Molecular Mechanisms Underlying the Link between Diet and DNA Methylation



Abstract
1. Introduction
2. Mechanisms of DNA Methylation
2.1. What is DNA Methylation?
2.2. Why Are DNA Methyltransferases Essential for DNA Methylation?
2.3. How DNMT1 Functions in DNA Methylation
2.4. How DNMT3 Functions in DNA Methylation
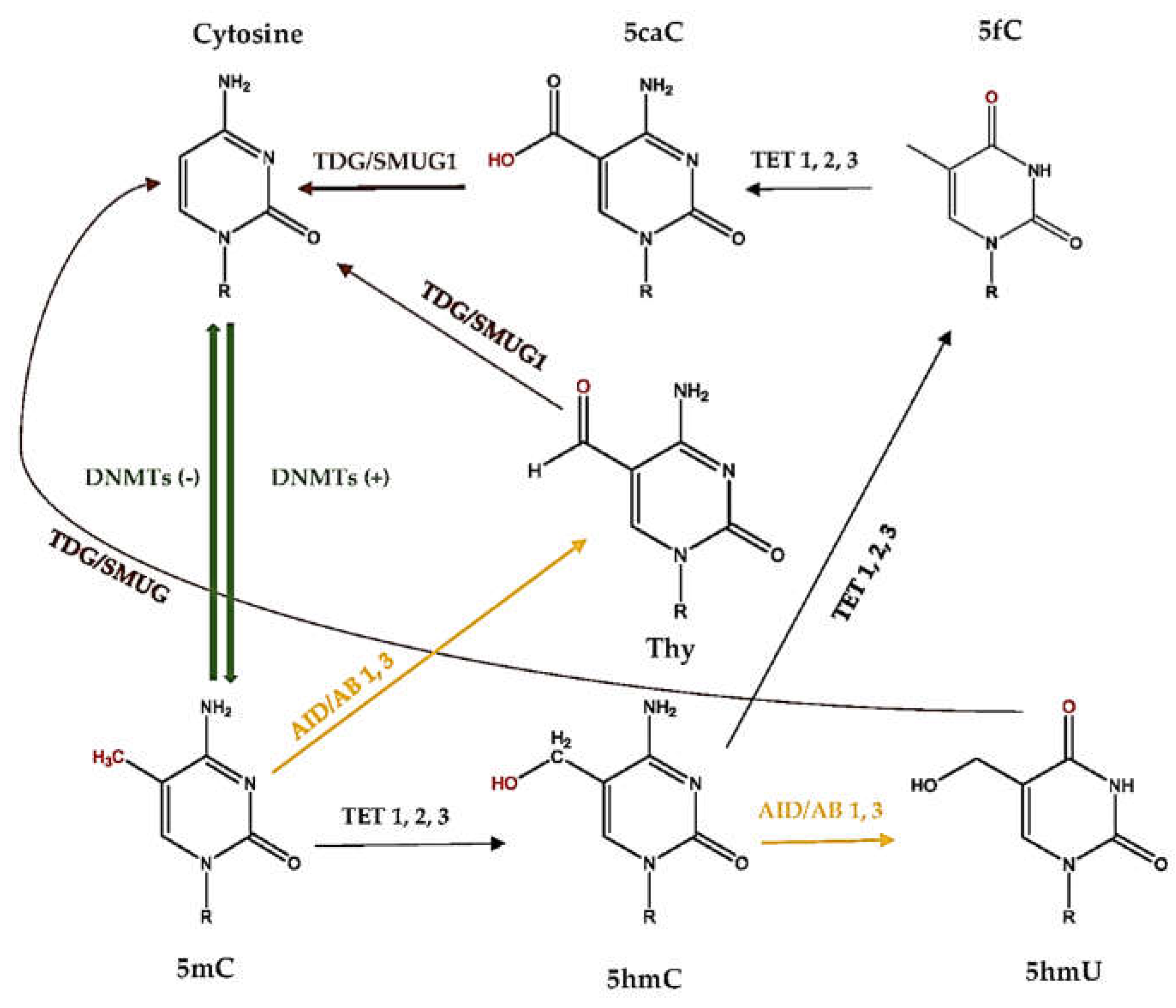
2.5. What Is DNA Demethylation?
2.6. How Active DNA Demethylation Occurs
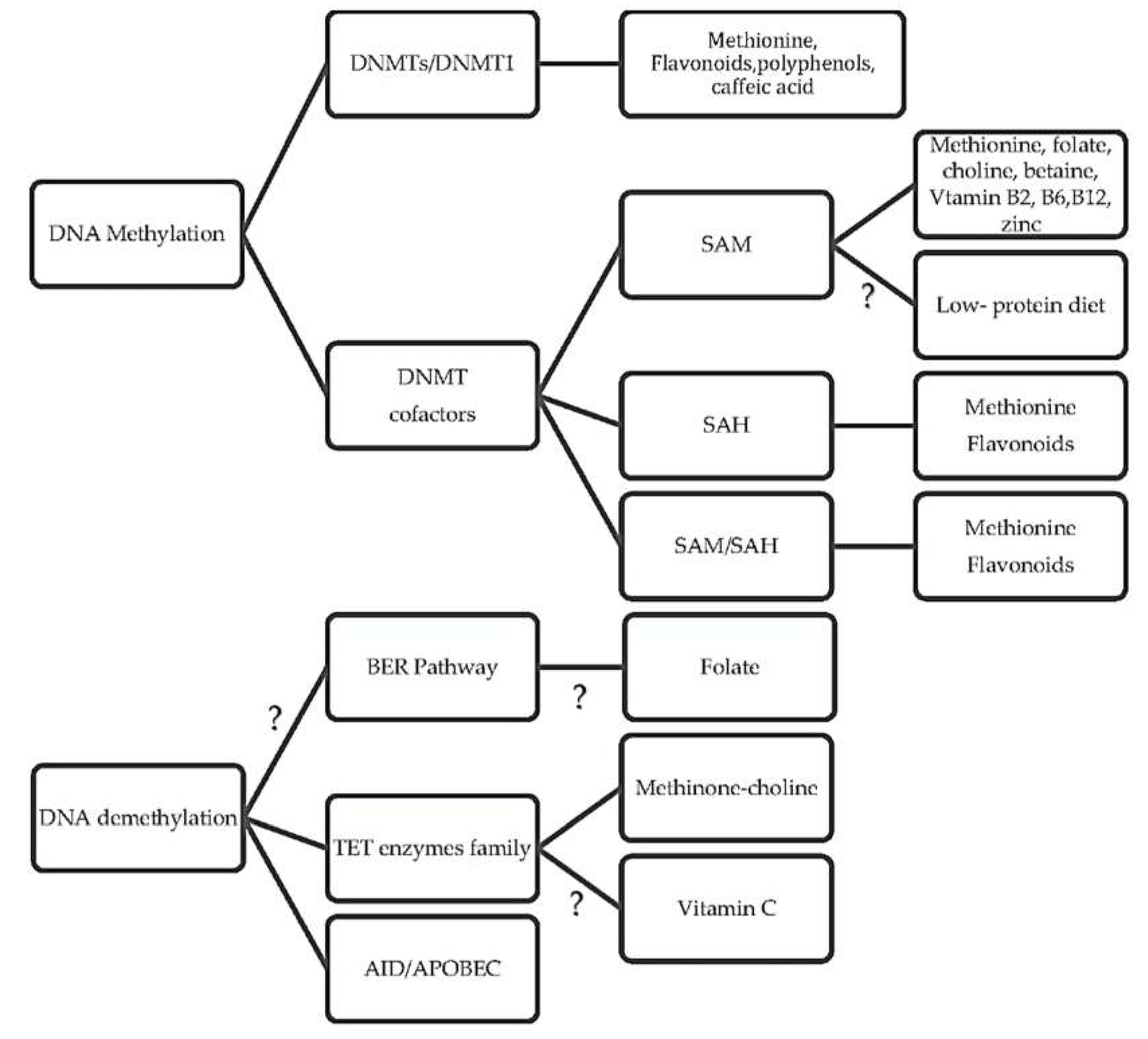
3. What Are the Underlying Mechanisms of Diet and DNA Methylation?
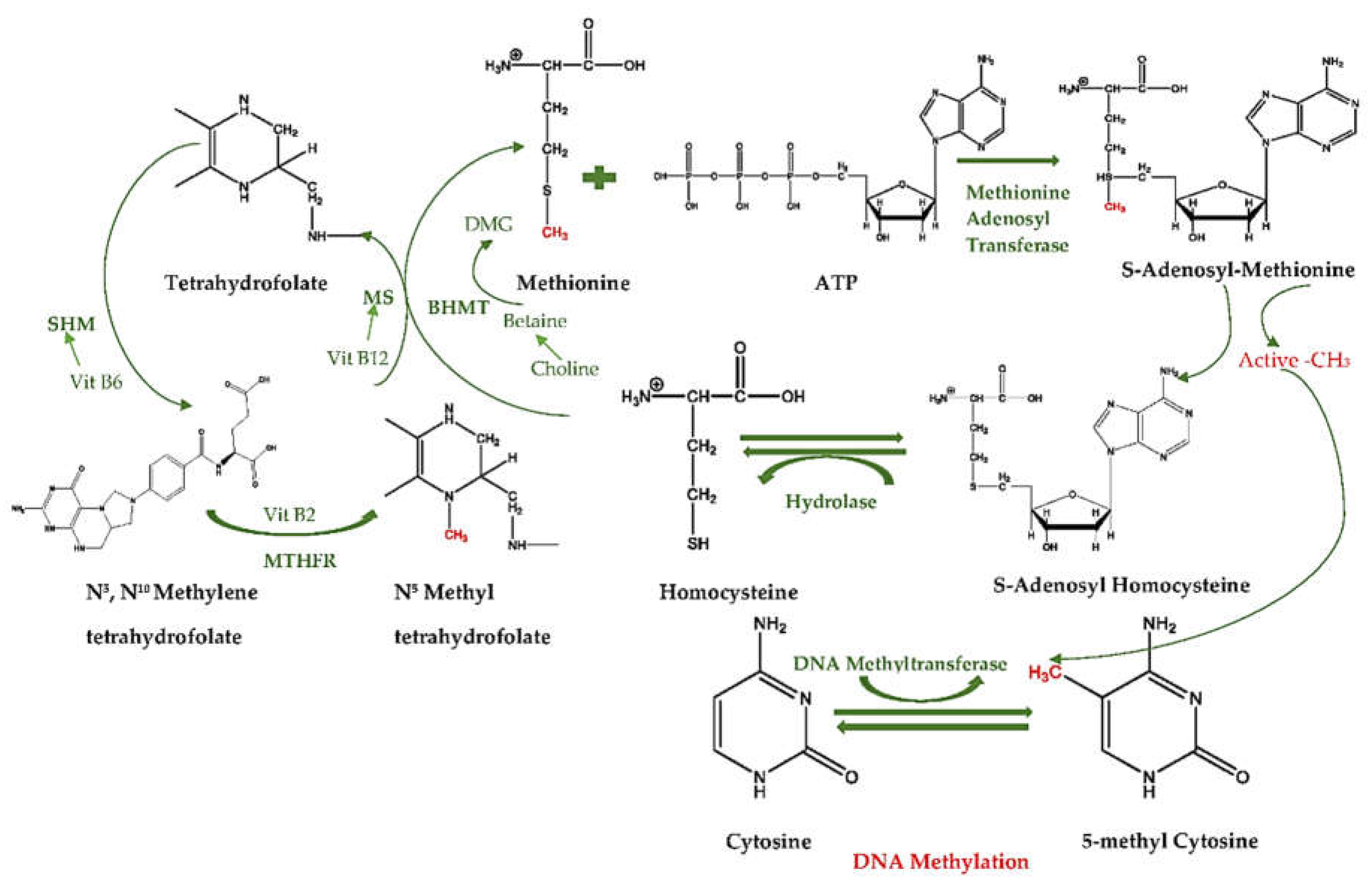
3.1. How Diet Influences Methylation Cycle and Methyl Donors
3.2. What Are the Diet-Related Cofactor and Enzyme Activities in One-Carbon Cycles?
3.3. How Diet Affects the DNA Methyltransferase Activity
3.4. Is There a Link between Diet and DNA Demethylation?
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| MTHFR | Methylenetetrahydrofolate reductase |
| SHMT | Serine hydroxyl methyltransferase |
| MS | Methionine synthase |
| BHMT | Betaine homocysteine methyltransferase |
| DMG | Dimethylglycine |
| 5mC | 5-methylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
| 5hmU | 5-hydroxymethyluracil |
| 5fC | 5-formylcytosine |
| 5caC | 5-carboxylcytosine |
| Thy | Thymine |
| DNMT | DNA methyltransferase |
| TET | Ten-eleven translocation |
| AID | Activation-induced deaminase |
| TDG | Thymine DNA glycosylase |
| SMUG1 | Single-strand selective monofunctional uracil DNA glycosylase |
| SAM | S-adenosyl-l-methionine |
| SAH | S-adenosylhomocysteine |
| BER | Base excision repair |
| RFTS | Replication foci targeting sequence |
| BAH | Bromo-adjacent homology |
| UHRF1 | Interacting protein E3 ubiquitin-protein ligase |
| NER | Nucleotide excision repair |
| dTTP | Deoxythymidine triphosphate |
| EGCG | Epigallocatechin-3-gallate |
References
- Zhang, N. Epigenetic modulation of DNA methylation by nutrition and its mechanisms in animals. Anim. Nutr. 2015, 1, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Bheemanaik, S.; Reddy, Y.V.; Rao, D.N. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem. J. 2006, 399, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Guo, J.U.; Ming, G.L.; Song, H. DNA excision repair proteins and Gadd45 as molecular players for active DNA demethylation. Cell Cycle 2009, 8, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, M.D. Nutritional epigenetics. ILAR J. 2012, 53, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Ratel, D.; Ravanat, J.-L.; Berger, F.; Wion, D. N6-methyladenine: The other methylated base of DNA. Bioessays 2006, 28, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Li, Y.; Robertson, K.D. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman and Co.: New York, NY, USA, 2002. [Google Scholar]
- Zydowsky, T.M.; Courtney, L.F.; Frasca, V.; Kobayashi, K.; Shimizu, H.; Yuen, L.D.; Matthews, R.G.; Benkovic, S.J.; Floss, H.G. Stereochemical analysis of the methyl transfer catalyzed by cobalamin-dependent methionine synthase from Escherichia coli B. J. Am. Chem. Soc. 1986, 108, 3152–3153. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Obeid, R. The Metabolic Burden of Methyl Donor Deficiency with Focus on the Betaine Homocysteine Methyltransferase Pathway. Nutrients 2013, 5, 3481–3495. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Martin, J.J.; Harris, B.J.; Kyle, W.E. Regulation of hepatic betaine-homocysteine methyltransferase by dietary betaine. J. Nutr. 1983, 113, 519–521. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- He, X.J.; Chen, T.; Zhu, J.K. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011, 21, 442–465. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, P.; Fang, L.; Zhu, H.; Xu, L.; Cheng, H.; Zhang, J.; Li, F.; Feng, Y.; Li, Y.; et al. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discov. 2016, 2, 16007. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, E. Structure and Function of Eukaryotic DNA Methyltransferases. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2004; Volume 60, pp. 55–89. [Google Scholar]
- Rhee, I.; Jair, K.-W.; Yen, R.-W.C.; Lengauer, C.; Herman, J.G.; Kinzler, K.W.; Vogelstein, B.; Baylin, S.B.; Schuebel, K.E. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000, 404, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Teplova, M.; Ishibe-Murakami, S.; Patel, D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012, 335, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Bacolla, A.; Wells, R.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase I. Expression, purification, and comparision of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Leonhardt, H.; Page, A.W.; Weier, H.U.; Bestor, T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef]
- Song, J.; Rechkoblit, O.; Bestor, T.H.; Patel, D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011, 331, 1036–1040. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef]
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2010, 17, 2141–2151. [Google Scholar] [CrossRef]
- Raddatz, G.; Gao, Q.; Bender, S.; Jaenisch, R.; Lyko, F. Dnmt3a protects active chromosome domains against cancer-associated hypomethylation. PLoS Genet. 2012, 8, e1003146. [Google Scholar] [CrossRef]
- Steine, E.J.; Ehrich, M.; Bell, G.W.; Raj, A.; Reddy, S.; van Oudenaarden, A.; Jaenisch, R.; Linhart, H.G. Genes methylated by DNA methyltransferase 3b are similar in mouse intestine and human colon cancer. J. Clin. Investig. 2011, 121, 1748–1752. [Google Scholar] [CrossRef]
- Linhart, H.G.; Lin, H.; Yamada, Y.; Moran, E.; Steine, E.J.; Gokhale, S.; Lo, G.; Cantu, E.; Ehrich, M.; He, T.; et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007, 21, 3110–3122. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Bochtler, M.; Kolano, A.; Xu, G.L. DNA demethylation pathways: Additional players and regulators. Bioessays 2017, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Qin, T.; Barton, M.C.; Jelinek, J.; Issa, J.-P.J. Minimal role of base excision repair in TET-induced global DNA demethylation in HEK293T cells. Epigenetics 2015, 10, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.; Dean, W.; Feng, S.; Cokus, S.J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105. [Google Scholar] [CrossRef]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Wang, K.Y.; Shen, C.K. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J. Biol. Chem. 2013, 288, 9084–9091. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.D.; Allegrucci, C.; Singh, R.; Gardner, D.S.; Sebastian, S.; Bispham, J.; Thurston, A.; Huntley, J.F.; Rees, W.D.; Maloney, C.A.; et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc. Natl. Acad. Sci. USA 2007, 104, 19351–19356. [Google Scholar] [CrossRef]
- Waterland, R.A. Assessing the effects of high methionine intake on DNA methylation. J. Nutr. 2006, 136, 1706S–1710S. [Google Scholar] [CrossRef]
- Amarasekera, M.; Martino, D.; Ashley, S.; Harb, H.; Kesper, D.; Strickland, D.; Saffery, R.; Prescott, S.L. Genome-wide DNA methylation profiling identifies a folate-sensitive region of differential methylation upstream of ZFP57-imprinting regulator in humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4068–4076. [Google Scholar] [CrossRef]
- Yu, H.L.; Dong, S.; Gao, L.F.; Li, L.; Xi, Y.D.; Ma, W.W.; Yuan, L.H.; Xiao, R. Global DNA methylation was changed by a maternal high-lipid, high-energy diet during gestation and lactation in male adult mice liver. Br. J. Nutr. 2015, 113, 1032–1039. [Google Scholar] [CrossRef]
- Altmann, S.; Murani, E.; Schwerin, M.; Metges, C.C.; Wimmers, K.; Ponsuksili, S. Dietary protein restriction and excess of pregnant German Landrace sows induce changes in hepatic gene expression and promoter methylation of key metabolic genes in the offspring. J. Nutr. Biochem. 2013, 24, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Zhou, D.; Moody, L.; Lezmi, S.; Chen, H.; Pan, Y.X. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol. Genom. 2015, 47, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Jirtle, R.L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef]
- Cravo, M.L.; Pinto, A.G.; Chaves, P.; Cruz, J.A.; Lage, P.; Nobre Leitao, C.; Costa Mira, F. Effect of folate supplementation on DNA methylation of rectal mucosa in patients with colonic adenomas: Correlation with nutrient intake. Clin. Nutr. 1998, 17, 45–49. [Google Scholar] [CrossRef]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. 2011, 202, 103–118. [Google Scholar] [CrossRef]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef] [PubMed]
- Sibani, S.; Melnyk, S.; Pogribny, I.P.; Wang, W.; Hiou-Tim, F.; Deng, L.; Trasler, J.; James, S.J.; Rozen, R. Studies of methionine cycle intermediates (SAM, SAH), DNA methylation and the impact of folate deficiency on tumor numbers in Min mice. Carcinogenesis 2002, 23, 61–65. [Google Scholar] [CrossRef]
- Bacolla, A.; Pradhan, S.; Roberts, R.J.; Wells, R.D. Recombinant human DNA (cytosine-5) methyltransferase. II. Steady-state kinetics reveal allosteric activation by methylated dna. J. Biol. Chem. 1999, 274, 33011–33019. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Poirier, L.A. Tissue levels of S-adenosylmethionine and S-adenosylhomocysteine in rats fed methyl-deficient, amino acid-defined diets for one to five weeks. Carcinogenesis 1983, 4, 1051–1057. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Ross, S.A.; Wise, C.; Pogribna, M.; Jones, E.A.; Tryndyak, V.P.; James, S.J.; Dragan, Y.P.; Poirier, L.A. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat. Res. 2006, 593, 80–87. [Google Scholar] [CrossRef]
- Jhaveri, M.S.; Wagner, C.; Trepel, J.B. Impact of Extracellular Folate Levels on Global Gene Expression. Mol. Pharmacol. 2001, 60, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Wallwork, J.C.; Duerre, J.A. Effect of zinc deficiency on methionine metabolism, methylation reactions and protein synthesis in isolated perfused rat liver. J. Nutr. 1985, 115, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Van Straten, E.M.; Bloks, V.W.; Huijkman, N.C.; Baller, J.F.; van Meer, H.; Lutjohann, D.; Kuipers, F.; Plosch, T. The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R275–282. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Slater-Jefferies, J.; Torrens, C.; Phillips, E.S.; Hanson, M.A.; Lillycrop, K.A. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 2007, 97, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Ingrosso, D.; Cimmino, A.; Perna, A.F.; Masella, L.; De Santo, N.G.; De Bonis, M.L.; Vacca, M.; D’Esposito, M.; D’Urso, M.; Galletti, P.; et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet 2003, 361, 1693–1699. [Google Scholar] [CrossRef]
- Farias, N.; Ho, N.; Butler, S.; Delaney, L.; Morrison, J.; Shahrzad, S.; Coomber, B.L. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 2015, 26, 818–826. [Google Scholar] [CrossRef]
- Tremolizzo, L.; Carboni, G.; Ruzicka, W.B.; Mitchell, C.P.; Sugaya, I.; Tueting, P.; Sharma, R.; Grayson, D.R.; Costa, E.; Guidotti, A. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc. Natl. Acad. Sci. USA 2002, 99, 17095–17100. [Google Scholar] [CrossRef]
- Dong, E.; Agis-Balboa, R.C.; Simonini, M.V.; Grayson, D.R.; Costa, E.; Guidotti, A. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 12578–12583. [Google Scholar] [CrossRef]
- Devlin, A.M.; Arning, E.; Bottiglieri, T.; Faraci, F.M.; Rozen, R.; Lentz, S.R. Effect of Mthfr genotype on diet-induced hyperhomocysteinemia and vascular function in mice. Blood 2004, 103, 2624–2629. [Google Scholar] [CrossRef]
- Finkelstein, J.D. The metabolism of homocysteine: Pathways and regulation. Eur. J. Pediatr. 1998, 157, S40–S44. [Google Scholar] [CrossRef]
- Dominguez-Salas, P.; Moore, S.E.; Cole, D.; da Costa, K.A.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Innis, S.M.; Waterland, R.A.; Zeisel, S.H.; et al. DNA methylation potential: Dietary intake and blood concentrations of one-carbon metabolites and cofactors in rural African women. Am. J. Clin. Nutr. 2013, 97, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.H.; Stabler, S.P.; Lindenbaum, J. Serum betaine, N,N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism 1993, 42, 1448–1460. [Google Scholar] [CrossRef]
- Jacobs, R.L.; Stead, L.M.; Devlin, C.; Tabas, I.; Brosnan, M.E.; Brosnan, J.T.; Vance, D.E. Physiological regulation of phospholipid methylation alters plasma homocysteine in mice. J. Biol. Chem. 2005, 280, 28299–28305. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Zola, T.; daCosta, K.A.; Pomfret, E.A. Effect of choline deficiency on S-adenosylmethionine and methionine concentrations in rat liver. Biochem. J. 1989, 259, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Imbard, A.; Smulders, Y.M.; Barto, R.; Smith, D.E.; Kok, R.M.; Jakobs, C.; Blom, H.J. Plasma choline and betaine correlate with serum folate, plasma S-adenosyl-methionine and S-adenosyl-homocysteine in healthy volunteers. Clin. Chem. Lab. Med. 2013, 51, 683–692. [Google Scholar] [CrossRef]
- Kim, Y.I.; Miller, J.W.; da Costa, K.A.; Nadeau, M.; Smith, D.; Selhub, J.; Zeisel, S.H.; Mason, J.B. Severe folate deficiency causes secondary depletion of choline and phosphocholine in rat liver. J. Nutr. 1994, 124, 2197–2203. [Google Scholar] [CrossRef]
- Horne, D.W.; Cook, R.J.; Wagner, C. Effect of dietary methyl group deficiency on folate metabolism in rats. J. Nutr. 1989, 119, 618–621. [Google Scholar] [CrossRef]
- Teng, Y.W.; Mehedint, M.G.; Garrow, T.A.; Zeisel, S.H. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J. Biol. Chem. 2011, 286, 36258–36267. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Shivapurkar, N.; Poirier, L.A. Hypomethylation of hepatic nuclear DNA in rats fed with a carcinogenic methyl-deficient diet. Biochem. J. 1984, 218, 987–990. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef]
- Wainfan, E.; Poirier, L.A. Methyl groups in carcinogenesis: Effects on DNA methylation and gene expression. Cancer Res. 1992, 52, 2071s–2077s. [Google Scholar] [PubMed]
- Pogribny, I.P.; James, S.J.; Beland, F.A. Molecular alterations in hepatocarcinogenesis induced by dietary methyl deficiency. Mol. Nutr. Food Res. 2012, 56, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Umegaki, K.; Higuchi, M.; Thomas, P.; Fenech, M. Methylenetetrahydrofolate reductase C677T polymorphism, folic acid and riboflavin are important determinants of genome stability in cultured human lymphocytes. J. Nutr. 2004, 134, 48–56. [Google Scholar] [CrossRef]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef]
- Hustad, S.; Ueland, P.M.; Vollset, S.E.; Zhang, Y.; Bjørke-Monsen, A.L.; Schneede, J. Riboflavin as a Determinant of Plasma Total Homocysteine: Effect Modification by the Methylenetetrahydrofolate Reductase C677T Polymorphism. Clin. Chem. 2000, 46, 1065–1071. [Google Scholar]
- Friso, S.; Choi, S.-W.; Girelli, D.; Mason, J.B.; Dolnikowski, G.G.; Bagley, P.J.; Olivieri, O.; Jacques, P.F.; Rosenberg, I.H.; Corrocher, R.; et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. USA 2002, 99, 5606–5611. [Google Scholar] [CrossRef]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Niculescu, M.D.; Haggarty, P. Nutrition in Epigenetics; Blackwell: Ames, IA, USA, 2011. [Google Scholar]
- James, S.J.; Pogribny, I.P.; Pogribna, M.; Miller, B.J.; Jernigan, S.; Melnyk, S. Mechanisms of DNA damage, DNA hypomethylation, and tumor progression in the folate/methyl-deficient rat model of hepatocarcinogenesis. J. Nutr. 2003, 133, 3740s–3747s. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Shpyleva, S.I.; Muskhelishvili, L.; Bagnyukova, T.V.; James, S.J.; Beland, F.A. Role of DNA damage and alterations in cytosine DNA methylation in rat liver carcinogenesis induced by a methyl-deficient diet. Mutat. Res. 2009, 669, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Kumar, A.P.; Ghosh, R. DNA Methylation and Flavonoids in Genitourinary Cancers. Curr. Pharmacol. Rep. 2015, 1, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Shim, J.Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (Ȓ)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Zhang, B.K.; Lai, Y.Q.; Niu, P.P.; Zhao, M.; Jia, S.J. Epigallocatechin-3-gallate inhibits homocysteine-induced apoptosis of endothelial cells by demethylation of the DDAH2 gene. Planta Med. 2013, 79, 1715–1719. [Google Scholar] [CrossRef]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter Demethylation and Chromatin Remodeling by Green Tea Polyphenols Leads to Re-expression of GSTP1 in Human Prostate Cancer Cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef]
- Shukla, S.; Trokhan, S.; Resnick, M.I.; Gupta, S. Epigallocatechin-3-gallate causes demethylation and activation of GSTP1 gene expression in human prostate cancer LNCaP cells. Cancer Res. 2005, 65, 369. [Google Scholar]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [PubMed]
- Day, J.K.; Bauer, A.M.; DesBordes, C.; Zhuang, Y.; Kim, B.E.; Newton, L.G.; Nehra, V.; Forsee, K.M.; MacDonald, R.S.; Besch-Williford, C.; et al. Genistein alters methylation patterns in mice. J. Nutr. 2002, 132, 2419S–2423S. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Zhu, B.T. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 2006, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Park, B.; Goel, A.; Aggarwal, B.B. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. 2011, 6, 93–108. [Google Scholar] [CrossRef]
- Liu, Z.; Xie, Z.; Jones, W.; Pavlovicz, R.E.; Liu, S.; Yu, J.; Li, P.K.; Lin, J.; Fuchs, J.R.; Marcucci, G.; et al. Curcumin is a potent DNA hypomethylation agent. Bioorg. Med. Chem. Lett. 2009, 19, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. Ther. 2010, 9, 8–14. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Lopez-Vallejo, F.; Kuck, D.; Lyko, F. Natural products as DNA methyltransferase inhibitors: A computer-aided discovery approach. Mol. Divers. 2011, 15, 293–304. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, S.; Xie, Z.; Pavlovicz, R.E.; Wu, J.; Chen, P.; Aimiuwu, J.; Pang, J.; Bhasin, D.; Neviani, P.; et al. Modulation of DNA Methylation by a Sesquiterpene Lactone Parthenolide. J. Pharmacol. Exp. Ther. 2009, 329, 505–514. [Google Scholar] [CrossRef]
- Nakahara, K.; Trakoontivakorn, G.; Alzoreky, N.S.; Ono, H.; Onishi-Kameyama, M.; Yoshida, M. Antimutagenicity of some edible Thai plants, and a bioactive carbazole alkaloid, mahanine, isolated from Micromelum minutum. J. Agric. Food Chem. 2002, 50, 4796–4802. [Google Scholar] [CrossRef]
- Ramsewak, R.S.; Nair, M.G.; Strasburg, G.M.; DeWitt, D.L.; Nitiss, J.L. Biologically active carbazole alkaloids from Murraya koenigii. J. Agric. Food Chem. 1999, 47, 444–447. [Google Scholar] [CrossRef]
- Sheikh, K.D.; Banerjee, P.P.; Jagadeesh, S.; Grindrod, S.C.; Zhang, L.; Paige, M.; Brown, M.L. Fluorescent epigenetic small molecule induces expression of the tumor suppressor ras-association domain family 1A and inhibits human prostate xenograft. J. Med. Chem. 2010, 53, 2376–2382. [Google Scholar] [CrossRef]
- Takumi, S.; Okamura, K.; Yanagisawa, H.; Sano, T.; Kobayashi, Y.; Nohara, K. The effect of a methyl-deficient diet on the global DNA methylation and the DNA methylation regulatory pathways. J. Appl. Toxicol. 2015, 35, 1550–1556. [Google Scholar] [CrossRef]
- Hon, G.C.; Song, C.X.; Du, T.; Jin, F.; Selvaraj, S.; Lee, A.Y.; Yen, C.A.; Ye, Z.; Mao, S.Q.; Wang, B.A.; et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 2014, 56, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.L.; Brena, R.M.; Kolle, G.; Grimmond, S.M.; Berman, B.P.; Laird, P.W.; Pera, M.F.; Wolvetang, E.J. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells 2010, 28, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Li, X.; Datta, J.; Bai, S.; Pogribny, I.; Pogribny, M.; Huang, Y.; Young, D.; Jacob, S.T. A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J. Nutr. 2006, 136, 1522–1527. [Google Scholar] [CrossRef]
- Dahl, C.; Grønbæk, K.; Guldberg, P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin. Chim. Acta 2011, 412, 831–836. [Google Scholar] [CrossRef]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007, 67, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Chanson, A.; Parnell, L.D.; Ciappio, E.D.; Liu, Z.; Crott, J.W.; Tucker, K.L.; Mason, J.B. Polymorphisms in uracil-processing genes, but not one-carbon nutrients, are associated with altered DNA uracil concentrations in an urban Puerto Rican population. Am. J. Clin. Nutr. 2009, 89, 1927–1936. [Google Scholar] [CrossRef]
- Choi, S.W.; Kim, Y.I.; Weitzel, J.N.; Mason, J.B. Folate depletion impairs DNA excision repair in the colon of the rat. Gut 1998, 43, 93–99. [Google Scholar] [CrossRef]
- Fenech, M. Recommended dietary allowances (RDAs) for genomic stability. Mutat. Res./Fund. Mol. Mech. Mutagen. 2001, 480–481, 51–54. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Studies | Dietary Components | Enzymes Inhibited or Expressed | Epigenetic Outcomes |
|---|---|---|---|
| Lee, W. J., et al. [98] | EGCG | DNMT1 | EGCG inhibited human DNMT1 activity by binding in the catalytic core region |
| Fang et al. [99] | EGC–EGCG | DNMT | EGC and EGCG showed competitive inhibition of DNMT1 and treatment of the KYSE 510 cell line. EGCG showed a dose and time-dependent reversal of hypermethylation and re-expression of mRNA of p16INK4a, RARβ, MGMT, and hMLH1 genes |
| Nandakumar, V., et al. [101] | EGC–EGCG | DNMTs | EGCG reduced the activity of DNMTs by decreasing the mRNA levels and protein expression of DNMTs. |
| Zhang, B. K., et al. [100] | EGCG | DNMT1 | EGCG inhibited the mRNA and protein expression activity of DNMT1 and downregulated binding to the promoter of DDAH2. |
| Shukla, S., et al. [103] | EGCG | DNMT | EGCG decreased the mRNA and protein expression activity of DNMT1, and increased the expression of unmethylation-specific GSTP1 promoter. |
| Pandey, M., et al. [102] | Green tea polyphenols, EGCG | DNMT1 | A dose and time-dependent inhibition of DNMT activity and protein expression was observed. |
| Day et al. [105] | Genistein | Genistein diet was positively correlated with alterations in prostate DNA methylation at CpG islands of specific mouse genes. | |
| Fang et al. [104] | Genistein | DNMT1 | Genistein showed a dose-dependent inhibitory effect on recombinant DNMT1 activity, and also decreased DNMT activity in nuclear extracts from KYSE cells. However, no effect on the mRNA expression levels of DNMTs and methyl-CpG binding domain 2 was observed. |
| Lee and Zhu [106] | Caffeic acid, Chlorogenic acid | DNMT1, M.Sssl DNMT | The caffeic acid and chlorogenic acid inhibited the DNA methylation that was catalyzed by prokaryotic M.Sssl DNMT and human DNMT1, and increased levels of SAH. |
| Liu, Z., et al. [108] | Curcumin | DNMT1, | Curcumin covalently blocks the catalytic thiolate of DNMT1 to exert its inhibitory effect on DNA methylation. |
| Liu, Z., et al. [111] | Parthenolide | DNMT1, M.Sssl DNMT | Dose-dependent parthenolide treatment decreased DNMT1 protein levels and induced a decrease in global DNA methylation. The same study showed that parthenolide inhibited M.SssI by blocking the functional thiolate of the enzyme. |
| Minor, E.A., et al. [117] | Ascorbate (Vitamin C) | DNMTs, TET2-TET3 | Ascorbate increased the expression of DNMT1, DNMT3a, and mRNA expression of Tet2 and Tet3. |
| Sheikh, K. D., et al. [114] | Mahanine | DNMT | Mahanine was associated with the inhibition of DNMT activity, and hence, prevented the hypermethylation of a specific gene in the prostate cancer cell line. However, mechanisms are not clarified. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadayifci, F.Z.; Zheng, S.; Pan, Y.-X. Molecular Mechanisms Underlying the Link between Diet and DNA Methylation. Int. J. Mol. Sci. 2018, 19, 4055. https://doi.org/10.3390/ijms19124055
Kadayifci FZ, Zheng S, Pan Y-X. Molecular Mechanisms Underlying the Link between Diet and DNA Methylation. International Journal of Molecular Sciences. 2018; 19(12):4055. https://doi.org/10.3390/ijms19124055
Chicago/Turabian StyleKadayifci, Fatma Zehra, Shasha Zheng, and Yuan-Xiang Pan. 2018. "Molecular Mechanisms Underlying the Link between Diet and DNA Methylation" International Journal of Molecular Sciences 19, no. 12: 4055. https://doi.org/10.3390/ijms19124055
APA StyleKadayifci, F. Z., Zheng, S., & Pan, Y.-X. (2018). Molecular Mechanisms Underlying the Link between Diet and DNA Methylation. International Journal of Molecular Sciences, 19(12), 4055. https://doi.org/10.3390/ijms19124055