Drosophila Jak/STAT Signaling: Regulation and Relevance in Human Cancer and Metastasis

Abstract
1. Introduction
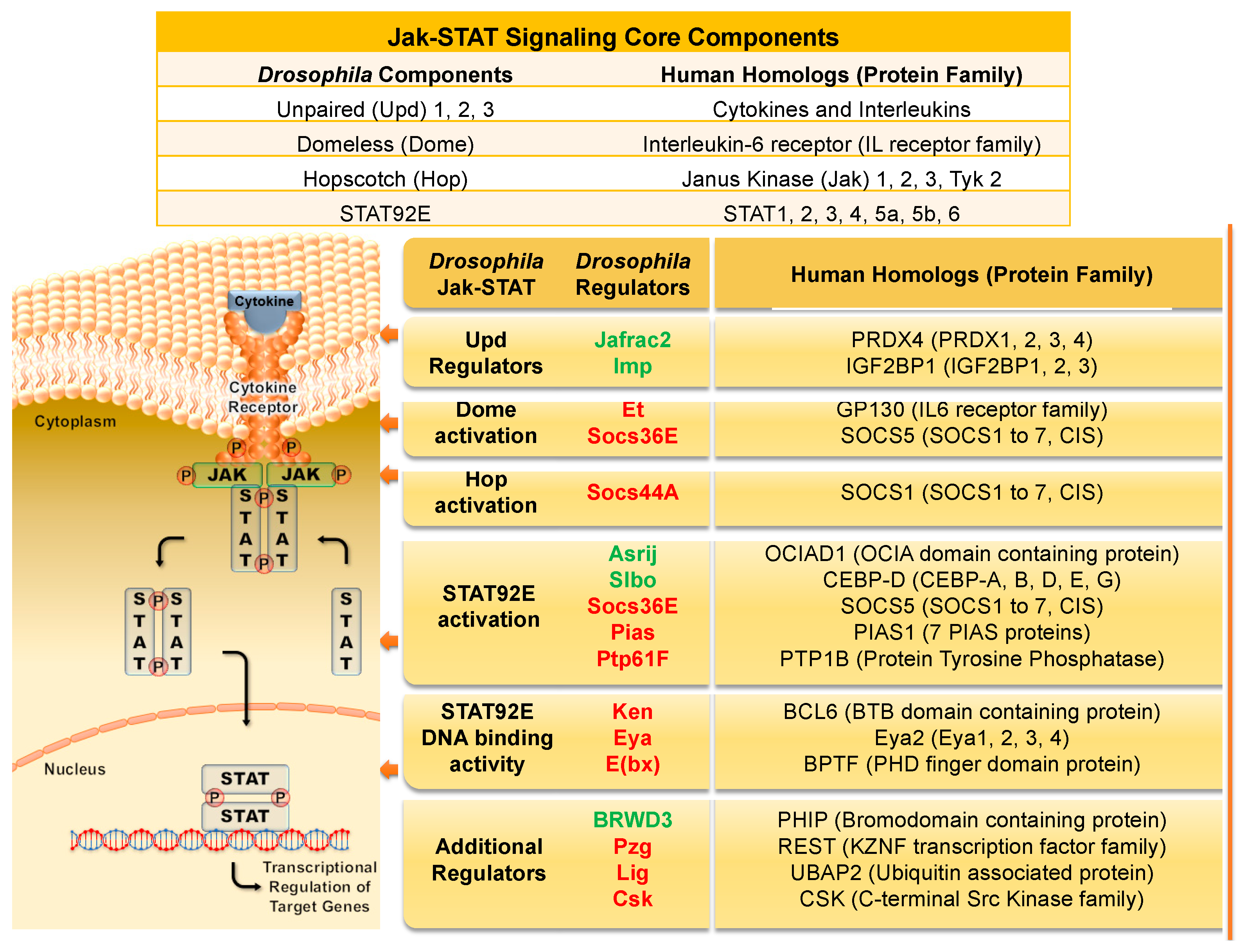
2. Jak/STAT Signaling Overview in Flies and Humans
2.1. Requirement of Jak/STAT Signaling in Development and Adulthood
2.2. Jak/STAT Activity Regulators
3. Jak/STAT Signaling in Blood Cell Proliferation and Cell Fate
3.1. Jak/STAT Signaling and Human Blood Cell Development and Disease
3.2. Drosophila Hemocytes as a Model for Blood Disorders
4. Jak/STAT Regulation of Stem Cell Character
4.1. Jak/STAT Signaling, Carcinomas, and Cancer Stem Cells
4.2. Drosophila Testes Stem Cells
5. Jak/STAT Signaling Promotes Cell Motility and Metastasis
5.1. Jak/STAT Signaling in Breast Cancer Metastasis
5.2. Jak/STAT Signaling in Prostate Cancer Metastasis
5.3. Jak/STAT Promotes Border Cell Migration in the Ovary
6. Drosophila-Jak/STAT Regulators Implicated in Human Metastatic Diseases
6.1. Positive Drosophila Jak/STAT Regulators in Metastasis
6.2. Negative Drosophila Jak/STAT Regulators in Metastasis
7. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Small, S.; Desplan, C.; Dearolf, C.R.; Darnell, J.E., Jr. Identification of a Stat gene that functions in Drosophila development. Cell 1996, 84, 421–430. [Google Scholar] [CrossRef]
- Harrison, D.A.; McCoon, P.E.; Binari, R.; Gilman, M.; Perrimon, N. Drosophila unpaired encodes a secreted protein that activates the JAK signaling pathway. Genes Dev. 1998, 12, 3252–3263. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Chen, X.; Oh, S.W.; Marinissen, M.J.; Gutkind, J.S.; Hou, S.X. mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev. 2002, 16, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Hu, N.; Hombria, J.C. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr. Biol. 2001, 11, 1700–1705. [Google Scholar] [CrossRef]
- Hou, X.S.; Melnick, M.B.; Perrimon, N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell 1996, 84, 411–419. [Google Scholar] [CrossRef]
- Luo, H.; Hanratty, W.P.; Dearolf, C.R. An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J. 1995, 14, 1412–1420. [Google Scholar] [CrossRef]
- Luo, H.; Rose, P.; Barber, D.; Hanratty, W.P.; Lee, S.; Roberts, T.M.; D’Andrea, A.D.; Dearolf, C.R. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol. Cell. Biol. 1997, 17, 1562–1571. [Google Scholar] [CrossRef]
- Hanratty, W.P.; Dearolf, C.R. The Drosophila Tumorous-lethal hematopoietic oncogene is a dominant mutation in the hopscotch locus. Mol. Gen. Genet. 1993, 238, 33–37. [Google Scholar]
- Harrison, D.A.; Binari, R.; Nahreini, T.S.; Gilman, M.; Perrimon, N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 1995, 14, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffe, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Peeters, P.; Raynaud, S.D.; Cools, J.; Wlodarska, I.; Grosgeorge, J.; Philip, P.; Monpoux, F.; Van Rompaey, L.; Baens, M.; Van den Berghe, H.; et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood 1997, 90, 2535–2540. [Google Scholar]
- Ward, A.C.; Touw, I.; Yoshimura, A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 2000, 95, 19–29. [Google Scholar] [PubMed]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Plumlee, C. Inteferons pen the JAK-STAT pathway. Semin. Cell Dev. Biol. 2008, 19, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Arbouzova, N.I.; Zeidler, M.P. JAK/STAT signalling in Drosophila: Insights into conserved regulatory and cellular functions. Development 2006, 133, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Hombria, J.C.; Brown, S.; Hader, S.; Zeidler, M.P. Characterisation of Upd2, a Drosophila JAK/STAT pathway ligand. Dev. Biol. 2005, 288, 420–433. [Google Scholar] [CrossRef]
- Wang, L.; Sexton, T.R.; Venard, C.; Giedt, M.; Guo, Q.; Chen, Q.; Harrison, D.A. Pleiotropy of the Drosophila JAK pathway cytokine Unpaired 3 in development and aging. Dev. Biol. 2014, 395, 218–231. [Google Scholar] [CrossRef]
- Wright, V.M.; Vogt, K.L.; Smythe, E.; Zeidler, M.P. Differential activities of the Drosophila JAK/STAT pathway ligands Upd, Upd2 and Upd3. Cell Signal. 2011, 23, 920–927. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Ghiglione, C.; Devergne, O.; Georgenthum, E.; Carballes, F.; Medioni, C.; Cerezo, D.; Noselli, S. The Drosophila cytokine receptor Domeless controls border cell migration and epithelial polarization during oogenesis. Development 2002, 129, 5437–5447. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.L.; Montell, D.J. Paracrine signaling through the JAK/STAT pathway activates invasive behavior of ovarian epithelial cells in Drosophila. Cell 2001, 107, 831–841. [Google Scholar] [CrossRef]
- Fisher, K.H.; Stec, W.; Brown, S.; Zeidler, M.P. Mechanisms of JAK/STAT pathway negative regulation by the short coreceptor Eye Transformer/Latran. Mol. Biol. Cell 2016, 27, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Kallio, J.; Myllymaki, H.; Gronholm, J.; Armstrong, M.; Vanha-aho, L.M.; Makinen, L.; Silvennoinen, O.; Valanne, S.; Ramet, M. Eye transformer is a negative regulator of Drosophila JAK/STAT signaling. FASEB J. 2010, 24, 4467–4479. [Google Scholar] [CrossRef] [PubMed]
- Makki, R.; Meister, M.; Pennetier, D.; Ubeda, J.M.; Braun, A.; Daburon, V.; Krzemien, J.; Bourbon, H.M.; Zhou, R.; Vincent, A.; et al. A short receptor downregulates JAK/STAT signalling to control the Drosophila cellular immune response. PLoS Biol. 2010, 8, e1000441. [Google Scholar] [CrossRef]
- Perrimon, N.; Mahowald, A.P. l(1)hopscotch, A larval-pupal zygotic lethal with a specific maternal effect on segmentation in Drosophila. Dev. Biol. 1986, 118, 28–41. [Google Scholar] [CrossRef]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E., 3rd; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef]
- Sweitzer, S.M.; Calvo, S.; Kraus, M.H.; Finbloom, D.S.; Larner, A.C. Characterization of a Stat-like DNA binding activity in Drosophila melanogaster. J. Biol. Chem. 1995, 270, 16510–16513. [Google Scholar] [CrossRef]
- Silver-Morse, L.; Li, W.X. JAK-STAT in heterochromatin and genome stability. JAKSTAT 2013, 2, e26090. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Zeidler, M.P. Unphosphorylated STATs go nuclear. Curr. Opin. Genet. Dev. 2008, 18, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Calhoun, H.C.; Xia, F.; Li, J.; Le, L.; Li, W.X. JAK signaling globally counteracts heterochromatic gene silencing. Nat. Genet. 2006, 38, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Stark, G.R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Casanova, J.L.; Holland, S.M.; Notarangelo, L.D. Inborn errors of human JAKs and STATs. Immunity 2012, 36, 515–528. [Google Scholar] [CrossRef]
- Valentino, L.; Pierre, J. JAK/STAT signal transduction: Regulators and implication in hematological malignancies. Biochem. Pharmacol. 2006, 71, 713–721. [Google Scholar] [CrossRef]
- Boudny, V.; Kovarik, J. JAK/STAT signaling pathways and cancer. Janus kinases/signal transducers and activators of transcription. Neoplasma 2002, 49, 349–355. [Google Scholar]
- Dorritie, K.A.; Redner, R.L.; Johnson, D.E. STAT transcription factors in normal and cancer stem cells. Adv. Biol. Regul. 2014, 56, 30–44. [Google Scholar] [CrossRef]
- Akira, S. Functional roles of STAT family proteins: Lessons from knockout mice. Stem Cells 1999, 17, 138–146. [Google Scholar] [CrossRef]
- Campbell, I.L. Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res. Brain Res. Rev. 2005, 48, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Robinson, G.W. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008, 22, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Del Valle Rodriguez, A.; Didiano, D.; Desplan, C. Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods 2011, 9, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Giedt, M.; Tang, L.; Harrison, D.A. Tools and methods for studying the Drosophila JAK/STAT pathway. Methods 2014, 68, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Jinks, T.M.; Polydorides, A.D.; Calhoun, G.; Schedl, P. The JAK/STAT signaling pathway is required for the initial choice of sexual identity in Drosophila melanogaster. Mol. Cell 2000, 5, 581–587. [Google Scholar] [CrossRef]
- Sefton, L.; Timmer, J.R.; Zhang, Y.; Beranger, F.; Cline, T.W. An extracellular activator of the Drosophila JAK/STAT pathway is a sex-determination signal element. Nature 2000, 405, 970–973. [Google Scholar] [CrossRef]
- Arbouzova, N.I.; Bach, E.A.; Zeidler, M.P. Ken & barbie selectively regulates the expression of a subset of Jak/STAT pathway target genes. Curr. Biol. 2006, 16, 80–88. [Google Scholar] [CrossRef]
- Wawersik, M.; Milutinovich, A.; Casper, A.L.; Matunis, E.; Williams, B.; Van Doren, M. Somatic control of germline sexual development is mediated by the JAK/STAT pathway. Nature 2005, 436, 563–567. [Google Scholar] [CrossRef]
- Agaisse, H.; Perrimon, N. The roles of JAK/STAT signaling in Drosophila immune responses. Immunol. Rev. 2004, 198, 72–82. [Google Scholar] [CrossRef]
- Minakhina, S.; Steward, R. Melanotic mutants in Drosophila: Pathways and phenotypes. Genetics 2006, 174, 253–263. [Google Scholar] [CrossRef]
- Ekas, L.A.; Baeg, G.H.; Flaherty, M.S.; Ayala-Camargo, A.; Bach, E.A. JAK/STAT signaling promotes regional specification by negatively regulating wingless expression in Drosophila. Development 2006, 133, 4721–4729. [Google Scholar] [CrossRef]
- Recasens-Alvarez, C.; Ferreira, A.; Milan, M. JAK/STAT controls organ size and fate specification by regulating morphogen production and signalling. Nat. Commun. 2017, 8, 13815. [Google Scholar] [CrossRef]
- Zeidler, M.P.; Perrimon, N.; Strutt, D.I. Polarity determination in the Drosophila eye: A novel role for unpaired and JAK/STAT signaling. Genes Dev. 1999, 13, 1342–1353. [Google Scholar] [CrossRef]
- Lengyel, J.A.; Iwaki, D.D. It takes guts: The Drosophila hindgut as a model system for organogenesis. Dev. Biol. 2002, 243, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Kovacs, L.; Sandor, G.O.; Juhasz, G. Stem-cell-specific endocytic degradation defects lead to intestinal dysplasia in Drosophila. Dis. Model. Mech. 2016, 9, 501–512. [Google Scholar] [CrossRef]
- Buchon, N.; Broderick, N.A.; Poidevin, M.; Pradervand, S.; Lemaitre, B. Drosophila intestinal response to bacterial infection: Activation of host defense and stem cell proliferation. Cell Host Microbe 2009, 5, 200–211. [Google Scholar] [CrossRef]
- Leatherman, J.L.; Dinardo, S. Germline self-renewal requires cyst stem cells and stat regulates niche adhesion in Drosophila testes. Nat. Cell Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Tulina, N.; Matunis, E. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 2001, 294, 2546–2549. [Google Scholar] [CrossRef]
- Kiger, A.A.; Jones, D.L.; Schulz, C.; Rogers, M.B.; Fuller, M.T. Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science 2001, 294, 2542–2545. [Google Scholar] [CrossRef]
- Beccari, S.; Teixeira, L.; Rorth, P. The JAK/STAT pathway is required for border cell migration during Drosophila oogenesis. Mech. Dev. 2002, 111, 115–123. [Google Scholar] [CrossRef]
- Silver, D.L.; Geisbrecht, E.R.; Montell, D.J. Requirement for JAK/STAT signaling throughout border cell migration in Drosophila. Development 2005, 132, 3483–3492. [Google Scholar] [CrossRef] [PubMed]
- Amoyel, M.; Bach, E.A. Functions of the Drosophila JAK-STAT pathway: Lessons from stem cells. JAKSTAT 2012, 1, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Hombria, J.C.; Brown, S. The fertile field of Drosophila Jak/STAT signalling. Curr. Biol. 2002, 12, R569–575. [Google Scholar] [CrossRef]
- Fossett, N. Signal transduction pathways, intrinsic regulators, and the control of cell fate choice. Biochim. Biophys. Acta 2013, 1830, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Bausek, N. JAK-STAT signaling in stem cells and their niches in Drosophila. JAKSTAT 2013, 2, e25686. [Google Scholar] [CrossRef] [PubMed]
- Amoyel, M.; Anderson, A.M.; Bach, E.A. JAK/STAT pathway dysregulation in tumors: A Drosophila perspective. Semin. Cell Dev. Biol. 2014, 28, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Morin-Poulard, I.; Vincent, A.; Crozatier, M. The Drosophila JAK-STAT pathway in blood cell formation and immunity. JAKSTAT 2013, 2, e25700. [Google Scholar] [CrossRef]
- Hombria, J.C.; Sotillos, S. JAK-STAT pathway in Drosophila morphogenesis: From organ selector to cell behavior regulator. JAKSTAT 2013, 2, e26089. [Google Scholar] [CrossRef]
- Zoranovic, T.; Grmai, L.; Bach, E.A. Regulation of proliferation, cell competition, and cellular growth by the Drosophila JAK-STAT pathway. JAKSTAT 2013, 2, e25408. [Google Scholar] [CrossRef]
- Keebaugh, E.S.; Schlenke, T.A. Insights from natural host-parasite interactions: The Drosophila model. Dev. Comp. Immunol. 2014, 42, 111–123. [Google Scholar] [CrossRef]
- Baeg, G.H.; Zhou, R.; Perrimon, N. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 2005, 19, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.H.; Wright, V.M.; Taylor, A.; Zeidler, M.P.; Brown, S. Advances in genome-wide RNAi cellular screens: A case study using the Drosophila JAK/STAT pathway. BMC Genom. 2012, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Kuttenkeuler, D.; Gesellchen, V.; Zeidler, M.P.; Boutros, M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 2005, 436, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, K.; Agrawal, D.K. Controlling cytokine signaling by constitutive inhibitors. Biochem. Pharmacol. 2005, 70, 649–657. [Google Scholar] [CrossRef]
- Starr, R.; Hilton, D.J. Negative regulation of the JAK/STAT pathway. Bioessays 1999, 21, 47–52. [Google Scholar] [CrossRef]
- Wormald, S.; Hilton, D.J. Inhibitors of cytokine signal transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jaaskelainen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.; Lampen, N.; Martinek, S.; Young, M.W.; Darnell, J.E., Jr. A Drosophila PIAS homologue negatively regulates stat92E. Proc. Natl. Acad. Sci. USA 2001, 98, 9563–9568. [Google Scholar] [CrossRef]
- Zeidler, M.P.; Bausek, N. The Drosophila JAK-STAT pathway. JAKSTAT 2013, 2, e25353. [Google Scholar] [CrossRef]
- Alexander, W.S.; Hilton, D.J. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 2004, 22, 503–529. [Google Scholar] [CrossRef]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rennebeck, G.; Harrison, S.M.; Xi, R.; Harrison, D.A. Two Drosophila suppressors of cytokine signaling (SOCS) differentially regulate JAK and EGFR pathway activities. BMC Cell Biol. 2004, 5, 38. [Google Scholar] [CrossRef]
- Callus, B.A.; Mathey-Prevot, B. SOCS36E, a novel Drosophila SOCS protein, suppresses JAK/STAT and EGF-R signalling in the imaginal wing disc. Oncogene 2002, 21, 4812–4821. [Google Scholar] [CrossRef] [PubMed]
- Karsten, P.; Hader, S.; Zeidler, M.P. Cloning and expression of Drosophila SOCS36E and its potential regulation by the JAK/STAT pathway. Mech. Dev. 2002, 117, 343–346. [Google Scholar] [CrossRef]
- Amoyel, M.; Anderson, J.; Suisse, A.; Glasner, J.; Bach, E.A. Socs36E Controls Niche Competition by Repressing MAPK Signaling in the Drosophila Testis. PLoS Genet. 2016, 12, e1005815. [Google Scholar] [CrossRef]
- Almudi, I.; Stocker, H.; Hafen, E.; Corominas, M.; Serras, F. SOCS36E specifically interferes with Sevenless signaling during Drosophila eye development. Dev. Biol. 2009, 326, 212–223. [Google Scholar] [CrossRef]
- Herranz, H.; Hong, X.; Hung, N.T.; Voorhoeve, P.M.; Cohen, S.M. Oncogenic cooperation between SOCS family proteins and EGFR identified using a Drosophila epithelial transformation model. Genes Dev. 2012, 26, 1602–1611. [Google Scholar] [CrossRef]
- Monahan, A.J.; Starz-Gaiano, M. Socs36E limits STAT signaling via Cullin2 and a SOCS-box independent mechanism in the Drosophila egg chamber. Mech. Dev. 2015, 138 Pt 3, 313–327. [Google Scholar] [CrossRef]
- Monahan, A.J.; Starz-Gaiano, M. Socs36E attenuates STAT signaling to optimize motile cell specification in the Drosophila ovary. Dev. Biol. 2013, 379, 152–166. [Google Scholar] [CrossRef]
- Matsumoto, A.; Masuhara, M.; Mitsui, K.; Yokouchi, M.; Ohtsubo, M.; Misawa, H.; Miyajima, A.; Yoshimura, A. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood 1997, 89, 3148–3154. [Google Scholar]
- Matsumoto, A.; Seki, Y.; Kubo, M.; Ohtsuka, S.; Suzuki, A.; Hayashi, I.; Tsuji, K.; Nakahata, T.; Okabe, M.; Yamada, S.; et al. Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine-inducible SH2-containing protein 1 transgenic mice. Mol. Cell. Biol. 1999, 19, 6396–6407. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.A.; Masuhara, M.; Yokouchi, M.; Suzuki, R.; Sakamoto, H.; Mitsui, K.; Matsumoto, A.; Tanimura, S.; Ohtsubo, M.; Misawa, H.; et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997, 387, 921–924. [Google Scholar] [CrossRef]
- Aoki, N.; Matsuda, T. A cytosolic protein-tyrosine phosphatase PTP1B specifically dephosphorylates and deactivates prolactin-activated STAT5a and STAT5b. J. Biol. Chem. 2000, 275, 39718–39726. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.P.; Andersen, J.N.; Cheng, A.; Tremblay, M.L.; Horvath, C.M.; Parisien, J.P.; Salmeen, A.; Barford, D.; Tonks, N.K. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 2001, 276, 47771–47774. [Google Scholar] [CrossRef] [PubMed]
- Mustelin, T.; Vang, T.; Bottini, N. Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol. 2005, 5, 43–57. [Google Scholar] [CrossRef]
- Saadin, A.; Starz-Gaiano, M. Identification of Novel Regulators of the JAK/STAT Signaling Pathway that Control Border Cell Migration in the Drosophila Ovary. G3 2016, 6, 1991–2002. [Google Scholar] [CrossRef]
- Strous, G.J.; van Kerkhof, P.; Govers, R.; Ciechanover, A.; Schwartz, A.L. The ubiquitin conjugation system is required for ligand-induced endocytosis and degradation of the growth hormone receptor. EMBO J. 1996, 15, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Vidal, O.M.; Stec, W.; Bausek, N.; Smythe, E.; Zeidler, M.P. Negative regulation of Drosophila JAK-STAT signalling by endocytic trafficking. J. Cell Sci. 2010, 123, 3457–3466. [Google Scholar] [CrossRef]
- Devergne, O.; Ghiglione, C.; Noselli, S. The endocytic control of JAK/STAT signalling in Drosophila. J. Cell Sci. 2007, 120, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Kurgonaite, K.; Gandhi, H.; Kurth, T.; Pautot, S.; Schwille, P.; Weidemann, T.; Bokel, C. Essential role of endocytosis for interleukin-4-receptor-mediated JAK/STAT signalling. J. Cell Sci. 2015, 128, 3781–3795. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zhang, Y.; Li, M.; Wu, L.; Wang, G.; Baeg, G.H.; You, J.; Li, Z.; Lin, X. Windpipe controls Drosophila intestinal homeostasis by regulating JAK/STAT pathway via promoting receptor endocytosis and lysosomal degradation. PLoS Genet. 2015, 11, e1005180. [Google Scholar] [CrossRef] [PubMed]
- Radtke, S.; Wuller, S.; Yang, X.P.; Lippok, B.E.; Mutze, B.; Mais, C.; de Leur, H.S.; Bode, J.G.; Gaestel, M.; Heinrich, P.C.; et al. Cross-regulation of cytokine signalling: Pro-inflammatory cytokines restrict IL-6 signalling through receptor internalisation and degradation. J. Cell Sci. 2010, 123, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Schepers, H.; Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. STAT5-mediated self-renewal of normal hematopoietic and leukemic stem cells. JAKSTAT 2012, 1, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Iwama, A.; Tadokoro, Y.; Shimoda, K.; Minoguchi, M.; Akira, S.; Tanaka, M.; Miyajima, A.; Kitamura, T.; Nakauchi, H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J. Exp. Med. 2005, 202, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Kallin, A.; Royer, Y.; Diaconu, C.C.; Dusa, A.; Demoulin, J.B.; Vainchenker, W.; Constantinescu, S.N. JAK2, the JAK2 V617F mutant and cytokine receptors. Pathol. Biol. 2007, 55, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Kallin, A.; Demoulin, J.B.; Vainchenker, W.; Constantinescu, S.N. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: Cross-talk with IGF1 receptor. J. Biol. Chem. 2005, 280, 41893–41899. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Ferrajoli, A.; Harris, D.; Van, Q.; Priebe, W.; Estrov, Z. WP-1034, a novel JAK-STAT inhibitor, with proapoptotic and antileukemic activity in acute myeloid leukemia (AML). Anticancer Res. 2005, 25, 1841–1850. [Google Scholar]
- Meydan, N.; Grunberger, T.; Dadi, H.; Shahar, M.; Arpaia, E.; Lapidot, Z.; Leeder, J.S.; Freedman, M.; Cohen, A.; Gazit, A.; et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 1996, 379, 645–648. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef]
- Lanot, R.; Zachary, D.; Holder, F.; Meister, M. Postembryonic hematopoiesis in Drosophila. Dev. Biol. 2001, 230, 243–257. [Google Scholar] [CrossRef]
- Krzemien, J.; Dubois, L.; Makki, R.; Meister, M.; Vincent, A.; Crozatier, M. Control of blood cell homeostasis in Drosophila larvae by the posterior signalling centre. Nature 2007, 446, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Bazzi, W.; Cattenoz, P.B.; Delaporte, C.; Dasari, V.; Sakr, R.; Yuasa, Y.; Giangrande, A. Embryonic hematopoiesis modulates the inflammatory response and larval hematopoiesis in Drosophila. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Munier, A.I.; Doucet, D.; Perrodou, E.; Zachary, D.; Meister, M.; Hoffmann, J.A.; Janeway, C.A., Jr.; Lagueux, M. PVF2, a PDGF/VEGF-like growth factor, induces hemocyte proliferation in Drosophila larvae. EMBO Rep. 2002, 3, 1195–1200. [Google Scholar] [CrossRef]
- Bruckner, K.; Kockel, L.; Duchek, P.; Luque, C.M.; Rorth, P.; Perrimon, N. The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev. Cell 2004, 7, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.M.; Bailetti, A.A.; Rodkin, E.; De, A.; Bach, E.A. A Genetic Screen Reveals an Unexpected Role for Yorkie Signaling in JAK/STAT-Dependent Hematopoietic Malignancies in Drosophila melanogaster. G3 2017, 7, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, R.P.; Tokusumi, T.; Schulz, R.A. The Friend of GATA protein U-shaped functions as a hematopoietic tumor suppressor in Drosophila. Dev. Biol. 2007, 311, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wu, X.; Fossett, N. Upregulation of the Drosophila Friend of GATA gene U-shaped by JAK/STAT signaling maintains lymph gland prohemocyte potency. Mol. Cell. Biol. 2009, 29, 6086–6096. [Google Scholar] [CrossRef]
- Minakhina, S.; Tan, W.; Steward, R. JAK/STAT and the GATA factor Pannier control hemocyte maturation and differentiation in Drosophila. Dev. Biol. 2011, 352, 308–316. [Google Scholar] [CrossRef]
- Inamdar, M.S. Drosophila asrij is expressed in pole cells, trachea and hemocytes. Dev. Genes Evol. 2003, 213, 134–137. [Google Scholar] [CrossRef]
- Khadilkar, R.J.; Rodrigues, D.; Mote, R.D.; Sinha, A.R.; Kulkarni, V.; Magadi, S.S.; Inamdar, M.S. ARF1-GTP regulates Asrij to provide endocytic control of Drosophila blood cell homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 4898–4903. [Google Scholar] [CrossRef]
- Bina, S.; Wright, V.M.; Fisher, K.H.; Milo, M.; Zeidler, M.P. Transcriptional targets of Drosophila JAK/STAT pathway signalling as effectors of haematopoietic tumour formation. EMBO Rep. 2010, 11, 201–207. [Google Scholar] [CrossRef]
- Ghosh, S.; Singh, A.; Mandal, S.; Mandal, L. Active hematopoietic hubs in Drosophila adults generate hemocytes and contribute to immune response. Dev. Cell 2015, 33, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, M.P.; Bach, E.A.; Perrimon, N. The roles of the Drosophila JAK/STAT pathway. Oncogene 2000, 19, 2598–2606. [Google Scholar] [CrossRef] [PubMed]
- Agaisse, H.; Petersen, U.M.; Boutros, M.; Mathey-Prevot, B.; Perrimon, N. Signaling role of hemocytes in Drosophila JAK/STAT-dependent response to septic injury. Dev. Cell 2003, 5, 441–450. [Google Scholar] [CrossRef]
- Dearolf, C.R. Fruit fly “leukemia”. Biochim. Biophys. Acta 1998, 1377, M13–23. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Hung, T.; Moroz, T.; Tran, Q.M.; Sidhu, S.; Cheney, M.D.; Speck, N.A.; Banerjee, U. Genetic manipulation of AML1-ETO-induced expansion of hematopoietic precursors in a Drosophila model. Blood 2010, 116, 4612–4620. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Oh, S.R.; Yin, C.H.; Lee, S.; Kim, E.A.; Kim, M.S.; Sandoval, C.; Jayabose, S.; Bach, E.A.; Lee, H.K.; et al. MS-1020 is a novel small molecule that selectively inhibits JAK3 activity. Br. J. Haematol. 2010, 148, 132–143. [Google Scholar] [CrossRef]
- Kim, B.H.; Yin, C.H.; Guo, Q.; Bach, E.A.; Lee, H.; Sandoval, C.; Jayabose, S.; Ulaczyk-Lesanko, A.; Hall, D.G.; Baeg, G.H. A small-molecule compound identified through a cell-based screening inhibits JAK/STAT pathway signaling in human cancer cells. Mol. Cancer Ther. 2008, 7, 2672–2680. [Google Scholar] [CrossRef]
- Thomas, S.; Fisher, K.; Snowden, J.; Danson, S.; Brown, S.; Zeidler, M. Effect of methotrexate on JAK/STAT pathway activation in myeloproliferative neoplasms. Lancet 2015, 385 (Suppl. 1). [Google Scholar] [CrossRef]
- Thomas, S.; Fisher, K.H.; Snowden, J.A.; Danson, S.J.; Brown, S.; Zeidler, M.P. Methotrexate Is a JAK/STAT Pathway Inhibitor. PLoS ONE 2015, 10, e0130078. [Google Scholar] [CrossRef]
- Eggert, U.S.; Kiger, A.A.; Richter, C.; Perlman, Z.E.; Perrimon, N.; Mitchison, T.J.; Field, C.M. Parallel chemical genetic and genome-wide RNAi screens identify cytokinesis inhibitors and targets. PLoS Biol. 2004, 2, e379. [Google Scholar] [CrossRef]
- Markstein, M.; Dettorre, S.; Cho, J.; Neumuller, R.A.; Craig-Muller, S.; Perrimon, N. Systematic screen of chemotherapeutics in Drosophila stem cell tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 4530–4535. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Gilliland, D.G. Divergent roles of STAT1 and STAT5 in malignancy as revealed by gene disruptions in mice. Oncogene 2000, 19, 2505–2510. [Google Scholar] [CrossRef] [PubMed]
- Calo, V.; Migliavacca, M.; Bazan, V.; Macaluso, M.; Buscemi, M.; Gebbia, N.; Russo, A. STAT proteins: From normal control of cellular events to tumorigenesis. J. Cell. Physiol. 2003, 197, 157–168. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Gopalan, V.; Smith, R.A.; Lam, A.K. Translational potential of cancer stem cells: A review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Exp. Cell Res. 2015, 335, 135–147. [Google Scholar] [CrossRef]
- Issigonis, M.; Matunis, E. SnapShot: Stem cell niches of the Drosophila testis and ovary. Cell 2011, 145, 994. [Google Scholar] [CrossRef]
- Singh, S.R.; Zheng, Z.; Wang, H.; Oh, S.W.; Chen, X.; Hou, S.X. Competitiveness for the niche and mutual dependence of the germline and somatic stem cells in the Drosophila testis are regulated by the JAK/STAT signaling. J. Cell. Physiol. 2010, 223, 500–510. [Google Scholar] [CrossRef]
- Issigonis, M.; Tulina, N.; de Cuevas, M.; Brawley, C.; Sandler, L.; Matunis, E. JAK-STAT signal inhibition regulates competition in the Drosophila testis stem cell niche. Science 2009, 326, 153–156. [Google Scholar] [CrossRef]
- Leatherman, J.L.; Dinardo, S. Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and nonautonomously influences germline stem cell self-renewal. Cell Stem Cell 2008, 3, 44–54. [Google Scholar] [CrossRef]
- Issigonis, M.; Matunis, E. The Drosophila BCL6 homolog Ken and Barbie promotes somatic stem cell self-renewal in the testis niche. Dev. Biol. 2012, 368, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Cherry, C.M.; Matunis, E.L. Epigenetic regulation of stem cell maintenance in the Drosophila testis via the nucleosome-remodeling factor NURF. Cell Stem Cell 2010, 6, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Shi, Z.; Chen, X. Enhancer of polycomb coordinates multiple signaling pathways to promote both cyst and germline stem cell differentiation in the Drosophila adult testis. PLoS Genet. 2017, 13, e1006571. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Shi, Z.; Xie, J.; Ma, B.; Chen, X. Enhancer of polycomb maintains germline activity and genome integrity in Drosophila testis. Cell Death Differ. 2018, 25, 1486–1502. [Google Scholar] [CrossRef] [PubMed]
- Tarayrah, L.; Herz, H.M.; Shilatifard, A.; Chen, X. Histone demethylase dUTX antagonizes JAK-STAT signaling to maintain proper gene expression and architecture of the Drosophila testis niche. Development 2013, 140, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Monahan, A.J.; Starz-Gaiano, M. Apontic regulates somatic stem cell numbers in Drosophila testes. BMC Dev. Biol. 2016, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Terry, N.A.; Tulina, N.; Matunis, E.; DiNardo, S. Novel regulators revealed by profiling Drosophila testis stem cells within their niche. Dev. Biol. 2006, 294, 246–257. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Yamashita, H.; Iwase, H.; Toyama, T.; Fujii, Y. Naturally occurring dominant-negative Stat5 suppresses transcriptional activity of estrogen receptors and induces apoptosis in T47D breast cancer cells. Oncogene 2003, 22, 1638. [Google Scholar] [CrossRef]
- Sp, N.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Kang, D.Y.; Kim, D.N.; Hwang, T.S.; Kim, S.Y.; Kim, W.S.; Lee, H.K.; et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer 2015, 15, 474. [Google Scholar] [CrossRef]
- Chang, R.; Song, L.; Xu, Y.; Wu, Y.; Dai, C.; Wang, X.; Sun, X.; Hou, Y.; Li, W.; Zhan, X.; et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat. Commun. 2018, 9, 3486. [Google Scholar] [CrossRef] [PubMed]
- Aysola, K.; Desai, A.; Welch, C.; Xu, J.; Qin, Y.; Reddy, V.; Matthews, R.; Owens, C.; Okoli, J.; Beech, D.J.; et al. Triple Negative Breast Cancer—An Overview. Hered. Genet. 2013, 2013. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Cook, R.S.; Estrada, M.V.; Giltnane, J.M.; Sanders, M.E.; Snchez, V.; Dean, P.T.; Wang, K.; Combs, S.E.; et al. Triple negative breast cancers with amplification of JAK2 at the 9p24 loci demonstrate JAK2-specific dependence. Sci. Transl. Med. 2016, 8, 334ra353. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 Feed-Forward Loop Drives Tumorigenesis and Metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. SOCS: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000, 113, 2813–2819. [Google Scholar] [PubMed]
- Qian, Q.; Lv, Y.; Li, P. SOCS1 is associated with clinical progression and acts as an oncogenic role in triple-negative breast cancer. IUBMB Life 2018, 70, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jeong, J.; Seo, J.; Kim, H.-S.; Kim, S.-J.; Jin, W. Dysregulated JAK2 expression by TrkC promotes metastasis potential, and EMT program of metastatic breast cancer. Sci. Rep. 2016, 6, 33899. [Google Scholar] [CrossRef]
- Yu, J.-M.; Sun, W.; Hua, F.; Xie, J.; Lin, H.; Zhou, D.-D.; Hu, Z.-W. BCL6 induces EMT by promoting the ZEB1-mediated transcription repression of E-cadherin in breast cancer cells. Cancer Lett. 2015, 365, 190–200. [Google Scholar] [CrossRef]
- Tran, T.H.; Utama, F.E.; Lin, J.; Yang, N.; Sjolund, A.B.; Ryder, A.; Johnson, K.J.; Neilson, L.M.; Liu, C.; Brill, K.L.; et al. Prolactin Inhibits Expression of the Proto-oncogene BCL6 in Breast Cancer through a Stat5a Dependent Mechanism. Cancer Res. 2010, 70, 1711. [Google Scholar] [CrossRef]
- Johnson, K.J.; Peck, A.R.; Liu, C.; Tran, T.H.; Utama, F.E.; Sjolund, A.B.; Schaber, J.D.; Witkiewicz, A.K.; Rui, H. PTP1B Suppresses Prolactin Activation of Stat5 in Breast Cancer Cells. Am. J. Pathol. 2010, 177, 2971–2983. [Google Scholar] [CrossRef]
- Liao, S.-C.; Li, J.-X.; Yu, L.; Sun, S.-R. Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J. Zhejiang Univ. Sci. B 2017, 18, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Dadakhujaev, S.; Salazar-Arcila, C.; Netherton, S.J.; Chandhoke, A.S.; Singla, A.K.; Jirik, F.R.; Bonni, S. A novel role for the SUMO E3 ligase PIAS1 in cancer metastasis. Oncoscience 2014, 1, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Gu, L.; Liao, Z.; Talati, P.G.; Shen, F.; Koptyra, M.; Tan, S.-H.; Ellsworth, E.; Gupta, S.; Montie, H.; et al. Inhibition of Stat5a/b enhances proteasomal degradation of androgen receptor liganded by antiandrogens in prostate cancer. Mol. Cancer Ther. 2015, 14, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Vogiatzi, P.; Puhr, M.; Dagvadorj, A.; Lutz, J.; Ryder, A.; Addya, S.; Fortina, P.; Cooper, C.; Leiby, B.; et al. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr.-Relat. Cancer 2010, 17, 481–493. [Google Scholar] [CrossRef]
- Santer, F.R.; Malinowska, K.; Culig, Z.; Cavarretta, I.T. Interleukin-6 trans-signalling differentially regulates proliferation, migration, adhesion and maspin expression in human prostate cancer cells. Endocr.-Relat. Cancer 2010, 17, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Talati, P.G.; Gu, L.; Ellsworth, E.M.; Girondo, M.A.; Trerotola, M.; Hoang, D.T.; Leiby, B.; Dagvadorj, A.; McCue, P.A.; Lallas, C.D.; et al. Jak2-Stat5a/b Signaling Induces Epithelial-to-Mesenchymal Transition and Stem-Like Cell Properties in Prostate Cancer. Am. J. Pathol. 2015, 185, 2505–2522. [Google Scholar] [CrossRef]
- Gu, L.; Liao, Z.; Hoang, D.; Dagvadorj, A.; Gupta, S.; Blackmon, S.; Ellsworth, E.; Talati, P.; Leiby, B.; Zinda, M.; et al. Pharmacological Inhibition of Jak2-Stat5 Signaling by Jak2 Inhibitor Azd1480 Potently Suppresses Growth of Both Primary and Castrate-Resistant Prostate Cancer. Clin. Cancer Res. 2013, 19, 5658–5674. [Google Scholar] [CrossRef]
- Da Silva, H.B.; Amaral, E.; Nolasco, E.L.; deVicto, N.; Atique, R.; Jank, C.C.; Anschau, V.; Zerbini, L.F.; Correa, R.G. Dissecting Major Signaling Pathways throughout the Development of Prostate Cancer. Prostate Cancer 2013, 2013, 23. [Google Scholar]
- Villalobos-Hernandez, A.; Bobbala, D.; Kandhi, R.; Khan, M.G.M.; Mayhue, M.; Dubois, C.M.; Ferbeyre, G.; Saucier, C.; Ramanathan, S.; Ilangumaran, S. SOCS1 inhibits migration and invasion of prostate cancer cells, attenuates tumor growth and modulates the tumor stroma. Prostate Cancer Prostatic Dis. 2016, 20, 36. [Google Scholar] [CrossRef]
- Shariat, S.F.; Chromecki, T.F.; Hoefer, J.; Barbieri, C.E.; Scherr, D.S.; Karakiewicz, P.I.; Roehrborn, C.G.; Montorsi, F.; Culig, Z.; Cavarretta, I.T. Soluble gp130 Regulates Prostate Cancer Invasion and Progression in an Interleukin-6 Dependent and Independent Manner. J. Urol. 2011, 186, 2107–2114. [Google Scholar] [CrossRef]
- Lessard, L.; Labb. PTP1B Is an Androgen Receptor–Regulated Phosphatase That Promotes the Progression of Prostate Cancer. Cancer Res. 2012, 72, 1529. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Dietrich, D.; van Leenders, G.; Uhl, B.; Hoogland, M.; Handle, F.; Schlick, B.; Neuwirt, H.; et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene 2016, 35, 2322–2332. [Google Scholar] [CrossRef]
- Duhart, J.C.; Parsons, T.T.; Raftery, L.A. The repertoire of epithelial morphogenesis on display: Progressive elaboration of Drosophila egg structure. Mech. Dev. 2017, 148, 18–39. [Google Scholar] [CrossRef]
- Denef, N.; Schupbach, T. Patterning: JAK-STAT signalling in the Drosophila follicular epithelium. Curr. Biol. 2003, 13, R388–390. [Google Scholar] [CrossRef]
- Van de Bor, V.; Zimniak, G.; Cerezo, D.; Schaub, S.; Noselli, S. Asymmetric localisation of cytokine mRNA is essential for JAK/STAT activation during cell invasiveness. Development 2011, 138, 1383–1393. [Google Scholar] [CrossRef]
- Borensztejn, A.; Boissoneau, E.; Fernandez, G.; Agnes, F.; Pret, A.M. JAK/STAT autocontrol of ligand-producing cell number through apoptosis. Development 2013, 140, 195–204. [Google Scholar] [CrossRef]
- Xi, R.; McGregor, J.R.; Harrison, D.A. A gradient of JAK pathway activity patterns the anterior-posterior axis of the follicular epithelium. Dev. Cell 2003, 4, 167–177. [Google Scholar] [CrossRef]
- Starz-Gaiano, M.; Melani, M.; Wang, X.; Meinhardt, H.; Montell, D.J. Feedback inhibition of Jak/STAT signaling by apontic is required to limit an invasive cell population. Dev. Cell 2008, 14, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Saadin, A.; Starz-Gaiano, M. Circuitous Genetic Regulation Governs a Straightforward Cell Migration. Trends Genet. 2016, 32, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Naora, H.; Montell, D.J. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat. Rev. Cancer 2005, 5, 355–366. [Google Scholar] [CrossRef]
- Yoshida, H.; Cheng, W.; Hung, J.; Montell, D.; Geisbrecht, E.; Rosen, D.; Liu, J.; Naora, H. Lessons from border cell migration in the Drosophila ovary: A role for myosin VI in dissemination of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 8144–8149. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, E.M.; Montell, D.J. Requirement for Par-6 and Bazooka in Drosophila border cell migration. Development 2004, 131, 5243–5251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qiu, Z.; Xu, Z.; Chen, S.J.; Luo, J.; Wang, X.; Chen, J. aPKC is a key polarity determinant in coordinating the function of three distinct cell polarities during collective migration. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Sotillos, S.; Krahn, M.; Espinosa-Vazquez, J.M.; Hombria, J.C. Src kinases mediate the interaction of the apical determinant Bazooka/PAR3 with STAT92E and increase signalling efficiency in Drosophila ectodermal cells. Development 2013, 140, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, P.; Godt, D.; Tepass, U. DE-Cadherin is required for intercellular motility during Drosophila oogenesis. J. Cell Biol. 1999, 144, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Chen, S.C.; Prasad, M.; He, L.; Wang, X.; Choesmel-Cadamuro, V.; Sawyer, J.K.; Danuser, G.; Montell, D.J. Mechanical feedback through E-cadherin promotes direction sensing during collective cell migration. Cell 2014, 157, 1146–1159. [Google Scholar] [CrossRef]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol. Cell. Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Haeger, A.; Wolf, K.; Zegers, M.M.; Friedl, P. Collective cell migration: Guidance principles and hierarchies. Trends Cell Biol. 2015, 25, 556–566. [Google Scholar] [CrossRef]
- Hegerfeldt, Y.; Tusch, M.; Brocker, E.B.; Friedl, P. Collective cell movement in primary melanoma explants: Plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar]
- Khalil, A.A.; Ilina, O.; Gritsenko, P.G.; Bult, P.; Span, P.N.; Friedl, P. Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin. Exp. Metastasis 2017, 34, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.H.; Meinhardt, H.; Montell, D. miRNA-mediated feedback inhibition of JAK/STAT morphogen signalling establishes a cell fate threshold. Nat. Cell Biol. 2011, 13, 1062. [Google Scholar] [CrossRef]
- Starz-Gaiano, M.; Melani, M.; Meinhardt, H.; Montell, D. Interpretation of the UPD/JAK/STAT morphogen gradient in Drosophila follicle cells. Cell Cycle 2009, 8, 2917–2925. [Google Scholar] [CrossRef]
- Maimon, I.; Popliker, M.; Gilboa, L. Without children is required for Stat-mediated zfh1 transcription and for germline stem cell differentiation. Development 2014, 141, 2602–2610. [Google Scholar] [CrossRef] [PubMed]
- Saadin, A.; Starz-Gaiano, M. Cytokine exocytosis and JAK/STAT activation in the Drosophila ovary requires the vesicle trafficking regulator α-Snap J. Cell Sci. 2018. [Google Scholar] [CrossRef]
- Sengupta, S.; Michener, C.M.; Escobar, P.; Belinson, J.; Ganapathi, R. Ovarian cancer immuno-reactive antigen domain containing 1 (OCIAD1), a key player in ovarian cancer cell adhesion. Gynecol. Oncol. 2008, 109, 226–233. [Google Scholar] [CrossRef]
- Kulkarni, V.; Khadilkar, R.J.; Magadi, S.S.; Inamdar, M.S. Asrij maintains the stem cell niche and controls differentiation during Drosophila lymph gland hematopoiesis. PLoS ONE 2011, 6, e27667. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Bheemsetty, V.A.; Inamdar, M.S. A double helical motif in OCIAD2 is essential for its localization, interactions and STAT3 activation. Sci. Rep. 2018, 8, 7362. [Google Scholar] [CrossRef] [PubMed]
- De Semir, D.; Nosrati, M.; Bezrookove, V.; Dar, A.A.; Federman, S.; Bienvenu, G.; Venna, S.; Rangel, J.; Climent, J.; Tamguney, T.M.; et al. Pleckstrin homology domain-interacting protein (PHIP) as a marker and mediator of melanoma metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 7067–7072. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, H.; Guo, X.; Zhu, Z.; Cai, H.; Kong, X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J. Hematol. Oncol. 2018, 11, 88. [Google Scholar] [CrossRef]
- Su, Y.; Xiong, J.; Hu, J.; Wei, X.; Zhang, X.; Rao, L. MicroRNA-140-5p targets insulin like growth factor 2 mRNA binding protein 1 (IGF2BP1) to suppress cervical cancer growth and metastasis. Oncotarget 2016, 7, 68397–68411. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Pan, S.; Kang, M.; Dong, R.; Zhao, J. MicroRNA-150 functions as a tumor suppressor in osteosarcoma by targeting IGF2BP1. Tumor Biol. 2016, 37, 5275–5284. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, R.; Zhang, J.; Sun, T.; Liu, X.; Yang, B. miR-506 inhibits the proliferation and invasion by targeting IGF2BP1 in glioblastoma. Am. J. Transl. Res. 2015, 7, 2007–2014. [Google Scholar] [PubMed]
- Zhou, X.; Zhang, C.Z.; Lu, S.X.; Chen, G.G.; Li, L.Z.; Liu, L.L.; Yi, C.; Fu, J.; Hu, W.; Wen, J.M.; et al. miR-625 suppresses tumour migration and invasion by targeting IGF2BP1 in hepatocellular carcinoma. Oncogene 2014, 34, 965. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Hayama, S.; Yamabuki, T.; Ishikawa, N.; Miyamoto, M.; Ito, T.; Tsuchiya, E.; Kondo, S.; Nakamura, Y.; Daigo, Y. Increased Expression of Insulin-like Growth Factor-II Messenger RNA–Binding Protein 1 Is Associated with Tumor Progression in Patients with Lung Cancer. Clin. Cancer Res. 2007, 13, 434. [Google Scholar] [CrossRef] [PubMed]
- Toledano, H.; D’Alterio, C.; Czech, B.; Levine, E.; Jones, D.L. The let-7-Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature 2012, 485, 605–610. [Google Scholar] [CrossRef]
- Hwang, J.A.; Song, J.S.; Yu, D.Y.; Kim, H.R.; Park, H.J.; Park, Y.S.; Kim, W.S.; Choi, C.M. Peroxiredoxin 4 as an independent prognostic marker for survival in patients with early-stage lung squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 6627–6635. [Google Scholar]
- Basu, A.; Banerjee, H.; Rojas, H.; Martinez, S.R.; Roy, S.; Jia, Z.; Lilly, M.B.a. Differential Expression of Peroxiredoxins in Prostate Cancer: Consistent Upregulation of PRDX3 and PRDX4. Prostate 2011, 71, 755–765. [Google Scholar] [CrossRef]
- Kim, T.H.; Song, J.a. Suppression of Peroxiredoxin 4 in Glioblastoma Cells Increases Apoptosis and Reduces Tumor Growth. PLoS ONE 2012, 7, e42818. [Google Scholar] [CrossRef]
- Radyuk, S.N.; Klichko, V.I.; Michalak, K.; Orr, W.C. The effect of peroxiredoxin 4 on fly physiology is a complex interplay of antioxidant and signaling functions. FASEB J. 2013, 27, 1426–1438. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Wu, W.-J.; Wang, W.-J.; Huang, H.-Y.; Li, W.-M.; Yeh, B.-W.; Wu, T.-F.; Shiue, Y.-L.; Sheu, J.J.-C.; Wang, J.-M.; et al. CEBPD amplification and overexpression in urothelial carcinoma: A driver of tumor metastasis indicating adverse prognosis. Oncotarget 2015, 6, 31069–31084. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Rashi, H.; Ofek, E.; Perelman, M.; Skarda, J.; Yaron, P.; Hajdch, M.; Jacob-Hirsch, J.; Amariglio, N.; Krupsky, M.; Simansky, D.A.; et al. The Expression of Three Genes in Primary Non–Small Cell Lung Cancer Is Associated with Metastatic Spread to the Brain. Clin. Cancer Res. 2009, 15, 1755. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Nosrati, M.; Bezrookove, V.; de Semir, D.; Majid, S.; Thummala, S.; Sun, V.; Tong, S.; Leong, S.P.L.; Minor, D.; et al. The Role of BPTF in Melanoma Progression and in Response to BRAF-Targeted Therapy. JNCI J. Natl. Cancer Inst. 2015, 107, djv034. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Majid, S.; Bezrookove, V.; Phan, B.; Ursu, S.; Nosrati, M.a. BPTF transduces MITF-driven prosurvival signals in melanoma cells. Proc. Natl. Acad. Sci. USA 2016, 113, 6254–6258. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Lu, J.-J.; Guo, W.; Yu, W.; Wang, Q.; Tang, R.; Tang, Z.; Xiao, Y.; Li, Z.; Sun, W.; et al. BPTF promotes tumor growth and predicts poor prognosis in lung adenocarcinomas. Oncotarget 2015, 6, 33878–33892. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Liu, L.; Fang, M.; Zhou, X.; Peng, X.; Long, J.; Lu, X. BPTF Associated with EMT Indicates Negative Prognosis in Patients with Hepatocellular Carcinoma. Dig. Dis. Sci. 2015, 60, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Liu, L.; Lu, X.; Long, J.; Zhou, X.; Fan, M. The prognostic significance of bromodomain PHD-finger transcription factor in colorectal carcinoma and association with vimentin and E-cadherin. J. Cancer Res. Clin. Oncol. 2015, 141, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Leinonen, K.A.; Grnlund, T.; Vessella, R.L.; Tammela, T.L.J.; Saramki, O.R.; Visakorpi, T. Amplification of the 9p13.3 chromosomal region in prostate cancer. Genes Chromosom. Cancer 2016, 55, 617–625. [Google Scholar] [CrossRef]
- Bai, D.-S.; Wu, C.; Yang, L.-X.; Zhang, C.; Zhang, P.-F.; He, Y.-Z.; Cai, J.-B.; Song, Z.-J.; Dong, Z.-R.; Huang, X.-Y.; et al. UBAP2 negatively regulates the invasion of hepatocellular carcinoma cell by ubiquitinating and degradating Annexin A2. Oncotarget 2016, 7, 32946–32955. [Google Scholar] [CrossRef]
- Baumgartner, R.; Stocker, H.; Hafen, E. The RNA-binding proteins FMR1, rasputin and caprin act together with the UBA protein lingerer to restrict tissue growth in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003598. [Google Scholar] [CrossRef]
- Gunsalus, K.T.W.; Wagoner, M.P.; Meyer, K.; Potter, W.B.; Schoenike, B.; Kim, S.; Alexander, C.M.; Friedl, A.; Roopra, A. Induction of the RNA Regulator LIN28A is Required for the Growth and Pathogenesis of RESTless Breast Tumors. Cancer Res. 2012, 72, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Studach, L.; Hullinger, R.L.; Xie, J.; Andrisani, O.M. Down-regulation of RE-1 Silencing Transcription Factor (REST) in advanced prostate cancer by hypoxia-induced miR-106b\25. Exp. Cell Res. 2014, 320, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Kugler, S.J.; Gehring, E.M.; Wallkamm, V.; Kruger, V.; Nagel, A.C. The Putzig-NURF nucleosome remodeling complex is required for ecdysone receptor signaling and innate immunity in Drosophila melanogaster. Genetics 2011, 188, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Zheng, S.; Li, Q.; Ran, P.; Sun, L.; Yuan, Y.; Xiao, C. Aberrant hypomethylation and overexpression of the eyes absent homologue 2 suppresses tumor cell growth of human lung adenocarcinoma cells. Oncol. Rep. 2015, 34, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xu, X.; Wang, T.; Li, Y.; You, W.; Fu, J.; Liu, Y.; Jin, S.; Ji, Q.; Zhao, W.; et al. The EGFR/miR-338-3p/EYA2 axis controls breast tumor growth and lung metastasis. Cell Death Dis. 2017, 8, e2928. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Hong, S.-M.; Hu, C.; Omura, N.; Young, A.; Kim, H.; Yu, J.; Knight, S.; Ayars, M.; Griffith, M.a. Epigenetic silencing of EYA2 in pancreatic adenocarcinomas promotes tumor growth. Oncotarget 2014, 5, 2575–2587. [Google Scholar] [CrossRef]
- Copf, T.; Goguel, V.; Lampin-Saint-Amaux, A.; Scaplehorn, N.; Preat, T. Cytokine signaling through the JAK/STAT pathway is required for long-term memory in Drosophila. Proc. Natl. Acad. Sci. USA 2011, 108, 8059–8064. [Google Scholar] [CrossRef]
- Johansen, K.A.; Iwaki, D.D.; Lengyel, J.A. Localized JAK/STAT signaling is required for oriented cell rearrangement in a tubular epithelium. Development 2003, 130, 135–145. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Calhoun, H.C.; Xia, F.; Gao, F.B.; Li, W.X. Patterns and functions of STAT activation during Drosophila embryogenesis. Mech. Dev. 2003, 120, 1455–1468. [Google Scholar] [CrossRef]
- Baksa, K.; Parke, T.; Dobens, L.L.; Dearolf, C.R. The Drosophila STAT protein, stat92E, regulates follicle cell differentiation during oogenesis. Dev. Biol. 2002, 243, 166–175. [Google Scholar] [CrossRef]
- Sheng, X.R.; Posenau, T.; Gumulak-Smith, J.J.; Matunis, E.; Van Doren, M.; Wawersik, M. Jak-STAT regulation of male germline stem cell establishment during Drosophila embryogenesis. Dev. Biol. 2009, 334, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xia, F.; Li, W.X. Coactivation of STAT and Ras is required for germ cell proliferation and invasive migration in Drosophila. Dev. Cell 2003, 5, 787–798. [Google Scholar] [CrossRef]
- Brown, S.; Zeidler, M.P.; Hombria, J.E. JAK/STAT signalling in Drosophila controls cell motility during germ cell migration. Dev. Dyn. 2006, 235, 958–966. [Google Scholar] [CrossRef]
- Cao, S.; Wang, C.; Zheng, Q.; Qiao, Y.; Xu, K.; Jiang, T.; Wu, A. STAT5 regulates glioma cell invasion by pathways dependent and independent of STAT5 DNA binding. Neurosci. Lett. 2011, 487, 228–233. [Google Scholar] [CrossRef]
- Wolf, Monika, J.; Hoos, A.; Bauer, J.; Boettcher, S.; Knust, M.; Weber, A.; Simonavicius, N.; Schneider, C.; Lang, M.; Strzl, M.; et al. Endothelial CCR2 Signaling Induced by Colon Carcinoma Cells Enables Extravasation via the JAK2-Stat5 and p38MAPK Pathway. Cancer Cell 2012, 22, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Su, W.-Y.; Liang, Q.-C.; Zhang, Z.-G.; Chen, H.-M.; Du, W.; Chen, Y.-X.; Fang, J.-Y. Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in human colorectal cancer cells. Lab. Investig. 2009, 89, 717. [Google Scholar] [CrossRef] [PubMed]
- Klupp, F.; Diers, J.; Kahlert, C.; Neumann, L.; Halama, N.; Franz, C.; Schmidt, T.; Lasitschka, F.; Warth, A.; Weitz, J.; et al. Expressional STAT3/STAT5 Ratio is an Independent Prognostic Marker in Colon Carcinoma. Ann. Surg. Oncol. 2015, 22, 1548–1555. [Google Scholar] [CrossRef]
- Wellbrock, C.; Weisser, C.; Hassel, J.C.; Fischer, P.; Becker, J.; Vetter, C.S.; Behrmann, I.; Kortylewski, M.; Heinrich, P.C.; Schartl, M. STAT5 Contributes to Interferon Resistance of Melanoma Cells. Curr. Biol. 2005, 15, 1629–1639. [Google Scholar] [CrossRef]
- Moser, C.; Ruemmele, P.; Gehmert, S.; Schenk, H.; Kreutz, M.P.; Mycielska, M.E.; Hackl, C.; Kroemer, A.; Schnitzbauer, A.A.; Stoeltzing, O.; et al. STAT5b as Molecular Target in Pancreatic Cancer—Inhibition of Tumor Growth, Angiogenesis, and Metastases. Neoplasia 2012, 14, 915–925. [Google Scholar] [CrossRef]
- Luo, H.; Asha, H.; Kockel, L.; Parke, T.; Mlodzik, M.; Dearolf, C.R. The Drosophila Jak kinase hopscotch is required for multiple developmental processes in the eye. Dev. Biol. 1999, 213, 432–441. [Google Scholar] [CrossRef]
- Liu, Y.H.; Jakobsen, J.S.; Valentin, G.; Amarantos, I.; Gilmour, D.T.; Furlong, E.E. A systematic analysis of Tinman function reveals Eya and JAK-STAT signaling as essential regulators of muscle development. Dev. Cell 2009, 16, 280–291. [Google Scholar] [CrossRef] [PubMed]
- McGregor, J.R.; Xi, R.; Harrison, D.A. JAK signaling is somatically required for follicle cell differentiation in Drosophila. Development 2002, 129, 705–717. [Google Scholar] [PubMed]
- Yun, H.M.; Park, K.R.; Quang, T.H.; Oh, H.; Hong, J.T.; Kim, Y.C.; Kim, E.C. 4-parvifuran inhibits metastatic and invasive actions through the JAK2/STAT3 pathway in osteosarcoma cells. Arch. Pharm. Res. 2017, 40, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.L.; Liu, Y.Q.; Wang, P.; Song, C.H.; Wang, K.J.; Dai, L.P.; Zhang, J.Y.; Ye, H. The effect of quercetin nanoparticle on cervical cancer progression by inducing apoptosis, autophagy and anti-proliferation via JAK2 suppression. Biomed. Pharmacother. 2016, 82, 595–605. [Google Scholar] [CrossRef]
- Liu, X.; Ji, Q.; Ye, N.; Sui, H.; Zhou, L.; Zhu, H.; Fan, Z.; Cai, J.; Li, Q. Berberine Inhibits Invasion and Metastasis of Colorectal Cancer Cells via COX-2/PGE(2) Mediated JAK2/STAT3 Signaling Pathway. PLoS ONE 2015, 10, e0123478. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Das, S.; Rachagani, S.; Torres-Gonzalez, M.P.; Lakshmanan, I.; Majhi, P.D.; Smith, L.M.; Wagner, K.U.; Batra, S.K. Carboxyl-terminal domain of MUC16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget 2015, 6, 5772–5787. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Sun, Y.H. Long-range effect of upd, a ligand for Jak/STAT pathway, on cell cycle in Drosophila eye development. Genesis 2004, 39, 141–153. [Google Scholar] [CrossRef]
- Medioni, C.; Noselli, S. Dynamics of the basement membrane in invasive epithelial clusters in Drosophila. Development 2005, 132, 3069–3077. [Google Scholar] [CrossRef]
- Tu, B.; Du, L.; Fan, Q.M.; Tang, Z.; Tang, T.T. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 2012, 325, 80–88. [Google Scholar] [CrossRef]
- Schneider, M.R.; Hoeflich, A.; Fischer, J.R.; Wolf, E.; Sordat, B.; Lahm, H. Interleukin-6 stimulates clonogenic growth of primary and metastatic human colon carcinoma cells. Cancer Lett. 2000, 151, 31–38. [Google Scholar] [CrossRef]
- Zeng, J.; Tang, Z.-H.; Liu, S.; Guo, S.-S. Clinicopathological significance of overexpression of interleukin-6 in colorectal cancer. World J. Gastroenterol. 2017, 23, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.Y.; Zheng, D.F.; Shen, A.; Gu, H.T.; Wei, X.F.; Mou, T.; Zhang, J.B.; Liu, R. IL-37b suppresses epithelial mesenchymal transition in hepatocellular carcinoma by inhibiting IL-6/STAT3 signaling. Hepatobiliary Pancreat Dis. Int. 2018, 17, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Zhang, X.; Xu, C. IL6-induced metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of 3,3′-diindolylmethane in ovarian cancer cells. Cell. Oncol. 2016, 39, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, B.; Vandooren, J.; Gerg, M.; Ahomaa, K.; Hunger, A.; Berchtold, S.; Akbareian, S.; Schaten, S.; Knolle, P.; Edwards, D.R.; et al. Systemic Ablation of MMP-9 Triggers Invasive Growth and Metastasis of Pancreatic Cancer via Deregulation of IL6 Expression in the Bone Marrow. Mol. Cancer Res. 2016, 14, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Lacreusette, A.; Nguyen, J.M.; Pandolfino, M.C.; Khammari, A.; Dreno, B.; Jacques, Y.; Godard, A.; Blanchard, F. Loss of oncostatin M receptor $\beta$ in metastatic melanoma cells. Oncogene 2006, 26, 881. [Google Scholar] [CrossRef] [PubMed]
- Kuhnlein, R.P.; Chen, C.K.; Schuh, R. A transcription unit at the ken and barbie gene locus encodes a novel Drosophila zinc finger protein. Mech. Dev. 1998, 79, 161–164. [Google Scholar] [CrossRef]
- Lukacsovich, T.; Yuge, K.; Awano, W.; Asztalos, Z.; Kondo, S.; Juni, N.; Yamamoto, D. The ken and barbie gene encoding a putative transcription factor with a BTB domain and three zinc finger motifs functions in terminalia development of Drosophila. Arch. Insect. Biochem. Physiol. 2003, 54, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Li, Y.; Li, Z.; Zhao, Q.; Fan, L. Overexpression of PTP1B in human colorectal cancer and its association with tumor progression and prognosis. J. Mol. Histol. 2014, 45, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-M.; Shang, L.; Zhang, Y.; Hao, J.-J.; Shi, F.; Luo, W.; Zhang, T.-T.; Wang, B.-S.; Yang, Y.; Liu, Z.-H.; et al. PTP1B Contributes to Calreticulin-Induced Metastatic Phenotypes in Esophageal Squamous Cell Carcinoma. Mol. Cancer Res. 2013, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338. [Google Scholar] [CrossRef]
- Liu, J.; Luan, W.; Zhang, Y.; Gu, J.; Shi, Y.; Yang, Y.; Feng, Z.; Qi, F. HDAC6 interacts with PTPN1 to enhance melanoma cells progression. Biochem. Biophys. Res. Commun. 2018, 495, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Lin, G.; Lucito, R.; Tonks, N.K. Protein-tyrosine phosphatase 1B antagonized signaling by insulin-like growth factor-1 receptor and kinase BRK/PTK6 in ovarian cancer cells. J. Biol. Chem. 2013, 288, 24923–24934. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, D.; Sun, Y.; Huang, L.; Zhang, S.; Yuan, Y. Protein inhibitor of activated STAT-1 is downregulated in gastric cancer tissue and involved in cell metastasis. Oncol. Rep. 2012, 28, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Naudin, C.; Letourneur, M.; Polrot, M.; Renoir, J.-M.; Lazar, V.; Dessen, P.; Roche, S.; Bertoglio, J.; Pierre, J. Suppressor of cytokine signaling 1 modulates invasion and metastatic potential of colorectal cancer cells. Mol. Oncol. 2014, 8, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Khan, M.G.M.; Bobbala, D.; Dubois, C.; Ramanathan, S.; Saucier, C.; Ilangumaran, S. Attenuation of MET-mediated migration and invasion in hepatocellular carcinoma cells by SOCS1. World J. Gastroenterol. 2017, 23, 6639–6649. [Google Scholar] [CrossRef] [PubMed]
- Scutti, J.A.B.; Matsuo, A.L.; Pereira, F.V.; Massaoka, M.H.; Figueiredo, C.R.; Moreira, D.F.; Belizrio, J.E.; Travassos, L.R. Role of SOCS-1 Gene on Melanoma Cell Growth and Tumor Development. Transl. Oncol. 2011, 4, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Qin, B.; Liu, F.; Chen, Y.; Zhang, R. miR-885-5p upregulation promotes colorectal cancer cell proliferation and migration by targeting suppressor of cytokine signaling. Oncol. Lett. 2018, 16, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejias, A.; Kwon, J.; Chew, X.H.; Siemens, A.; Sohn, H.S.; Jing, G.; Zhang, B.; Yang, H.; Tay, Y. A novel SOCS5/miR-18/miR-25 axis promotes tumorigenesis in liver cancer. Int. J. Cancer 2018, 0. [Google Scholar] [CrossRef]
- Kwon, S.Y.; Xiao, H.; Wu, C.; Badenhorst, P. Alternative splicing of NURF301 generates distinct NURF chromatin remodeling complexes with altered modified histone binding specificities. PLoS Genet. 2009, 5, e1000574. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Pazman, C.; Sinsimer, K.S.; Wong, L.C.; McLeod, I.; Yates, J., 3rd; Haynes, S.; Schedl, P. Rasputin functions as a positive regulator of orb in Drosophila oogenesis. PLoS ONE 2013, 8, e72864. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Ahrens, C.H.; Alayari, N.N.; Qeli, E.; Rucker, J.; Reedy, M.C.; Zmasek, C.M.; Gucek, M.; Cole, R.N.; Van Eyk, J.E.; et al. A mighty small heart: The cardiac proteome of adult Drosophila melanogaster. PLoS ONE 2011, 6, e18497. [Google Scholar] [CrossRef] [PubMed]
- Bonini, N.M.; Leiserson, W.M.; Benzer, S. The eyes absent gene: Genetic control of cell survival and differentiation in the developing Drosophila eye. Cell 1993, 72, 379–395. [Google Scholar] [CrossRef]
- Rayapureddi, J.P.; Kattamuri, C.; Steinmetz, B.D.; Frankfort, B.J.; Ostrin, E.J.; Mardon, G.; Hegde, R.S. Eyes absent represents a class of protein tyrosine phosphatases. Nature 2003, 426, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Dabbouseh, N.M.; Rebay, I. Interactions with the Abelson tyrosine kinase reveal compartmentalization of eyes absent function between nucleus and cytoplasm. Dev. Cell 2009, 16, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Vining, M.S.; Bradley, P.L.; Comeaux, C.A.; Andrew, D.J. Organ positioning in Drosophila requires complex tissue-tissue interactions. Dev. Biol. 2005, 287, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Montell, D. Eyes absent, a key repressor of polar cell fate during Drosophila oogenesis. Development 2002, 129, 5377–5388. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, J.J.; Boyle, M.; DiNardo, S. A somatic role for eyes absent (eya) and sine oculis (so) in Drosophila spermatocyte development. Dev. Biol. 2003, 258, 117–128. [Google Scholar] [CrossRef]
- Wen, Z.; Liang, C.; Pan, Q.; Wang, Y. Eya2 overexpression promotes the invasion of human astrocytoma through the regulation of ERK/MMP9 signaling. Int. J. Mol. Med. 2017, 40, 1315–1322. [Google Scholar] [CrossRef]
- Farabaugh, S.M.; Micalizzi, D.S.; Jedlicka, P.; Zhao, R.; Ford, H.L. Eya2 Is Required to Mediate the Pro-Metastatic Functions of Six1 Via the Induction of TGF-β Signaling, Epithelial-Mesenchymal Transition, and Cancer Stem Cell Properties. Oncogene 2012, 31, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.B.; Drasin, D.J.; Lea, W.A.; Patrick, A.N.; Patnaik, S.; Backos, D.S.; Matheson, C.J.; Hu, X.; Barnaeva, E.; Holliday, M.J.; et al. Allosteric inhibitors of the Eya2 phosphatase are selective and inhibit Eya2-mediated cell migration. J. Biol. Chem. 2014, 289, 16349–16361. [Google Scholar] [CrossRef] [PubMed]
- Aradska, J.; Bulat, T.; Sialana, F.J.; Birner-Gruenberger, R.; Erich, B.; Lubec, G. Gel-free mass spectrometry analysis of Drosophila melanogaster heads. Proteomics 2015, 15, 3356–3360. [Google Scholar] [CrossRef]
- Chen, W.Y.; Shih, H.T.; Liu, K.Y.; Shih, Z.S.; Chen, L.K.; Tsai, T.H.; Chen, M.J.; Liu, H.; Tan, B.C.; Chen, C.Y.; et al. Intellectual disability-associated dBRWD3 regulates gene expression through inhibition of HIRA/YEM-mediated chromatin deposition of histone H3.3. EMBO Rep. 2015, 16, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Bashirullah, A. Genetic control of specificity to steroid-triggered responses in Drosophila. Genetics 2014, 196, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Wasbrough, E.R.; Dorus, S.; Hester, S.; Howard-Murkin, J.; Lilley, K.; Wilkin, E.; Polpitiya, A.; Petritis, K.; Karr, T.L. The Drosophila melanogaster sperm proteome-II (DmSP-II). J. Proteom. 2010, 73, 2171–2185. [Google Scholar] [CrossRef] [PubMed]
- Bezrookove, V.a. Prognostic Impact of PHIP Copy Number in Melanoma: Linkage to Ulceration. J. Investig. Dermatol. 2014, 134, 783–790. [Google Scholar] [CrossRef]
- Munro, T.P.; Kwon, S.; Schnapp, B.J.; St Johnston, D. A repeated IMP-binding motif controls oskar mRNA translation and anchoring independently of Drosophila melanogaster IMP. J. Cell Biol. 2006, 172, 577–588. [Google Scholar] [CrossRef]
- Jiang, T.; Li, M.; Li, Q.; Guo, Z.; Sun, X.; Zhang, X.; Liu, Y.; Yao, W.; Xiao, P. MicroRNA-98-5p Inhibits Cell Proliferation and Induces Cell Apoptosis in Hepatocellular Carcinoma via Targeting IGF2BP1. Oncol. Res. 2017, 25, 1117–1127. [Google Scholar] [CrossRef]
- Yuan, P.; Meng, L.; Wang, N. SOX12 upregulation is associated with metastasis of hepatocellular carcinoma and increases CDK4 and IGF2BP1 expression. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3821–3826. [Google Scholar]
- Zhang, J.; Cheng, J.; Zeng, Z.; Wang, Y.; Li, X.; Xie, Q.; Jia, J.; Yan, Y.; Guo, Z.; Gao, J.; et al. Comprehensive profiling of novel microRNA-9 targets and a tumor suppressor role of microRNA-9 via targeting IGF2BP1 in hepatocellular carcinoma. Oncotarget 2015, 6, 42040–42052. [Google Scholar] [CrossRef] [PubMed]
- Montell, D.J.; Rorth, P.; Spradling, A.C. slow border cells, a locus required for a developmentally regulated cell migration during oogenesis, encodes Drosophila C/EBP. Cell 1992, 71, 51–62. [Google Scholar] [CrossRef]
- Min, Y.; Ghose, S.; Boelte, K.; Li, J.; Yang, L.; Lin, P.C. C/EBP-β regulates VEGF-C autocrine signaling in lymphangiogenesis and metastasis of lung cancer through HIF-1α. Oncogene 2011, 30, 4901–4909. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-W.; Li, C.-F.; Chi, J.-Y.; Tseng, J.T.; Chang, Y.; Hsu, L.-J.; Lee, C.-H.; Chang, T.-H.; Wang, S.-M.; Wang, D.D.H.; et al. CCAAT/Enhancer Binding Protein β in Macrophages Contributes to Immunosuppression and Inhibits Phagocytosis in Nasopharyngeal Carcinoma. Sci. Signal. 2013, 6, ra59. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Larson, D.E.; Cagan, R.L. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell 2006, 10, 33–44. [Google Scholar] [CrossRef]
- Read, R.D.; Bach, E.A.; Cagan, R.L. Drosophila C-terminal Src kinase negatively regulates organ growth and cell proliferation through inhibition of the Src, Jun N-terminal kinase, and STAT pathways. Mol. Cell. Biol. 2004, 24, 6676–6689. [Google Scholar] [CrossRef]
- Gerlach, S.U.; Eichenlaub, T.; Herranz, H. Yorkie and JNK Control Tumorigenesis in Drosophila Cells with Cytokinesis Failure. Cell Rep. 2018, 23, 1491–1503. [Google Scholar] [CrossRef]
- O’Reilly, A.M.; Ballew, A.C.; Miyazawa, B.; Stocker, H.; Hafen, E.; Simon, M.A. Csk differentially regulates Src64 during distinct morphological events in Drosophila germ cells. Development 2006, 133, 2627–2638. [Google Scholar] [CrossRef]
- Rengifo-Cam, W.; Konishi, A.; Morishita, N.; Matsuoka, H.; Yamori, T.; Nada, S.; Okada, M. Csk defines the ability of integrin-mediated cell adhesion and migration in human colon cancer cells: Implication for a potential role in cancer metastasis. Oncogene 2004, 23, 289–297. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tanaka, S.; Suzuki, H.; Takayanagi, H.; Miyazaki, T.; Nakamura, K.; Tsuruo, T. Overexpression of the csk gene suppresses tumor metastasis in vivo. Int. J. Cancer 2000, 88, 384–391. [Google Scholar] [CrossRef]


{kind=link}
| Drosophila Jak/STAT Component | Drosophila Cell Type/Tissue | Human Homolog | Metastatic Cancer Type |
|---|---|---|---|
| Signal Transducer and Activator of Transcription92E (STAT92E) | Brain [227] Embryo [3,7] Eyes [51,53] Hindgut [228,229] Lymph Glands [117] Ovaries [24,60,176,177,230] Primordial Germ Cells [231,232,233] Testes [58,59] Trachea [229] Wing Disc [52] | Signal Transducer and Activator of Transcription 5b (STAT5b) | Brain Cancer [234] Breast Cancer [149,150] Colorectal Cancer [235,236,237] Melanoma [238] Pancreatic Cancer [239] Prostate Cancer [163,164] |
| Hopscotch (Hop) | Brain [227] Embryo [7,28] Eyes [53,240] Haltere Disc [52] Hemocytes [10] Hindgut [229] Leg Disc [52] Lymph Glands [116] Somatic Muscle [241] Ovaries [23,24,60,177,242] Trachea [229] Testes [58] Wing Disc [52] | Janus Kinase 2 (Jak2) | Breast Cancer [151,153,157] Bone Cancer [243] Cervical Cancer [244] Colorectal Cancer [245] Melanoma [246] Pancreatic Cancer [247] Prostate Cancer [166,167] |
| Domeless (Dome) | Brain [227] Embryo [5,6] Eye [248] Hindgut [228] Lymph Gland [120,126] Ovaries [23,177,249] Trachea [6] Wing [52] | Interleukin 6 Receptor (IL-6R) | Breast Cancer [154] Bone Cancer [250] Colorectal Cancer [251,252,253] Hepatocellular Carcinoma [254] Ovarian Cancer [255] Pancreatic Cancer [256] Prostate Cancer [165] |
| Eye Transformer (Et) | Eye [26] Intestines [26] Lymph Gland [27] | Glycoprotein 130 (GP130) | Melanoma [257] Prostate Cancer [170] |
| Ken and Barbie (Ken) | Embryo [258] Eyes [259] Genitalia [258,259] Ovaries [192] Testes [141] | B Cell CLL/Lymphoma 6 (BCL6) | Breast Cancer [159] |
| Protein Tyrosine Phosphatase 61F (Ptp61F) | Eyes [78] Ovaries [96] Testes [141] | Protein Tyrosine Phosphatase 1B (PTP1B) | Breast Cancer [160,161] Colorectal Cancer [260] Esophageal Cancer [261] Lung Cancer [262] Melanoma [263] Ovarian Cancer [264] Prostate Cancer [171] |
| Protein Inhibitor of Activated STAT (Pias) (Su(var) 2-10) | Eyes [78] Hemocytes [78] Ovaries [23] | Protein Inhibitor of Activated STAT 1 (PIAS1) | Breast Cancer [162] Gastric Cancer [265] Prostate Cancer [172] |
| Suppressor of Cytokine Signaling 44A (Socs44A) | Wings [82] | Suppressor of Cytokine Signaling 1 (SOCS1) | Breast Cancer [156] Colorectal Cancer [266] Hepatocellular Carcinoma [267] Melanoma [268] Prostate Cancer [169] |
| Suppressor of Cytokine Signaling 36E (Socs36E) | Notum [83] Ovaries [61,89] Testes [85,138,139] Wing Disc [82,83,84,87] | Suppressor of Cytokine Signaling 5 (SOCS5) | Colorectal Cancer [269] Liver Cancer [270] |
| Enhancer of Bithorax (E(Bx)) | Testes [271] | Bromodomain PHD Finger Transcription Factor (BPTF) | Brain Cancer [212] Colorectal Cancer [217] Hepatocellular Carcinoma [216] Lung Cancer [215] Melanoma [213,214] |
| Lingerer (Lig) | Eyes [220] Imaginal Discs [220] Ovaries [272] | Ubiquitin Associated Protein 2 (UBAP2) | Hepatocellular Carcinoma [219] Prostate Cancer [218] |
| Putzig (Pzg) | Heart [273] Wing Disc [223] | RE1 Silencing Transcription Factor (REST) | Breast Cancer [221] Prostate Cancer [222] |
| Eyes Absent (Eya) | Eyes [274,275,276] Salivary Gland [277] Somatic Muscle [241] Ovaries [193,249,278] Testes [279] | Eyes Absent 2 (EYA2) | Brain Cancer [280] Breast Cancer [281,282] |
| Asrij (Arj) | Head [283] Hemocyte [119,120] Lymph Gland [197] Trachea [119] | Ovarian Cancer Immunoreactive Antigen Domain Containing 1 (OCIAD1) | Ovarian Cancer [196] |
| Bromodomain and WD Repeat Containing Protein 3 (BRWD3) | Eyes [284] Heart [273] Midgut [285] Salivary Gland [285] Testes [286] | Pleckstrin Homology Domain Interacting Protein (PHIP) | Melanoma [199,287] |
| IGF-II-mRNA-Binding Protein (Imp) | Head [283] Ovaries [288] Testes [206,279] | Insulin-like Growth Factor II Binding Protein 1 (IGF2BP1) | Bone Cancer [202] Cervical Cancer [201] Hepatocellular Carcinoma [289,290,291] Lung Cancer [205] |
| Thioredoxin Peroxidase 2 (Jafrac2) | Head [283] Heart [273] Hemolymph [210] | Peroxiredoxin 4 (PRDX4) | Brain Cancer [209] Lung Cancer [207] |
| Slow Border Cells (Slbo) | Ovaries [292] | CCAAT Enhancer Binding Protein Delta (CEBPD) | Lung Cancer [293] Nasopharyngeal Carcinoma [294] Urothelial Cancer [211] |
| C-Terminal Src Kinase (Csk) | Eyes [295,296] Imaginal Discs [297] Ovaries [298] | C-Terminal Src Kinase (CSK) | Colon Cancer [299,300] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, S.; Starz-Gaiano, M. Drosophila Jak/STAT Signaling: Regulation and Relevance in Human Cancer and Metastasis. Int. J. Mol. Sci. 2018, 19, 4056. https://doi.org/10.3390/ijms19124056
Trivedi S, Starz-Gaiano M. Drosophila Jak/STAT Signaling: Regulation and Relevance in Human Cancer and Metastasis. International Journal of Molecular Sciences. 2018; 19(12):4056. https://doi.org/10.3390/ijms19124056
Chicago/Turabian StyleTrivedi, Sunny, and Michelle Starz-Gaiano. 2018. "Drosophila Jak/STAT Signaling: Regulation and Relevance in Human Cancer and Metastasis" International Journal of Molecular Sciences 19, no. 12: 4056. https://doi.org/10.3390/ijms19124056
APA StyleTrivedi, S., & Starz-Gaiano, M. (2018). Drosophila Jak/STAT Signaling: Regulation and Relevance in Human Cancer and Metastasis. International Journal of Molecular Sciences, 19(12), 4056. https://doi.org/10.3390/ijms19124056