligomers, which affect β cells by inducing apoptosis and amyloidogenesis in T2DM [53,54] (Figure 2A). A recent computational analysis showed that zinc concentration determines insulin oligomer equilibrium and that the hIAPP monomer preferentially binds to both the insulin monomer and dimer, compared to the formation of hIAPP homodimer. Therefore, regarding the loss of ZnT8 function, the zinc deficiency shifts the equilibrium of the insulin oligomers toward monomers and dimers, which isolate hIAPP monomers (nontoxic form) and prevent hIAPP from self-association and subsequent aggregation (toxic forms), thereby reducing the risk of T2DM (Figure 2B) [55]. The theory of altered hIAPP aggregation in β cells in response to altered ZnT8 function is a promising but still correlative hypothesis at this point. Therefore, this hypothesis can be validated by creating hIAPP transgenic (hIAPP-Tg) mice from ZnT8-KO mice, to investigate whether hIAPP cytotoxicity can be ameliorated by the deletion of ZnT8.

Figure 2.
Schematic model of the relationship among hIAPP, insulin, and zinc. (A) hIAPP can easily form toxic oligomers that induce apoptosis and amyloidogenesis in β cells in T2DM patients; (B) When the zinc concentration in insulin secretory granules is high, zinc is used to form zinc-insulin-hexamer, and hIAPP can easily form toxic oligomers. On the other hand, when the zinc concentration is low in insulin secretory granules (such as insulin secretory granule in ZnT8-KO mice), insulin exists as monomer or dimer, which preferentially binds to hIAPP monomer and prevents hIAPP from self-associating and aggregating.
4. Zinc Distribution Affects Adipocyte Metabolism
4.1. Zinc Distribution in Obesity
Chronic low intake of zinc is associated with an increased risk of diabetes. Hence, zinc supplementation is expected to be an effective method for preventing metabolic syndrome and diabetes. A previous study analyzed the effect of zinc supplementation to prepubertal obese children on insulin resistance and metabolic syndrome. Zinc supplementation was suggested to be a useful and safe additional intervention treatment [56]. However, to our knowledge, the effectiveness of zinc supplementation for the treatment of obesity and diabetes has not been demonstrated in large-scale studies, particularly in adults. Therefore, it is important to establish the safety, efficacy, and effective dose of zinc supplementation in adults.
Nevertheless, zinc might be referred to as the insulin-mimetic, since zinc stimulates lipogenesis and glucose uptake in isolated adipocytes, and zinc ion acts as an insulin-mimetics through their direct effect on the insulin-signaling pathway [2]. The insulin-sensitizing effect of zinc has been attributed to the inhibition of the tyrosine phosphatase activity of protein tyrosine phosphatase 1B (PTP1B) (Figure 3) [57,58]. Zinc ion inactivates PTP1B by non-covalent binding to its cysteine residues which is crucial for the enzymatic activity and reactive oxygen species is also known to inactivate the enzyme in a similar manner. In fact, oxidative stress is upregulated in the most of the patients with diabetes and diabetic animals, further mechanistic analysis is needed to examine the risks and benefits of zinc supplementation as a mean for mitigating obesity and type 2 diabetes.
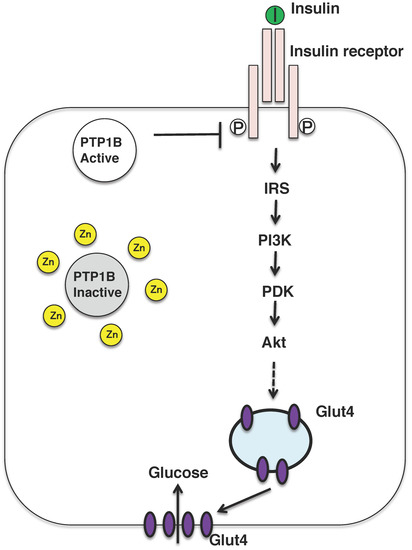
Figure 3.
Insulin signaling pathway and insulin mimicking function of zinc ions. Insulin binds to the insulin receptor located in the plasma membrane in the peripheral tissues, such as liver and muscle. The insulin-signaling pathway is activated and the glucose transporter GLUT4 is translocated to the plasma membrane. Zinc might inhibit the activity of PTP1B, which activates the insulin-signaling pathway. PI3K, phosphatidylinositol-3-kinase; IRS, insulin receptor substrate; PKD, protein kinase D.
4.2. Association between Adipocyte Metabolism and Zinc Homeostasis
Obesity and its associated metabolic diseases develop when energy intake exceeds energy expenditure; this can be caused by decreased physical activity, the inability of the CNS to downregulate appetite, or the ingestion of high-calorie foods [59]. Adipose tissue is involved in energy storage and also functions as an endocrine organ to release free fatty acids (FFA) and adipokines, such as leptin, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and adiponectin [60]. In addition, adipose tissue comprises numerous types of stromal cells, including preadipocytes, endothelial cells, immune cells, and fibroblasts. During the course of obesity, adipocyte cells and stromal cells in adipose tissue change in number and characteristics. Invasion of macrophages into the adipose tissue of obese individuals is associated with increased secretion of inflammatory adipocytokines, including TNF-α and IL-6, which leads to insulin resistance [61]. Macrophages have recently been reported to be involved in adipose tissue inflammation as well as in the regulation of adipose metabolism through the disrupted modulation of adipocytokines production [61].
Several reports have addressed the association between zinc transporters and adipose metabolism. For example, in the adipose tissue of Zip14-KO mice, hypertrophy together with enhanced proinflammatory signaling is observed through activation of nuclear factor-kappaB (NF-κB) and the Janus-activating kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) pathway, and this might contribute to obesity-induced insulin resistance [62]. Consistent with this idea, Zip14 expression is significantly reduced in obese individuals compared with non-obese individuals, and is increased markedly following weight loss [63].
Adipose tissue plays a crucial role in controlling energy balance. It comprises white and brown adipocytes, which perform different functions. White adipocytes store excess energy, whereas brown adipocytes play a role in energy expenditure [59]. In mammals, brown adipose tissue (BAT) dissipates energy in the form of heat and acts as a defense mechanism against hypothermia. Brown adipocytes are unique in that they have a very large number of mitochondria and are also able to metabolize glucose and fats to produce heat rather than ATP. This thermogenic activity of brown adipocytes is mediated largely via the actions of uncoupling protein-1 (UCP1) [64]. Furthermore, two distinct types of thermogenic adipocytes have recently been identified, namely, the “classical brown adipocytes” and the “beige adipocytes”. Beige adipocytes are induced in white adipose tissue (WAT), particularly in inguinal WAT upon various external factors, including exercise, chronic cold exposure, and bariatric surgery [65]. Identification and implementation of therapies based on beige fat require a detailed understanding of the differences in the developmental mechanisms and functions of white, brown, and beige adipocytes.
Many transcriptional regulators and transcription factors are used for differentiation into these various fat cell types, such as peroxisome proliferator-activated receptor gamma (PPARγ) and the CCAAT/enhancer binding protein (C/EBP) family of transcription factors, respectively [66]. The induced differentiation of preadipocytes triggers DNA replication and reentry into the cell cycle (mitotic clonal expansion). Mitotic clonal expansion involves a transcription factor cascade, followed by the expression of adipocyte genes. A critical event is phosphorylation of C/EBP-β, which is a form of activated C/EBP-β, which then triggers PPARγ and C/EBP-α, which in turn coordinately activate genes whose expression produces the adipocyte phenotype, such as aP2 [67]. Several studies have analyzed the roles of zinc and the zinc transporter in the differentiation process of white adipocytes. Expression of the Zip14 gene was found to be upregulated during early adipocyte differentiation [63,68]. During mitotic cell expansion, zinc and MT levels within cells are rapidly increased. This elevation is essential for the transition from G0/G1- to S-phase of the cell cycle [69] (Figure 4). ZnT7-KO mice show a mild zinc deficiency, with low body weight gain as well as body fat accumulation. The underlying mechanism of these characteristics in ZnT7-KO mice is that ZnT7 is likely to be involved in lipogenesis in adipocytes, rather than in the early adipocyte differentiation process, such as mitotic clonal expansion [70].

Figure 4.
The expression of zinc transporters during white adipocyte differentiation. Preadipocytes are differentiated and trigger DNA replication and reentry into the cell cycle (mitotic clonal expansion, MCE). The expression of several genes related to zinc homeostasis is altered.
Most of the transcription factors that are known to direct cells toward a brown/beige adipocyte lineage instead of a white adipocyte lineage act via the core transcriptional machinery of adipogenesis. PR domain containing 16 (PRDM16), which is an essential transcriptional coregulators of brown/beige adipocyte differentiation [71], determines the brown/beige adipocyte lineage mainly via its interaction with various transcriptional factors, including PPARγ, peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α), C/EBP-β, and zinc finger protein 516 [65,72]. Among them, many zinc-containing transcriptional factors participate in brown/beige adipocyte differentiation and function.
4.3. ZIP13 Regulates Beige Adipocyte Biogenesis and Energy Expenditure
As explained above, we have been investigating the roles of zinc homeostasis in the health and disease of endocrine organs by focusing on the biological functions of zinc transporters. Zip13-KO mice were reported to show impaired bone formation and growth retardation [73]. Importantly, ZIP13 is also plays a crucial role in connective tissue development in humans. Patients with a loss-of-function mutation in ZIP13 showed a similar phenotype to Zip13-KO mice and were diagnosed as having a novel type of Ehlers-Danlos syndrome. Interestingly, human patients of Ehlers-Danlos syndrome were reported to display lipoatrophy [73]. Therefore, we aimed to clarify the roles of ZIP13 in fat tissue. During our investigation, we found that Zip13-KO inguinal WAT had a high number of functional beige adipocytes [74]. Furthermore, Zip13-KO mice showed a significantly higher oxygen consumption rate than wild-type mice, although there were no differences in food intake, suggesting that Zip13-KO mice have a tendency to not gain weight. Consistent with this idea, Zip13-KO mice showed resistance to high fat diet-induced obesity [74].
Furthermore, both gain-of-function and loss-of-function experiments have demonstrated that the accumulation of C/EBP-β, which is involved in determining brown/beige adipocyte lineage in cooperation with the dominant transcriptional coregulator PRDM16, is crucial for the increased adipocyte browning resulting from the loss of ZIP13 [74], implying most likely that ZIP13-mediated zinc transport is required for the inhibition of adipocyte browning (Figure 5), and that ZIP13 may deliver zinc ions to specific molecular targets that control the function of C/EBP-β or/and other target proteins. Further analyses of Zip13-deficient cells will clarify the specific roles of ZIP13 in beige adipocyte biogenesis.
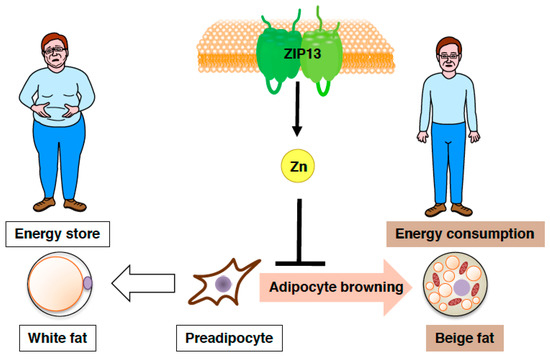
Figure 5.
Zinc transporter ZIP13 inhibits adipocyte browning. Schematic model of the role of ZIP13 in adipocyte browning.
Importantly, the presence and activity of thermogenic beige adipocytes are associated with improved global metabolic fitness, such as improvements in insulin resistance and glucose homeostasis [65]. Indeed, Zip13-KO mice reportedly have improved glucose tolerance and insulin tolerance compared with control mice in addition to increased energy expenditure [74]. Furthermore, given that recent studies demonstrated that adult human brown adipocytes share biological characteristics with rodent beige adipocytes, rather than rodent brown adipocyte [75]. The results of our study may hence contribute to the establishment of novel treatments for obese diabetic patients via the manipulation of adipocyte identity.
5. Involvement of Zinc in the Insulin Signaling Pathway
5.1. Zinc Transporters Affect Skeletal Muscle Insulin Signaling
Skeletal muscle plays a key role in insulin-stimulated glucose uptake in the postprandial state. Under insulin-resistant conditions, there is a decrease in insulin signaling via IRS-1, PI3K, and Akt, resulting in decreased translocation of the GLUT4 glucose transporter, to the plasma membrane, as well as decreased insulin-stimulated glucose transport into cells (Figure 3).
Skeletal muscle is the major reservoir for zinc, containing approximately 60% of the total whole-body zinc [76]. Zinc transporter SLC39A7 (ZIP7) has been reported to be involved in glycemic control within skeletal muscle. The knockdown of Zip7 in the mouse skeletal muscle cell line C2C12 demonstrated that glucose metabolism is enhanced by ZIP7 via Akt phosphorylation, in which ZIP7-mediated zinc activates insulin receptor signaling via its binding to PTP1B [77]. A different study reported that ZnT7-KO mice demonstrate impaired glucose tolerance and insulin sensitivity, resulting from a decrease in the insulin signaling pathway, such as decreased levels of Akt phosphorylation in skeletal muscle and adipocytes, as described above, which thereby reduce glucose uptake [78]. Although many studies have been performed to analyze the insulin-mimetic actions of zinc [79], the specific zinc transporters that are responsible for initiating these signaling processes remain unclear. In particular, given that ZIP7 acts as the gatekeeper of zinc release from the Golgi apparatus [80] and that insulin-signaling cascades in skeletal muscle are affected by ZIP7-mediated zinc, it will be very important to analyze the metabolic phenotypes of skeletal muscle in tissue-specific Zip7-KO mice.
5.2. Zinc Homeostasis and Sarcopenia
Sarcopenia is the age-associated degenerative loss of skeletal muscle mass, quality, and strength. In older people, sarcopenia is often accompanied by diabetes [81]. Some of the mechanisms involved in the development of sarcopenia, such as insulin resistance, mitochondrial dysfunction, and chronic inflammation, are also thought to play roles in the pathogenesis of diabetes [82]. Recently, the zinc/zinc transporter and MT expression levels have been reported to be upregulated during skeletal muscle atrophy. For example, the blocking of MTs 1 and 2 has been shown to increase skeletal muscle mass and strength [83], suggesting the potential of MTs as therapeutic targets for sarcopenia. Furthermore, muscle zinc levels and Zip14 expression levels are known to increase with age [84], although the underlying mechanisms and physiological meaning of these observations remain to be clarified. Further analyses are required to understand the precise roles of zinc transporters in sarcopenia that occurs in association with diabetes.
6. Summary
Since human patients with Zip13 deficiency have been reported to show lipoatrophy, we expected Zip13-KO mice to be resistant to insulin [73]. However, on the contrary, Zip13-KO mice showed improved insulin sensitivity owing to the acceleration of adipocyte browning. To our knowledge, our data are the first to show that the Golgi-to-cytoplasm transport of zinc by ZIP13 is necessary for the regulation of beige adipocyte metabolism and insulin sensitivity, because simple zinc supplementation cannot inhibit adipocyte browning. On the other hand, a study of ZnT8-KO mice also showed that the release of adequate amounts of zinc from β cells together with insulin is important for the regulation of insulin clearance, suggesting that the zinc transported by ZnT8 acts as a signaling molecule between organs and mediates pancreas-to-liver organ communication. ZnT7-KO mice demonstrate mild zinc deficiency accompanied with low body weights and very little accumulation of body fat. Dietary zinc supplementation in ZnT7-KO mice cannot rescue these phenotypes. The main tissue responsible for the phenotypes of ZnT7-KO mice might be adipose tissue, which results in impaired glucose tolerance and insulin sensitivity.
Considering the aforementioned phenotypes of zinc transporter KO mice, we would like to suggest a new perspective; that adequate local zinc delivery by zinc transporters are important, and their disruption leads to the pathogenesis of a variety of diseases. In particular, as sarcopenia is an important issue in aging societies, zinc transporters or factors associated with zinc homeostasis might be candidate biomarkers or therapeutic targets. Further analyses are required toward the development of zinc transporter-mediated therapies against both obesity and diabetes.
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/19/2/476/s1.
Acknowledgments
We thank H. Akiko Popiel for helpful advice and criticism. This work was supported by grants from the Ministry of Education, Sports and Culture of Japan (to AF [16K09764, 26860700, and 24790936] and YF [25461364]), Fumi Yamamura Memorial Foundation for Female Natural Scientists, Suzuken Memorial Foundation, Banyu Life Science Foundation International, Front Runner of Future Diabetes Research, Astellas Foundation of Research on Metabolic Disorders, and Japan Diabetes Foundation (to Ayako Fukunaka).
Author Contributions
Ayako Fukunaka wrote the manuscript and Ayako Fukunaka and Yoshio Fujitani reviewed the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Simcox, J.A.; McClain, D.A. Iron and diabetes risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in Pancreatic Islet Biology, Insulin Sensitivity, and Diabetes. Prev. Nutr. Food Sci. 2017, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Leapman, R.D.; Li, M.X.; Atwater, I. Elemental composition of secretory granules in pancreatic islets of Langerhans. Biophys. J. 1993, 64, 525–532. [Google Scholar] [CrossRef]
- Hutton, J.C.; Penn, E.J.; Peshavaria, M. Low-molecular-weight constituents of isolated insulin-secretory granules. Bivalent cations, adenine nucleotides and inorganic phosphate. Biochem. J. 1983, 210, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takeda, T.A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of zinc transporters: Molecular and genetic importance in zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Chimienti, F.; Devergnas, S.; Pattou, F.; Schuit, F.; Garcia-Cuenca, R.; Vandewalle, B.; Kerr-Conte, J.; Van Lommel, L.; Grunwald, D.; Favier, A.; et al. In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J. Cell Sci. 2006, 119, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.; Steiner, D. The role of assembly in insulin’s biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194. [Google Scholar] [CrossRef]
- Dodson, E.J.; Dodson, G.G.; Hodgkin, D.C.; Reynolds, C.D. Structural relationships in the two-zinc insulin hexamer. Can. J. Biochem. 1979, 57, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.F. Zinc-ligand interactions modulate assembly and stability of the insulin hexamer—A review. Biometals 2005, 18, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Havu, N.; Lundgren, G.; Falkmer, S. Zinc and manganese contents of micro-dissected pancreatic islets of some rodents. A microchemical study in adult and newborn guinea pigs, rats, Chinese hamsters and spiny mice. Acta Endocrinol. 1977, 86, 570–577. [Google Scholar] [PubMed]
- Steiner, D.F.; Rouille, Y.; Gong, Q.; Martin, S.; Carroll, R.; Chan, S.J. The role of prohormone convertases in insulin biosynthesis: Evidence for inherited defects in their action in man and experimental animals. Diabetes Metab. 1996, 22, 94–104. [Google Scholar] [PubMed]
- Emdin, S.O.; Dodson, G.G.; Cutfield, J.M.; Cutfield, S.M. Role of zinc in insulin biosynthesis. Some possible zinc-insulin interactions in the pancreatic B-cell. Diabetologia 1980, 19, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hutton, J.C. The insulin secretory granule. Diabetologia 1989, 32, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.C.; Min, L.; Pessin, J.E. Insulin granule biogenesis, trafficking and exocytosis. Vitam. Horm. 2009, 80, 473–506. [Google Scholar] [PubMed]
- Tamaki, M.; Fujitani, Y.; Hara, A.; Uchida, T.; Tamura, Y.; Takeno, K.; Kawaguchi, M.; Watanabe, T.; Ogihara, T.; Fukunaka, A.; et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J. Clin. Investig. 2013, 123, 4513–4524. [Google Scholar] [CrossRef] [PubMed]
- Boquist, L.; Lernmark, A. Effects on the endocrine pancreas in Chinese hamsters fed zinc deficient diets. Acta Pathol. Microbiol. Scand. 1969, 76, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.M.; Gershoff, S.N. Effect of zinc deficiency in rats on insulin release from the pancreas. J. Nutr. 1973, 103, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes 2005, 54 (Suppl. S2), S108–S113. [Google Scholar] [CrossRef] [PubMed]
- Mysore, T.B.; Shinkel, T.A.; Collins, J.; Salvaris, E.J.; Fisicaro, N.; Murray-Segal, L.J.; Johnson, L.E.; Lepore, D.A.; Walters, S.N.; Stokes, R.; et al. Overexpression of glutathione peroxidase with two isoforms of superoxide dismutase protects mouse islets from oxidative injury and improves islet graft function. Diabetes 2005, 54, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.L.; Westman, N.G.; Lundgren, E.; Roos, G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982, 42, 1955–1961. [Google Scholar] [PubMed]
- Sun, Q.; van Dam, R.M.; Willett, W.C.; Hu, F.B. Prospective study of zinc intake and risk of type 2 diabetes in women. Diabetes Care 2009, 32, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Bao, W.; Zhang, Y.; Rong, Y.; Wang, X.; Jin, Y.; Song, Y.; Yao, P.; Sun, C.; Hu, F.B.; et al. Interactions between zinc transporter-8 gene (SLC30A8) and plasma zinc concentrations for impaired glucose regulation and type 2 diabetes. Diabetes 2014, 63, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Sun, W.; Fu, Y.; Miao, L.; Cai, L. Zinc homeostasis in the metabolic syndrome and diabetes. Front. Med. 2013, 7, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular zinc in insulin secretion and action: A determinant of diabetes risk? Proc. Nutr. Soc. 2016, 75, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Chabosseau, P.; Rutter, G.A. Zinc and diabetes. Arch. Biochem. Biophys. 2016, 611, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Dai, F.F.; Hardy, A.B.; Giglou, P.R.; Bhattacharjee, A.; Koshkin, V.; Chimienti, F.; Gaisano, H.Y.; Rutter, G.A.; Wheeler, M.B. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010, 53, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.K.; Hu, M.; Chabosseau, P.L.; Cane, M.C.; Meur, G.; Bellomo, E.A.; Carzaniga, R.; Collinson, L.M.; Li, W.H.; Hodson, D.J.; et al. Molecular Genetic Regulation of SLC30A8/ZnT8 Reveals a Positive Association With Glucose Tolerance. Mol. Endocrinol. 2016, 30, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Pound, L.D.; Sarkar, S.A.; Ustione, A.; Dadi, P.K.; Shadoan, M.K.; Lee, C.E.; Walters, J.A.; Shiota, M.; McGuinness, O.P.; Jacobson, D.A.; et al. The physiological effects of deleting the mouse SLC30A8 gene encoding zinc transporter-8 are influenced by gender and genetic background. PLoS ONE 2012, 7, e40972. [Google Scholar] [CrossRef] [PubMed]
- Pound, L.D.; Sarkar, S.A.; Benninger, R.K.; Wang, Y.; Suwanichkul, A.; Shadoan, M.K.; Printz, R.L.; Oeser, J.K.; Lee, C.E.; Piston, D.W.; et al. Deletion of the mouse SLC30A8 gene encoding zinc transporter-8 results in impaired insulin secretion. Biochem. J. 2009, 421, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Holst, J.J.; Schmidt, W.E.; Nauck, M.A. Reduction of hepatic insulin clearance after oral glucose ingestion is not mediated by glucagon-like peptide 1 or gastric inhibitory polypeptide in humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E849–E856. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Yang, Y.; Rezaei, K.; Fernstrom, M.A.; Lee, A.D.; Kido, Y.; Erickson, S.K.; Najjar, S.M. CEACAM1 regulates insulin clearance in liver. Nat. Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Boesgaard, T.W.; Zilinskaite, J.; Vanttinen, M.; Laakso, M.; Jansson, P.A.; Hammarstedt, A.; Smith, U.; Stefan, N.; Fritsche, A.; Haring, H.; et al. The common SLC30A8 Arg325Trp variant is associated with reduced first-phase insulin release in 846 non-diabetic offspring of type 2 diabetes patients—The EUGENE2 study. Diabetologia 2008, 51, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Malito, E.; Hedouin, S.; Reinstatler, L.; Sahara, T.; Abdul-Hay, S.O.; Choudhry, S.; Maharvi, G.M.; Fauq, A.H.; Huzarska, M.; et al. Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS ONE 2010, 5, e10504. [Google Scholar] [CrossRef] [PubMed]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, H.J.; Cole, T.B.; Born, D.E.; Schwartzkroin, P.A.; Palmiter, R.D. Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc. Natl. Acad. Sci. USA 1997, 94, 12676–12681. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose regulates free cytosolic Zn2+ concentration, Slc39 (ZiP), and metallothionein gene expression in primary pancreatic islet beta-cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef] [PubMed]
- Smidt, K.; Jessen, N.; Petersen, A.B.; Larsen, A.; Magnusson, N.; Jeppesen, J.B.; Stoltenberg, M.; Culvenor, J.G.; Tsatsanis, A.; Brock, B.; et al. SLC30A3 responds to glucose- and zinc variations in beta-cells and is critical for insulin production and in vivo glucose-metabolism during beta-cell stress. PLoS ONE 2009, 4, e5684. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Narita, H.; Yamaguchi-Iwai, Y.; Hirose, J.; Amano, T.; Sugiura, N.; Sasaki, R.; Mori, K.; Iwanaga, T.; Nagao, M. Cloning and characterization of a novel mammalian zinc transporter, zinc transporter 5, abundantly expressed in pancreatic beta cells. J. Biol. Chem. 2002, 277, 19049–19055. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yan, M.; Kirschke, C.P. Over-expression of ZnT7 increases insulin synthesis and secretion in pancreatic beta-cells by promoting insulin gene transcription. Exp. Cell Res. 2010, 316, 2630–2643. [Google Scholar] [CrossRef] [PubMed]
- Syring, K.E.; Boortz, K.A.; Oeser, J.K.; Ustione, A.; Platt, K.A.; Shadoan, M.K.; McGuinness, O.P.; Piston, D.W.; Powell, D.R.; O’Brien, R.M. Combined Deletion of Slc30a7 and SLC30A8 Unmasks a Critical Role for ZnT8 in Glucose-Stimulated Insulin Secretion. Endocrinology 2016, 157, 4534–4541. [Google Scholar] [CrossRef] [PubMed]
- Merriman, C.; Huang, Q.; Rutter, G.A.; Fu, D. Lipid-tuned Zinc Transport Activity of Human ZnT8 Protein Correlates with Risk for Type-2 Diabetes. J. Biol. Chem. 2016, 291, 26950–26957. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.P.; Allen, N.B.; Meyers, M.S.; Link, E.O.; Zhang, X.; MacRenaris, K.W.; El Muayed, M. Exploring the Association Between Demographics, SLC30A8 Genotype, and Human Islet Content of Zinc, Cadmium, Copper, Iron, Manganese and Nickel. Sci. Rep. 2017, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chimienti, F. SLC30A8 mutations in type 2 diabetes. Diabetologia 2015, 58, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Cole, T.B.; Palmiter, R.D.; Suh, S.W.; Koh, J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7705–7710. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Langen, R.; Gurlo, T.; Matveyenko, A.V.; Butler, P.C. beta-Cell failure in type 2 diabetes: A case of asking too much of too few? Diabetes 2013, 62, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Nedumpully-Govindan, P.; Ding, F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci. Rep. 2015, 5, 8240. [Google Scholar] [CrossRef] [PubMed]
- Hashemipour, M.; Kelishadi, R.; Shapouri, J.; Sarrafzadegan, N.; Amini, M.; Tavakoli, N.; Movahedian-Attar, A.; Mirmoghtadaee, P.; Poursafa, P. Effect of zinc supplementation on insulin resistance and components of the metabolic syndrome in prepubertal obese children. Hormones 2009, 8, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Protein tyrosine phosphatases as targets of the combined insulinomimetic effects of zinc and oxidants. Biometals 2005, 18, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in Cellular Regulation: The Nature and Significance of “Zinc Signals”. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Kahn, C.R. Brown fat as a therapy for obesity and diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Troche, C.; Aydemir, T.B.; Cousins, R.J. Zinc transporter Slc39a14 regulates inflammatory signaling associated with hypertrophic adiposity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E258–E268. [Google Scholar] [CrossRef] [PubMed]
- Maxel, T.; Smidt, K.; Larsen, A.; Bennetzen, M.; Cullberg, K.; Fjeldborg, K.; Lund, S.; Pedersen, S.B.; Rungby, J. Gene expression of the zinc transporter ZIP14 (SLC39a14) is affected by weight loss and metabolic status and associates with PPARgamma in human adipose tissue and 3T3-L1 pre-adipocytes. BMC Obes. 2015, 2, 46. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Saito, M. A new era in brown adipose tissue biology: Molecular control of brown fat development and energy homeostasis. Annu. Rev. Physiol. 2014, 76, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, X.; Tang, Q.Q. Transcriptional regulation of adipocyte differentiation: A central role for CCAAT/enhancer-binding protein (C/EBP) beta. J. Biol. Chem. 2015, 290, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Kagata, T.; Johmura, Y.; Hishida, T.; Nishizuka, M.; Imagawa, M. SLC39A14, a LZT protein, is induced in adipogenesis and transports zinc. FEBS J. 2005, 272, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Beyersmann, D. Transient peaks in zinc and metallothionein levels during differentiation of 3T3L1 cells. Arch. Biochem. Biophys. 1999, 364, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tepaamorndech, S.; Kirschke, C.P.; Pedersen, T.L.; Keyes, W.R.; Newman, J.W.; Huang, L. Zinc transporter 7 deficiency affects lipid synthesis in adipocytes by inhibiting insulin-dependent Akt activation and glucose uptake. FEBS J. 2016, 283, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Seale, P. Transcriptional Regulatory Circuits Controlling Brown Fat Development and Activation. Diabetes 2015, 64, 2369–2375. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Fukunaka, A.; Fukada, T.; Bhin, J.; Suzuki, L.; Tsuzuki, T.; Takamine, Y.; Bin, B.H.; Yoshihara, T.; Ichinoseki-Sekine, N.; Naito, H.; et al. Zinc transporter ZIP13 suppresses beige adipocyte biogenesis and energy expenditure by regulating C/EBP-beta expression. PLoS Genet. 2017, 13, e1006950. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, K.; Luijten, I.H.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Physiology of Zinc: General aspects. In Zinc in Human Biology; Mills, C.F., Ed.; Springer: London, UK, 1989; pp. 1–14. [Google Scholar]
- Myers, S.A.; Nield, A.; Chew, G.S.; Myers, M.A. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PLoS ONE 2013, 8, e79316. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kirschke, C.P.; Lay, Y.A.; Levy, L.B.; Lamirande, D.E.; Zhang, P.H. Znt7-null mice are more susceptible to diet-induced glucose intolerance and insulin resistance. J. Biol. Chem. 2012, 287, 33883–33896. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Fukunaka, A.; Hagihara, M.; Watanabe, K.; Kamino, S.; Kambe, T.; Enomoto, S.; Hiromura, M. Essential role of the zinc transporter ZIP9/SLC39A9 in regulating the activations of Akt and Erk in B-cell receptor signaling pathway in DT40 cells. PLoS ONE 2013, 8, e58022. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci. Signal. 2012, 5, ra11. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; Williams, J.; Atherton, P.; Larvin, M.; Lund, J.; Narici, M. Sarcopenia, dynapenia, and the impact of advancing age on human skeletal muscle size and strength; a quantitative review. Front. Physiol. 2012, 3, 260. [Google Scholar] [CrossRef] [PubMed]
- Cleasby, M.E.; Jamieson, P.M.; Atherton, P.J. Insulin resistance and sarcopenia: Mechanistic links between common co-morbidities. J. Endocrinol. 2016, 229, R67–R81. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Bouzan, A.; Pierrel, E.; Melly, S.; Stauffer, D.; Gutzwiller, S.; Nolin, E.; Dornelas, C.; Fryer, C.; Leighton-Davies, J.; et al. Blockade of Metallothioneins 1 and 2 Increases Skeletal Muscle Mass and Strength. Mol. Cell. Biol. 2017, 37, e00305–e00316. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Troche, C.; Kim, J.; Kim, M.H.; Teran, O.Y.; Leeuwenburgh, C.; Cousins, R.J. Aging amplifies multiple phenotypic defects in mice with zinc transporter Zip14 (Slc39a14) deletion. Exp. Gerontol. 2016, 85, 88–94. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).