Progression of Repair and Injury in Human Liver Slices


Abstract
1. Introduction
2. Results
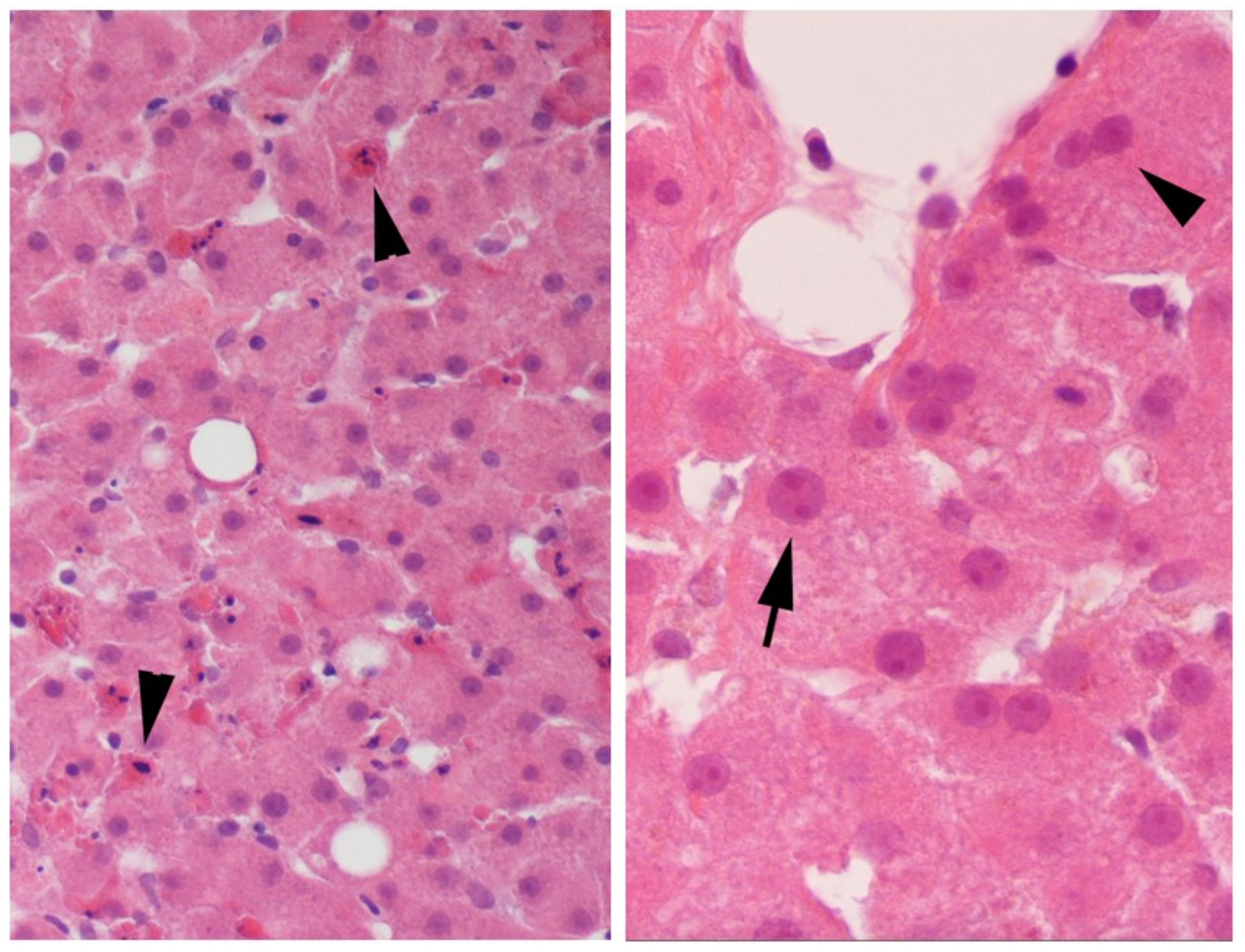
2.1. Evidence of Regeneration
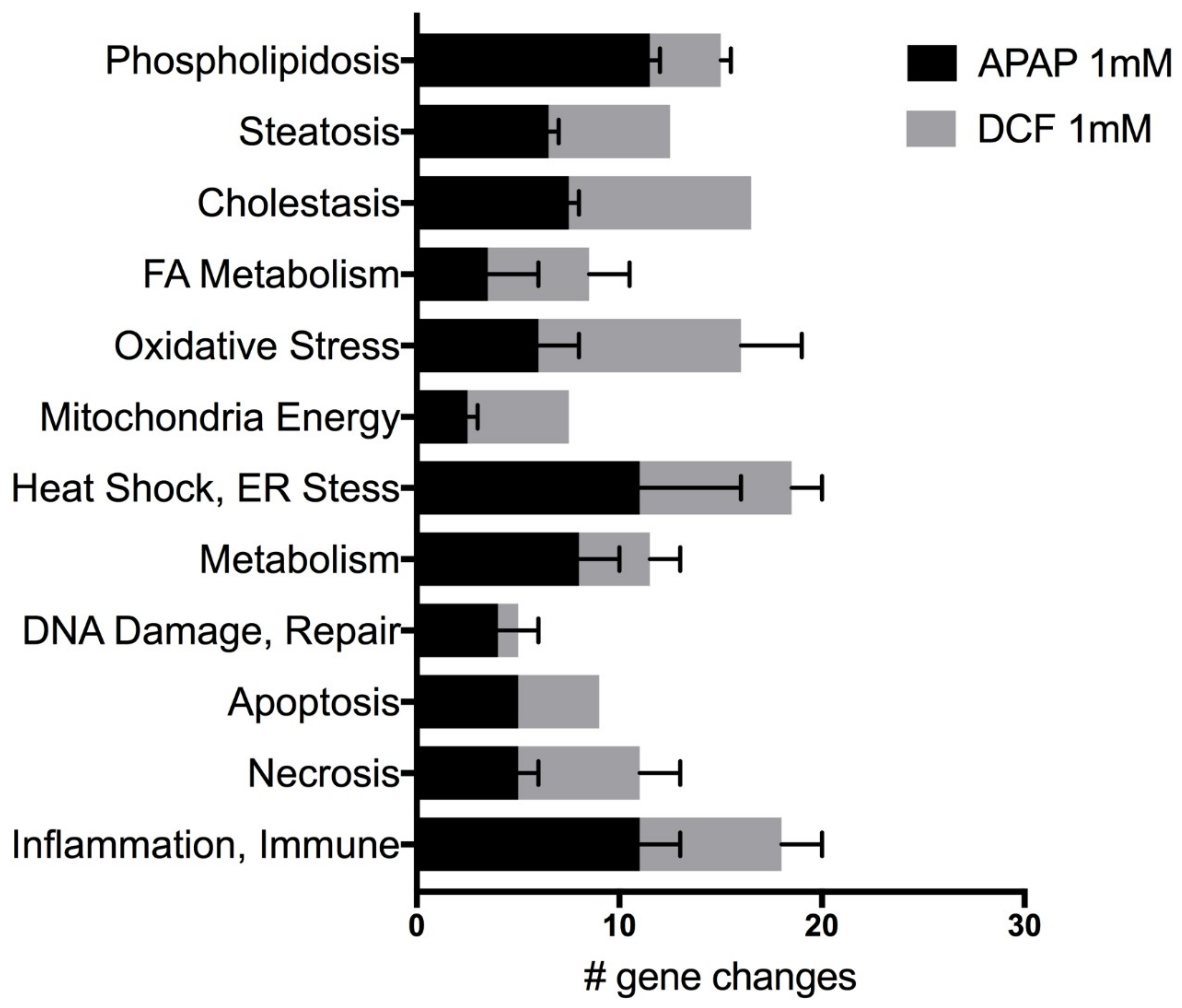
2.2. APAP and DCF Toxicity Profiling
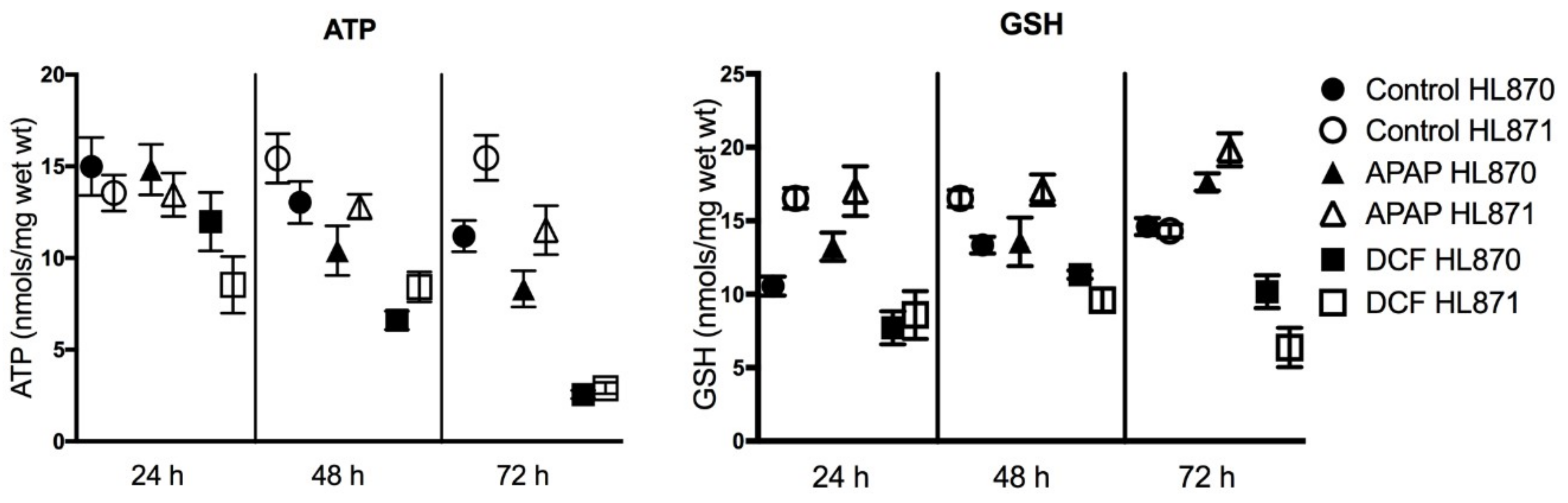
2.3. Functional Assays
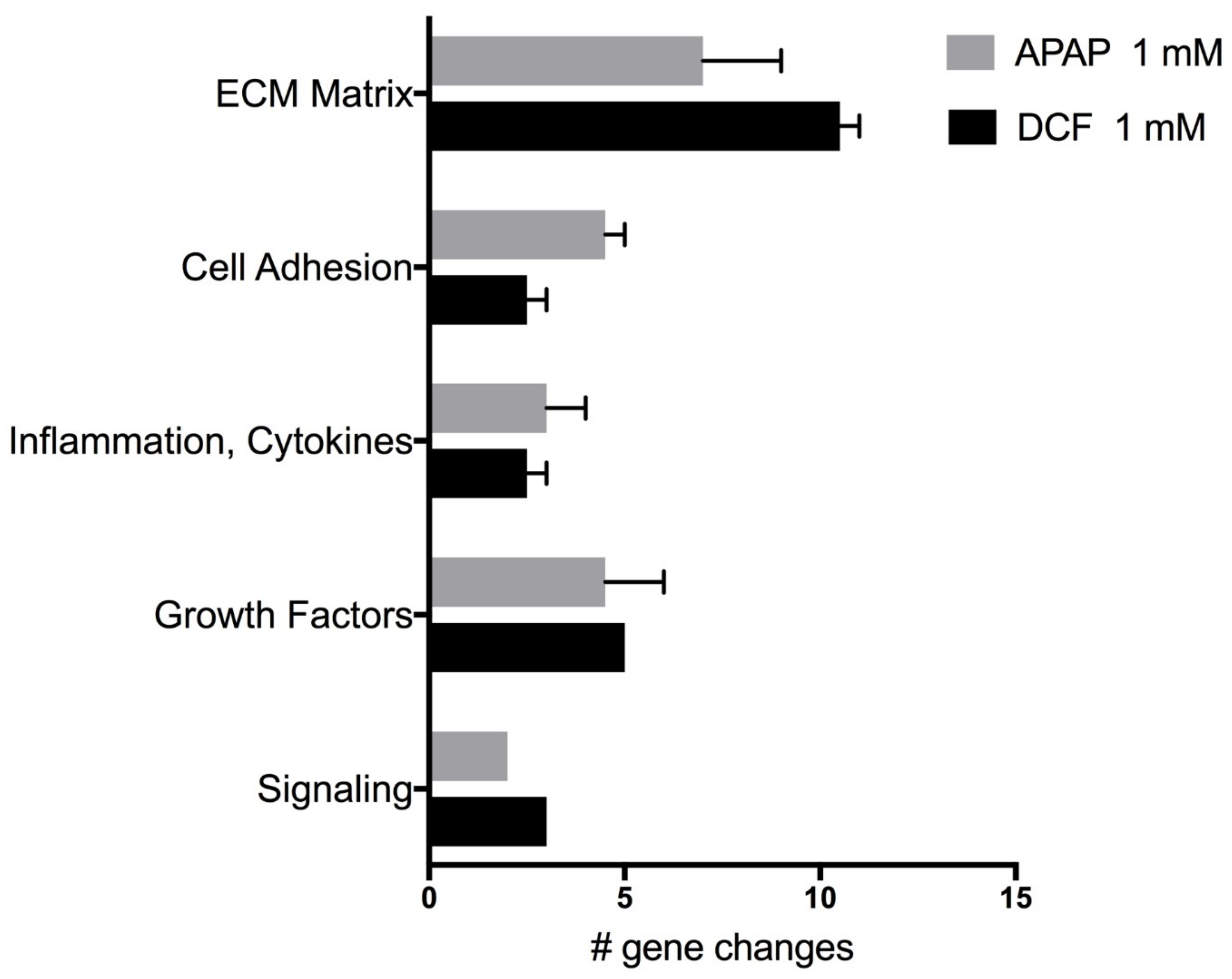
2.4. Wound Repair
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Biologicals
4.3. Human Liver Slice Cultures
4.4. Functional Assays
4.5. Morphology
4.6. Gene Arrays
4.7. PCR Arrays
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vickers, A.E.M.; Fisher, R.L. Human liver slices to investigate injury and repair. Appl. In Vitro Toxicol. 2018, 4, 280–287. [Google Scholar] [CrossRef]
- Vickers, A.E.M.; Fisher, R.L. Evaluation of drug-induced injury and human response in precision-cut tissue slices. Xenobiotica 2013, 43, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Hillier, C.; Bunton, D. Functional human tissue assays. Drug Discov. Today 2007, 12, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.E.; Marciniak, S.J. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 2. Protein misfolding and ER stress. Am. J. Physiol. Cell Physiol. 2014, 307, C657–C670. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Erbay, E. ER-mitochondrial communication gets stressful. Sci. Transl. Med. 2014, 6, 267ec213. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; Maggs, J.L.; Pirmohamed, M.; Williams, D.P. The role of metabolic activation in drug-induced hepatotoxicity. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 177–202. [Google Scholar] [CrossRef]
- Tang, W. Drug metabolite profiling and elucidation of drug-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2007, 3, 407–420. [Google Scholar] [CrossRef]
- O’Brien, P.J. Peroxidases. Chem. Biol. Interact. 2000, 129, 113–139. [Google Scholar] [CrossRef]
- Tafazoli, S.; O’Brien, P.J. Peroxidases: A role in the metabolism and side effects of drugs. Drug Discov. Today 2005, 10, 617–625. [Google Scholar] [CrossRef]
- Erve, J.C. Chemical toxicology: Reactive intermediates and their role in pharmacology and toxicology. Expert Opin. Drug Metab. Toxicol. 2006, 2, 923–946. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.; Fischer, V.; Connors, S.; Fisher, R.L.; Baldeck, J.P.; Maurer, G.; Brendel, K. Cyclosporin A metabolism in human liver, kidney, and intestine slices. Comparison to rat and dog slices and human cell lines. Drug Metab. Dispos. 1992, 20, 802–809. [Google Scholar] [PubMed]
- Vickers, A.E. Tissue slices for the evaluation of metabolism-based toxicity with the example of diclofenac. Chem. Biol. Interact. 2009, 179, 9–16. [Google Scholar] [CrossRef]
- Mehendale, H.M. Tissue repair: An important determinant of final outcome of toxicant-induced injury. Toxicol. Pathol. 2005, 33, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem. Res. Toxicol. 2009, 22, 1376–1385. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol. In Vitro 2016, 31, 93–102. [Google Scholar] [CrossRef]
- Regenthal, R.; Krueger, M.; Koeppel, C.; Preiss, R. Drug levels: Therapeutic and toxic serum/plasma concentrations of common drugs. J. Clin. Monit. 1999, 15, 529–544. [Google Scholar] [CrossRef]
- Watkins, P.B.; Kaplowitz, N.; Slattery, J.T.; Colonese, C.R.; Colucci, S.V.; Stewart, P.W.; Harris, S.C. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: A randomized controlled trial. JAMA 2006, 296, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-induced hepatotoxicity: A comprehensive update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar]
- Miyatake, S.; Ichiyama, H.; Kondo, E.; Yasuda, K. Randomized clinical comparisons of diclofenac concentration in the soft tissues and blood plasma between topical and oral applications. Br. J. Clin. Pharmacol. 2008, 67, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.L.; Vickers, A.E.M. Preparation and culture of precision-cut organ slices from human and animal. Xenobiotica 2013, 43, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.M.; Saulnier, M.; Cruz, E.; Merema, M.T.; Rose, K.; Bentley, P.; Olinga, P. Organ slice viability extended for pathway characterization: An in vitro model to investigate fibrosis. Toxicol. Sci. 2004, 82, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.E.; Fisher, R.; Olinga, P.; Dial, S. Repair pathways evident in human liver organ slices. Toxicol. In Vitro 2011, 25, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Miyajima, A. Liver regeneration and fibrosis after inflammation. Inflamm. Regen. 2016, 36, 19–24. [Google Scholar] [CrossRef]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Invest. 2018, 128, 85–96. [Google Scholar] [CrossRef]
- Chou, Y.-H.; Pan, S.-Y.; Yang, C.-H.; Lin, S.-L. Stem cells and kidney regeneration. J. Formosan. Med. Assoc. 2014, 113, 201–209. [Google Scholar] [CrossRef]
- Vickers, A.E.M.; Ulyanov, A.V.; Fisher, R.L. Liver effects of clinical drugs differentiated in human liver slices. Int. J. Mol. Sci. 2017, 18, 574. [Google Scholar] [CrossRef]
- LiverTox. Available online: https://livertox.nlm.nih.gov/ (accessed on 15 September 2018).
- Nguyen, K.D.; Sundaram, V.; Ayoub, W.S. Atypicial causes of cholestasis. World J. Gastroenterol. 2014, 20, 9418–9426. [Google Scholar] [PubMed]
- Sriuttha, P.; Sirichanchuen, B.; Permsuwan, U. Hepatotoxicity of nonsteroidal anti-inflammatory drugs: A systemic review of randomized controlled trials. Int. J. Hepatol. 2018. [CrossRef] [PubMed]
- Pessayre, D.; Mansouri, A.; Haouzi, D.; Fromenty, B. Hepatotoxicity due to mitochondrial dysfunction. Cell Biol. Toxicol. 1999, 15, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, M.I.; Manjabacas, M.C.; Garcia-Carmona, F.; Valero, E. Mechanism of acetaminophen oxidation by the peroxidase-like activity of methemoglobin. Chem. Res. Toxicol. 2009, 22, 1841–1850. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Lim, P.L. Mitochondrial abnormalities—a link to idiosyncratic drug hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources pathophysiological role and therapeutic potential. Redox. Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Liebler, D.C. Mitochondrial protein targets of thiol-reactive electrophiles. Chem. Res. Toxicol. 2008, 21, 796–804. [Google Scholar] [CrossRef]
- Kretz-Rommel, A.; Boelsterli, U.A. Selective protein adducts to membrane proteins in cultured rat hepatocytes exposed to diclofenac: Radiochemical and immunochemical analysis. Mol. Pharmacol. 1994, 45, 237–244. [Google Scholar]
- Bajt, M.L.; Ramachandran, A.; Yan, H.M.; Lebofsky, M.; Farhood, A.; Lemasters, J.J.; Jaeschke, H. Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicol. Sci 2011, 122, 598–605. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- Baines, C.P. Role of mitochondrion in programmed necrosis. Front. in Physiol. 2010, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 291, G1–G7. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Acetaminophen Information. Available online: http://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm165107.htm (accessed on 15 September 2018).
- Dragomir, A.C.; Sun, R.; Mishin, V.; Hall, L.B.; Laskin, J.D.; Laskin, D.L. Role of galectin-3 in acetaminophen-induced hepatotoxicity and inflammatory mediator production. Toxicol. Sci 2012, 127, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Herrman, J.E.; Fisher, R.L.; Vickers, A.E.M. The delay of corneal wound healing by diclofenac in a human ex vivo front of the eye model and rabbit models. Appl. In Vitro Toxicol. 2016, 2, 37–48. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Category | Human Liver | Human Kidney | ||||
|---|---|---|---|---|---|---|
| Day 2 | Day 3 | Day 4 | Day 2 | Day 3 | Day 4 | |
| Extracellular Matrix | ||||||
| Collagen type IV α1 | 2.3 | 3.4 | 4.6 | |||
| Collagen type IV α2 | 1.9 | 3.3 | 4.0 | |||
| Collagen type V α1 | 2.0 | 2.7 | 3.7 | 1.1 | 1.1 | 1.8 |
| Collagen type V α2 | 2.1 | 3.4 | 4.9 | 1.2 | 1.9 | 3.7 |
| Collagen type VI α3 | 2.6 | 5.8 | 7.3 | 1.8 | 2.6 | 4.0 |
| Collagen type XV α1 | 3.8 | 8.3 | 9.9 | 1.9 | 2.5 | 4.0 |
| Laminin β1 | 1.6 | 2.4 | 2.9 | |||
| Laminin γ1 | 1.6 | 1.4 | 2.5 | |||
| Collagen catabolism | ||||||
| Matrix Metalloproteinase 1 | 6.8 | 14.0 | 19.4 | |||
| Matrix Metalloproteinase 2 | 1.4 | 1.6 | 2.4 | |||
| Matrix Metalloproteinase 3 | 1.6 | 2.6 | 3.6 | |||
| Matrix Metalloproteinase 7 | 1.6 | 4.5 | 5.7 | 1.1 | 1.8 | 2.2 |
| Matrix Metalloproteinase 12 | 1.6 | 1.8 | 4.9 | |||
| Matrix metalloproteinase 14 | 1.5 | 1.5 | 2.0 | |||
| TIMP-1 | 1.2 | 1.3 | 1.5 | |||
| Renin | 1.1 | 1.8 | 3.5 | |||
| Cell Growth, Cell Cycle | ||||||
| EGF | 2.4 | 2.6 | 3.0 | |||
| Epiregulin | 11.1 | 8.4 | 6.8 | |||
| TGF-β | 1.8 | 2.3 | 3.9 | 1.3 | 1.5 | 2.0 |
| Cell division cycle 2 | 4.9 | 21.6 | 18.2 | |||
| Cyclin A2 | 3.1 | 8.2 | 6.0 | |||
| Cyclin B2 | 1.6 | 4.6 | 5.7 | |||
| MCM4 | 2.9 | 3.1 | 2.5 | |||
| MCM5 | 6.5 | 4.9 | 5.2 | |||
| NIMA | 4.8 | 23.8 | 25.7 | |||
| Kinesin family member 11 | 3.6 | 5.1 | 4.7 | |||
| Kinesin family member 14 | 1.9 | 4.5 | 3.7 | |||
| Polymerase, epsilon gamma | 2.6 | 3.1 | 3.1 | |||
| Proliferating cell nuclear antigen | 1.6 | 2.3 | 2.1 | 2.0 | 1.8 | 1.7 |
| Signal Transduction | ||||||
| IL-1β | 1.4 | 1.5 | 1.8 | |||
| IL-24 | 3.6 | 4.5 | 10.5 | |||
| Genes | APAP (1 mM) | DCF (1 mM) | ||||
|---|---|---|---|---|---|---|
| HL870 | HL871 | HL870 | HL871 | HL870 | HL871 | |
| FDR < 15% | FDR < 15% | FDR < 30% | FDR < 30% | FDR < 30% | FDR < 30% | |
| Phospholipidosis | ||||||
| ALDH1A1 | 2.2 | |||||
| ASNS | 7.1 | 7.1 | ||||
| CES2 | −3.3 | |||||
| CTSB | −4.3 | −5.5 | ||||
| FABP1 | −1.8 | −25.2 | −54.7 | |||
| GSTM4 | −3.6 | −4.2 | ||||
| HPN | −3.6 | |||||
| INHBE | −4.7 | −6.4 | −4.6 | |||
| NR0B2 | −5.2 | |||||
| S100A8 | −2.8 | −2.9 | ||||
| SERPINA3 | −1.5 | |||||
| TAGLN | −6.9 | |||||
| TIMM10B | 5.3 | |||||
| WIPI1 | 3.6 | 4.0 | ||||
| Steatosis | ||||||
| ACACA | −2.1 | |||||
| ADK | 3.5 | |||||
| ALDH1A1 | 2.2 | −6.7 | ||||
| DNM1 | −2.0 | |||||
| FASN | −2.7 | −1.5 | −19.3 | |||
| GPD1 | −5.7 | |||||
| KHK | −3.2 | −6.4 | ||||
| LPL | 1.8 | |||||
| MAPK8 | 1.2 | 4.6 | ||||
| MTTP | −14.1 | −17.7 | ||||
| PNPLA3 | −2.0 | |||||
| SCD | −2.7 | −2.5 | −11.1 | −14.2 | ||
| SREBF1 | −2.0 | −2.9 | −5.5 | −7.4 | ||
| Cholestasis | ||||||
| ABCB1 | 1.8 | 3.0 | ||||
| ABCB4 | −2.3 | |||||
| ABCC1 | −1.8 | |||||
| ABCC2 | 2.9 | 2.0 | ||||
| ABCC3 | 1.5 | |||||
| APOA5 | −1.8 | |||||
| APOE | −1.6 | |||||
| APOF | −2.5 | −1.9 | −6.7 | −12.2 | ||
| CYP7A1 | 4.5 | 3.3 | −23.1 | |||
| CYP7B1 | −8.9 | −8.2 | ||||
| ESR1 | 1.9 | |||||
| HLA−DRB1 | −12.8 | |||||
| JAG1 | −1.5 | |||||
| NR1H4 | −1.4 | −4.2 | −4.9 | |||
| NUP210 | −7.0 | |||||
| PDYN | −2.1 | |||||
| SLC10A | −8.3 | |||||
| SLC51A | −2.8 | −8.8 | ||||
| SLCO1A2 | −19.2 | −54.6 | ||||
| UGT1A1 | −5.6 | −8.8 | ||||
| UGT2B4 | 3.2 | 2.7 | ||||
| Fatty Acid Metabolism | ||||||
| ACAA1 | 1.4 | |||||
| ACAD11 | 1.5 | |||||
| ACADSB | −6.2 | |||||
| ACAT1 | −1.5 | −1.7 | ||||
| ACAT2 | −20.7 | |||||
| ACOT1 | −1.7 | |||||
| ACOX2 | −20.7 | |||||
| ADH1C | 3.2 | −30.6 | ||||
| CPT1B | −1.6 | |||||
| CPT2 | 1.9 | −1.7 | ||||
| ECHS1 | −4.1 | |||||
| EHHADH | −5.0 | |||||
| GCDH | 1.6 | |||||
| Oxidative Stress | ||||||
| CAT | −3.5 | |||||
| DHCR24 | −5.6 | |||||
| DUOX1 | −2.8 | −2.9 | −45.5 | |||
| DUOX2 | −2.6 | −2.3 | −16.2 | |||
| EPHX1 | 2.6 | −7.6 | ||||
| GPX1 | 1.9 | |||||
| GPX2 | 1.5 | −31.2 | −24.6 | |||
| GPX4 | 1.6 | 2.2 | ||||
| GPX7 | −1.5 | −1.5 | ||||
| GSTA3 | 2.5 | |||||
| MPO | −2.6 | |||||
| NQO1 | 1.7 | 1.7 | ||||
| POR | 1.4 | 4.1 | 2.8 | |||
| PPP1R15B | 4.5 | 3.7 | ||||
| PRDX1 | 1.5 | |||||
| PRDX2 | 1.3 | |||||
| PRDX6 | 1.4 | 1.5 | 3.0 | |||
| TXNIP | −1.5 | −1.8 | −9.1 | |||
| Mitochondria Energy | ||||||
| ACLY | −1.6 | |||||
| COX6B1 | 1.9 | |||||
| CYC1 | 1.4 | |||||
| IDH1 | 1.5 | −5.3 | ||||
| IDH2 | −2.0 | |||||
| IDH3G | 1.3 | |||||
| MDH1B | −2.4 | −2.4 | ||||
| SDHA | 1.5 | |||||
| SDHC | −3.4 | |||||
| SUCLG1 | 3.0 | |||||
| UCP2 | −7.5 | −6.0 | ||||
| Heat Shock | ||||||
| CRYAA | −5.0 | |||||
| DNAJA2 | 2.5 | |||||
| DNAJB1 | 3.1 | |||||
| DNAJB6 | 2.4 | |||||
| DNAJC3 | −1.5 | |||||
| HSF1 | 1.6 | |||||
| HSF2 | 4.1 | 4.2 | ||||
| HSP90AA1 | 1.5 | 1.3 | ||||
| HSPA1A | 1.9 | |||||
| HSPA1B | 2.1 | |||||
| HSPA9 | 3.1 | 2.8 | ||||
| HSPBAP1 | −1.6 | |||||
| HSPB6 | −1.5 | −1.6 | ||||
| ER Stress | ||||||
| AMFR | −1.4 | |||||
| ATF4 | 5.6 | 4.6 | ||||
| ATF6 | 4.4 | 2.9 | ||||
| DDIT3 | −1.5 | |||||
| DERL1 | 4.7 | |||||
| EDEM1 | −1.5 | |||||
| EIF2AK3 | −1.7 | 2.9 | 3.6 | |||
| ERO1LB | 3.5 | |||||
| FBXO6 | 2.7 | |||||
| HTRA4 | −2.3 | |||||
| SERP1 | −1.5 | 2.7 | ||||
| VCP | 1.5 | |||||
| VIMP | 4.5 | |||||
| XBP1 | −1.6 | |||||
| Metabolism | ||||||
| CYP1A2 | −47.1 | −246.2 | ||||
| CYP2B6 | 4.4 | 1.8 | −19.9 | |||
| CYP2C9 | 2.6 | |||||
| CPY2C19 | 2.0 | −33.7 | ||||
| CYP2D6 | −1.4 | −4.0 | ||||
| CYP2E1 | −1.8 | |||||
| CYP3A4 | 3.2 | |||||
| GSTA3 | −14.5 | −77.1 | ||||
| FMO2 | −8.4 | |||||
| FMO3 | −5.8 | |||||
| FMO4 | −5.6 | −4.7 | ||||
| FMO5 | −19.9 | |||||
| UGT2B4 | −4.4 | −9.4 | ||||
| DNA damage/repair | ||||||
| CDKN1A | −1.4 | |||||
| DDIT3(GADD153) | 13.8 | 17.0 | ||||
| ERCC1 | 3.8 | 4.3 | ||||
| ERCC5 | 2.3 | |||||
| MDM2 | 4.1 | |||||
| MKI67 | −52.1 | |||||
| PCNA | 1.6 | |||||
| XRCC1 | 2.5 | |||||
| Apoptosis | ||||||
| AKT1 | −1.5 | |||||
| BAD | −1.5 | |||||
| BAK1 | 3.2 | |||||
| BCL2 | −1.6 | |||||
| BCL2L11 | −2.3 | −1.4 | ||||
| CASP1 | −8.0 | |||||
| FASLG | −1.8 | |||||
| GADD45A | 23.4 | 22.9 | ||||
| MCL1 | −1.3 | |||||
| TNFSF10A | −1.3 | 2.8 | 1.9 | |||
| TNFRSF10B | 5.2 | 3.3 | ||||
| TP53 | 2.7 | |||||
| Necrosis | ||||||
| ATP6V1G2 | −2.4 | |||||
| BMF | −2.4 | −2.0 | ||||
| CCDC103 | −3.0 | |||||
| CLEC18A | −2.6 | |||||
| CYLD | 3.9 | 4.0 | ||||
| GALNT5 | −2.5 | |||||
| HSPBAP1 | −1.7 | |||||
| MAG | 1.4 | −1.6 | ||||
| PARP2 | 3.1 | 2.9 | ||||
| RAB25 | −1.8 | −3.8 | −6.3 | |||
| SPATA2 | 2.1 | 6.3 | ||||
| TNFAIP8L1 | −7.1 | −11.0 | ||||
| TXNL4B | 3.2 | 2.8 | ||||
| Inflammation/Immunotoxicity | ||||||
| AHSG | −1.5 | −4.1 | ||||
| C3 | −3.0 | −12.4 | −16.0 | |||
| C9 | −1.7 | −1.6 | −14.9 | −15.2 | ||
| CD4 | −36.7 | |||||
| CD36 | −25.8 | −23.3 | ||||
| CD80 | −29.1 | |||||
| CD86 | −1.7 | −1.6 | −62.4 | −9.5 | ||
| CTSE | −2.4 | −17.8 | ||||
| F2 | −1.4 | |||||
| HRG | 3.0 | −9.6 | −56.1 | |||
| IL1B | −43.7 | −16.6 | ||||
| IL4 | −2.1 | |||||
| IL6 | −2.3 | −2.4 | ||||
| IL10 | −1.8 | |||||
| ITGAX | −24.0 | −12.3 | ||||
| KLF1 | 4.0 | |||||
| LYZ | −1.7 | −1.5 | −20.1 | |||
| METAP2 | 1.8 | |||||
| PON1 | −8.2 | |||||
| POU3F3 | 1.4 | |||||
| PTPRC | −1.6 | |||||
| TNF | 2.1 | −10.1 | ||||
| Gene Categories | APAP (1 mM) % Genes/Category | DCF (1 mM) % Genes/Category | ||||||
|---|---|---|---|---|---|---|---|---|
| HL870 48 h 72 h | HL871 48 h 72 h | HL870 48 h 72 h | HL871 48 h 72 h | |||||
| Cholestasis, Steatosis, Phospholipidosis | 29.0 | 27.3 | 19.6 | 33.3 | 20.9 | 29.2 | 19.6 | 33.8 |
| Fatty acid Metabolism | 9.7 | 9.1 | 6.5 | 4.8 | 7.0 | 1.1 | 5.9 | 8.1 |
| Oxidative Stress | 12.9 | 14.3 | 8.7 | 11.1 | 9.3 | 9.0 | 5.9 | 5.4 |
| Mitochondria Energy | 9.7 | 6.5 | 4.3 | 5.2 | 2.3 | 2.3 | 2.0 | 4.1 |
| Heat Shock, ER Stress | 12.9 | 11.7 | 15.2 | 14.3 | 25.6 | 18.0 | 27.5 | 8.1 |
| Metabolism | 9.7 | 6.5 | 6.5 | 3.2 | 7.0 | 6.7 | 9.8 | 13.5 |
| DNA Damage/Repair | 0 | 2.6 | 4.4 | 0 | 4.7 | 6.7 | 3.9 | 2.7 |
| Apoptosis | 3.2 | 5.2 | 10.9 | 6.4 | 14.0 | 5.6 | 15.7 | 6.8 |
| Necrosis | 6.5 | 5.2 | 15.2 | 12.7 | 0 | 6.7 | 2.0 | 5.4 |
| Inflammation/Immunotoxicity | 6.5 | 11.7 | 8.7 | 7.9 | 9.3 | 14.6 | 7.8 | 12.2 |
| Number of significant gene changes | 31 | 77 | 46 | 63 | 43 | 89 | 51 | 74 |
| Genes | APAP (1 mM) | DCF (1 mM) | ||||
|---|---|---|---|---|---|---|
| HL871 | HL870 | HL871 | HL871 | HL870 | HL871 | |
| FDR < 15% | FDR < 30% | FDR < 30% | FDR < 15% | FDR < 30% | FDR < 30% | |
| ECM Structural | ||||||
| COL1A1 | −2.4 | −73.9 | −38.3 | |||
| COL1A2 | −1.6 | −20.1 | −178.8 | |||
| COL3A1 | −1.8 | −1.8 | −22.8 | −140.2 | ||
| COL5A2 | −1.4 | −1.8 | −8.2 | |||
| COL5A3 | −2.9 | −2.2 | ||||
| VTN | −2.2 | −3.3 | ||||
| ECM Remodeling | ||||||
| FGA | −12.9 | −6.4 | ||||
| MMP1 | 12.8 | 25.1 | ||||
| MMP7 | −1.6 | −1.9 | −56.4 | −1.3 | ||
| MMP9 | −17.3 | |||||
| PLAU | −1.7 | −11.4 | ||||
| PLAUR | −1.7 | −4.5 | ||||
| PLG | −10.5 | |||||
| SERPINE1 | −1.5 | |||||
| TIMP1 | −4.0 | |||||
| Cell Adhesion | ||||||
| ITGA1 | −2.1 | −1.6 | ||||
| ITGA2 | −1.8 | −1.4 | ||||
| ITGA3 | −1.8 | |||||
| ITGA4 | −20.8 | −26.2 | ||||
| ITGA5 | −1.9 | −1.7 | −3.6 | −3.5 | ||
| ITGB6 | −3.1 | |||||
| TAGLN | −11.2 | −4.2 | ||||
| Inflammation, Cytokines | ||||||
| CCL2 | −22.7 | |||||
| CXCL1 | −1.8 | |||||
| CXCL2 | −5.7 | |||||
| CXCL5 | −2.0 | −38.7 | −12.2 | |||
| CXCL11 | 2.2 | |||||
| IFNG | 2.3 | |||||
| IL1B | −56.0 | −21.7 | ||||
| IL6 | −3.6 | −1.9 | ||||
| Growth Factors | ||||||
| CSF3 | 53.9 | 50.3 | ||||
| EGF | 2.1 | |||||
| FGF2 | 9.0 | 5.3 | ||||
| FGF7 | −1.7 | −13.5 | ||||
| HBEGF | 4.5 | |||||
| HGF | −1.6 | |||||
| PDGA | 3.8 | |||||
| TGFB1 | −4.7 | |||||
| TNF | 1.8 | −10.3 | ||||
| VEGFA | −1.6 | −1.6 | ||||
| Signaling | ||||||
| PTGS2 | −1.7 | −2.2 | 25.4 | 29.0 | ||
| WISP1 | −15.2 | −11.1 | ||||
| Donor | Age/Sex/Race | Cold Ischemia (h) | K+ μmols/g Wet Weight 24 h | ATP nmols/mg Wet Weight 24 h | GSH nmols/mg Wet Weight 24 h |
|---|---|---|---|---|---|
| HL714 | M/C/46 | 12 | 75.9 | 16.8 | 5.6 |
| HK3 | F/C/49 | 12.5 | 85.0 | 8.3 | 21.8 |
| HL870 | M/C/21 | 14 | 80.4 | 15.0 | 10.6 |
| HL871 | F/A/28 | 16 | 78.9 | 13.5 | 16.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vickers, A.E.M.; Ulyanov, A.V.; Fisher, R.L. Progression of Repair and Injury in Human Liver Slices. Int. J. Mol. Sci. 2018, 19, 4130. https://doi.org/10.3390/ijms19124130
Vickers AEM, Ulyanov AV, Fisher RL. Progression of Repair and Injury in Human Liver Slices. International Journal of Molecular Sciences. 2018; 19(12):4130. https://doi.org/10.3390/ijms19124130
Chicago/Turabian StyleVickers, Alison E. M., Anatoly V. Ulyanov, and Robyn L. Fisher. 2018. "Progression of Repair and Injury in Human Liver Slices" International Journal of Molecular Sciences 19, no. 12: 4130. https://doi.org/10.3390/ijms19124130
APA StyleVickers, A. E. M., Ulyanov, A. V., & Fisher, R. L. (2018). Progression of Repair and Injury in Human Liver Slices. International Journal of Molecular Sciences, 19(12), 4130. https://doi.org/10.3390/ijms19124130