Peripheral Acid Sphingomyelinase Activity Is Associated with Biomarkers and Phenotypes of Alcohol Use and Dependence in Patients and Healthy Controls

 ,
, 
Abstract
1. Introduction
2. Results
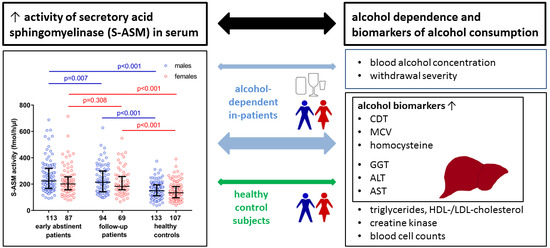
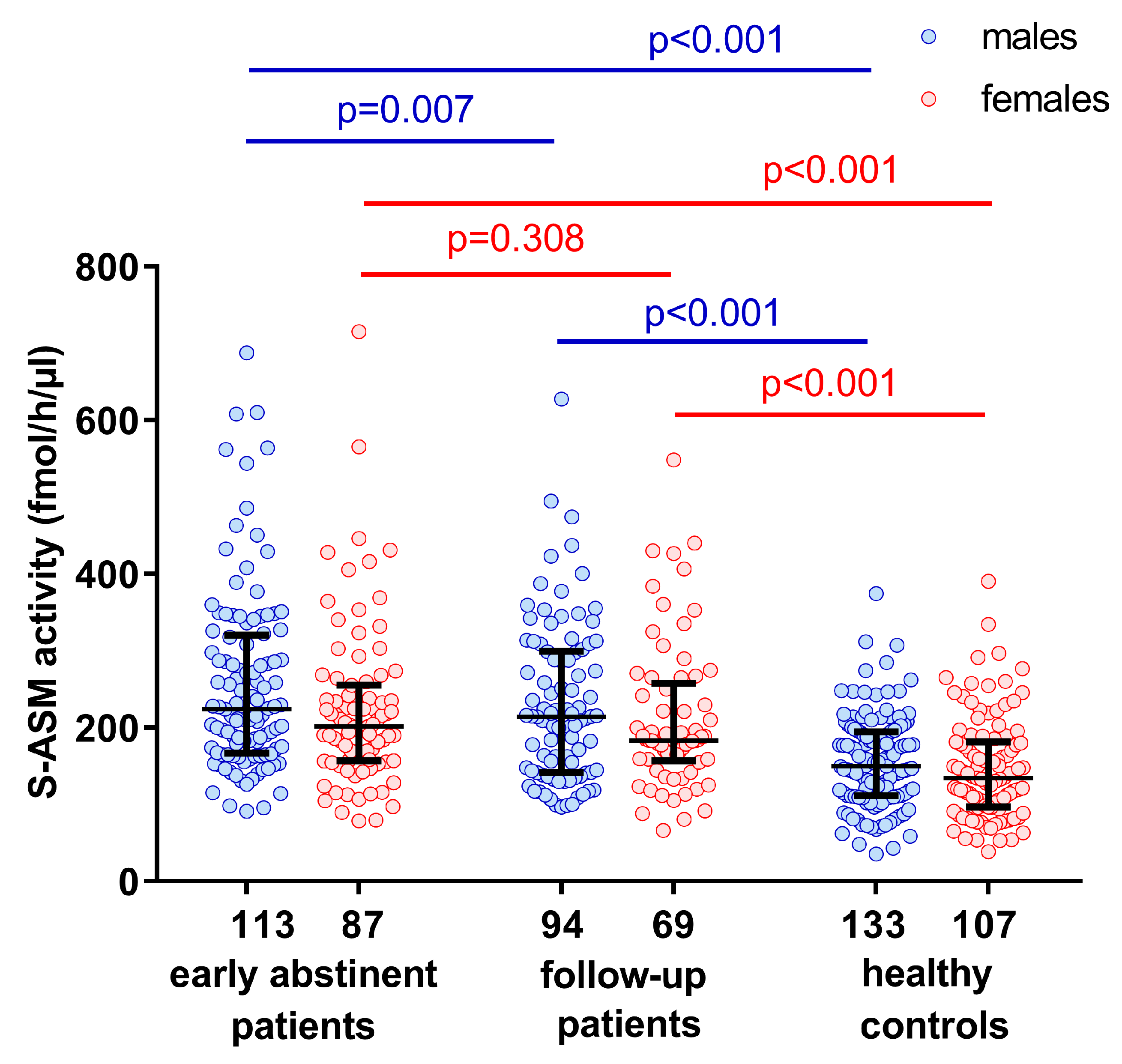
2.1. Elevated S-ASM Activity in Early-Abstinent Alcohol-Dependent Patients
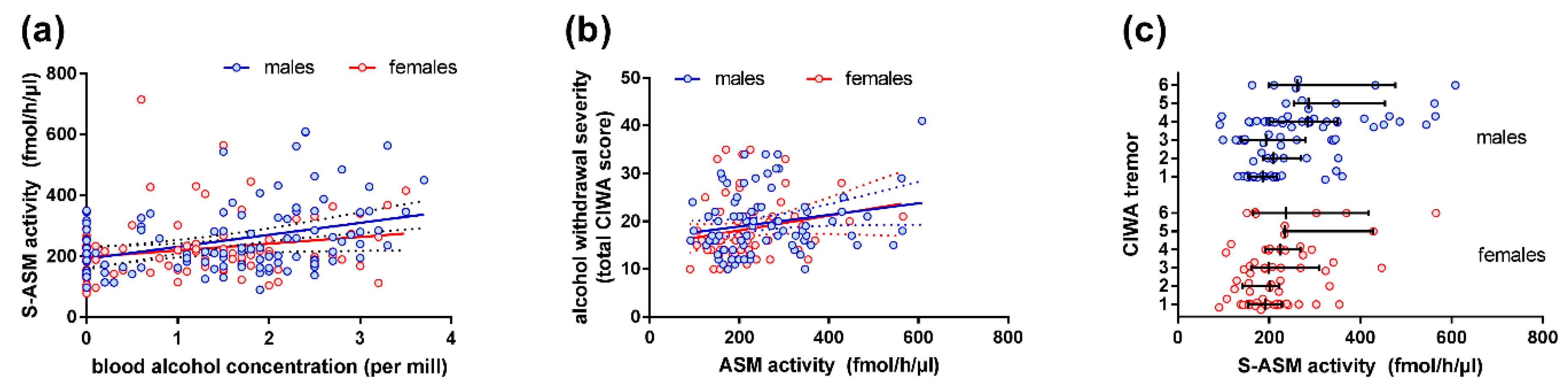
2.2. S-ASM Activity Is Positively Associated with Alcohol Levels at Admission and Withdrawal Severity
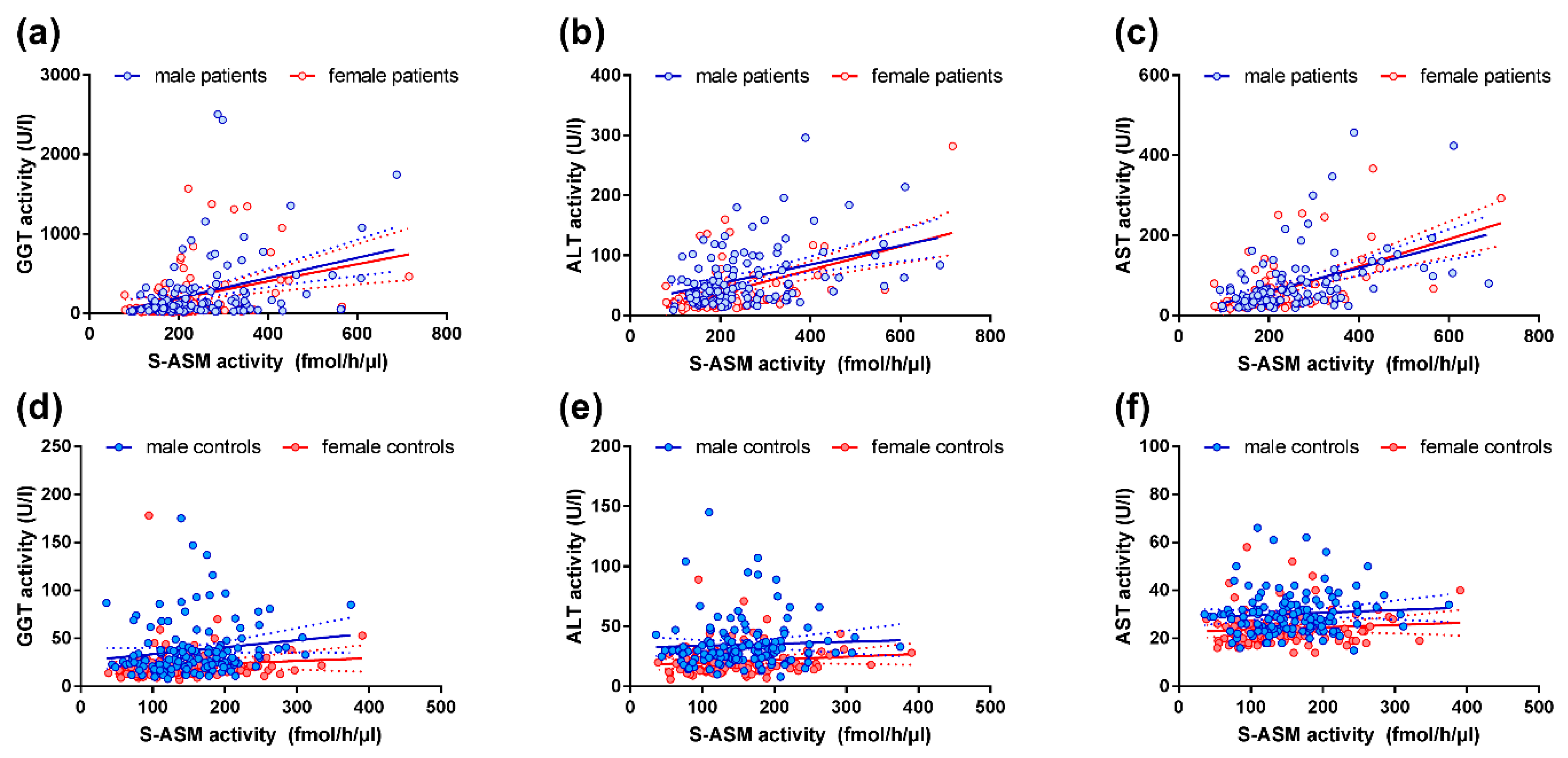
2.3. S-ASM Activity Is Strongly Associated with Liver Enzymes in Alcohol-Dependent Patients
2.4. S-ASM Activity Is Associated with GGT and ALT Activity in Healthy Controls
2.5. Comparison of S-ASM Activity with Additional Biomarkers of Alcohol Dependence
2.6. S-ASM Activity Is Differentially Associated with Myelosuppression in Patients and Controls
2.7. S-ASM Activity Is Associated with Alterations in Triglycerides in HDL Cholesterol in Patients
2.8. S-ASM Activity Does Not Predict Alcohol-Related Readmission for Patients
3. Discussion
4. Materials and Methods
4.1. Cohort Characteristics
4.2. Blood Analysis
4.3. Determination of S-ASM Activity
4.4. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | alanine aminotransferase (glutamic-pyruvic transaminase, GPT) |
| AST | aspartate aminotransferase (glutamic-oxaloacetic transaminase, GOT) |
| AUC | area under the curve |
| AUDIT | Alcohol Use Disorders Identification Test |
| BMI | body mass index |
| CAGE | acronym for 4-item questionnaire indicating potential problems with alcohol abuse |
| CDT | carbohydrate-deficient transferrin |
| CIWA-Ar score | Clinical Institute Withdrawal Assessment for Alcohol revised score |
| CK | creatine kinase |
| GGT | gamma-glutamyl transferase |
| Hcy | homocysteine |
| HDL | high-density lipoprotein |
| IQR | interquartile range |
| LDL | low-density lipoprotein |
| MCV | mean corpuscular volume |
| S-ASM | secretory acid sphingomyelinase |
| SMPD1 | sphingomyelin phosphodiesterase 1 gene encoding ASM |
References
- Tabakoff, B.; Hoffman, P.L. The neurobiology of alcohol consumption and alcoholism: An integrative history. Pharmacol. Biochem. Behav. 2013, 113, 20–37. [Google Scholar] [CrossRef]
- Foroud, T.; Edenberg, H.; Crabbe, J.C. Genetic Research—Who is at risk for alcoholism? Alcohol Res. Health 2010, 33, 64–75. [Google Scholar]
- Lenz, B.; Bouna-Pyrrou, P.; Mühle, C.; Kornhuber, J. Low digit ratio (2D:4D) and late pubertal onset indicate prenatal hyperandrogenziation in alcohol binge drinking. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 370–378. [Google Scholar] [CrossRef]
- Huber, S.E.; Zoicas, I.; Reichel, M.; Mühle, C.; Büttner, C.; Ekici, A.B.; Eulenburg, V.; Lenz, B.; Kornhuber, J.; Müller, C.P. Prenatal androgen receptor activation determines adult alcohol and water drinking in a sex-specific way. Addict. Biol. 2018, 23, 904–920. [Google Scholar] [CrossRef]
- Mühle, C.; Reichel, M.; Gulbins, E.; Kornhuber, J. Sphingolipids in psychiatric disorders and pain syndromes. Handb. Exp. Pharmacol. 2013, 431–456. [Google Scholar] [CrossRef]
- Huston, J.P.; Kornhuber, J.; Mühle, C.; Japtok, L.; Komorowski, M.; Mattern, C.; Reichel, M.; Gulbins, E.; Kleuser, B.; Topic, B.; et al. A sphingolipid mechanism for behavioral extinction. J. Neurochem. 2016, 137, 589–603. [Google Scholar] [CrossRef]
- Müller, C.P.; Reichel, M.; Mühle, C.; Rhein, C.; Gulbins, E.; Kornhuber, J. Brain membrane lipids in major depression and anxiety disorders. Biochim. Biophys. Acta 2015, 1851, 1052–1065. [Google Scholar] [CrossRef]
- Kornhuber, J.; Reichel, M.; Tripal, P.; Groemer, T.W.; Henkel, A.W.; Mühle, C.; Gulbins, E. The role of ceramide in major depressive disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259 (Suppl. 2), 199–204. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Boil. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Boil. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Kornhuber, J.; Rhein, C.; Müller, C.P.; Mühle, C. Secretory sphingomyelinase in health and disease. Biol. Chem. 2015, 396, 707–736. [Google Scholar] [CrossRef]
- Reichel, M.; Greiner, E.; Richter-Schmidinger, T.; Yedibela, O.; Tripal, P.; Jacobi, A.; Bleich, S.; Gulbins, E.; Kornhuber, J. Increased acid sphingomyelinase activity in peripheral blood cells of acutely intoxicated patients with alcohol dependence. Alcohol. Clin. Exp. Res. 2010, 34, 46–50. [Google Scholar] [CrossRef]
- Reichel, M.; Beck, J.; Mühle, C.; Rotter, A.; Bleich, S.; Gulbins, E.; Kornhuber, J. Activity of secretory sphingomyelinase is increased in plasma of alcohol-dependent patients. Alcohol. Clin. Exp. Res. 2011, 35, 1852–1859. [Google Scholar] [CrossRef]
- Mühle, C.; Amova, V.; Biermann, T.; Bayerlein, K.; Richter-Schmidinger, T.; Kraus, T.; Reichel, M.; Gulbins, E.; Kornhuber, J. Sex-dependent decrease of sphingomyelinase activity during alcohol withdrawal treatment. Cell. Physiol. Biochem. 2014, 34, 71–81. [Google Scholar] [CrossRef]
- Reichel, M.; Hönig, S.; Liebisch, G.; Lüth, A.; Kleuser, B.; Gulbins, E.; Schmitz, G.; Kornhuber, J. Alterations of plasma glycerophospholipid and sphingolipid species in male alcohol-dependent patients. Biochim. Biophys. Acta 2015, 1851, 1501–1510. [Google Scholar] [CrossRef]
- Deaciuc, I.V.; Nikolova-Karakashian, M.; Fortunato, F.; Lee, E.Y.; Hill, D.B.; McClain, C.J. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol. Clin. Exp. Res. 2000, 24, 1557–1565. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Rahmini, Y.; Ross, R.A.; Zhao, Z.; Xu, Y.; Crabb, D.W. Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. American journal of physiology. Gastrointest. Liver Physiol. 2012, 302, G515–G523. [Google Scholar] [CrossRef]
- Müller, C.P.; Kalinichenko, L.S.; Tiesel, J.; Witt, M.; Stöckl, T.; Sprenger, E.; Fuchser, J.; Beckmann, J.; Praetner, M.; Huber, S.E.; et al. Paradoxical antidepressant effects of alcohol are related to acid sphingomyelinase and its control of sphingolipid homeostasis. Acta Neuropathol. 2017, 133, 463–483. [Google Scholar] [CrossRef]
- Liu, J.J.; Wang, J.Y.; Hertervig, E.; Cheng, Y.; Nilsson, A.; Duan, R.D. Activation of neutral sphingomyelinase participates in ethanol-induced apoptosis in Hep G2 cells. Alcohol Alcohol. 2000, 35, 569–573. [Google Scholar] [CrossRef]
- Pascual, M.; Valles, S.L.; Renau-Piqueras, J.; Guerri, C. Ceramide pathways modulate ethanol-induced cell death in astrocytes. J. Neurochem. 2003, 87, 1535–1545. [Google Scholar] [CrossRef]
- Yang, L.; Jin, G.H.; Zhou, J.Y. The role of ceramide in the pathogenesis of alcoholic liver disease. Alcohol Alcohol. 2016, 51, 251–257. [Google Scholar] [CrossRef]
- Grammatikos, G.; Mühle, C.; Ferreiros, N.; Schroeter, S.; Bogdanou, D.; Schwalm, S.; Hintereder, G.; Kornhuber, J.; Zeuzem, S.; Sarrazin, C.; et al. Serum acid sphingomyelinase is upregulated in chronic hepatitis C infection and non alcoholic fatty liver disease. Biochim. Biophys. Acta 2014, 1841, 1012–1020. [Google Scholar] [CrossRef]
- Fernandez, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Cholesterol and sphingolipids in alcohol-induced liver injury. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 1), S9–S15. [Google Scholar] [CrossRef]
- Setshedi, M.; Longato, L.; Petersen, D.R.; Ronis, M.; Chen, W.C.; Wands, J.R.; de la Monte, S.M. Limited therapeutic effect of N-acetylcysteine on hepatic insulin resistance in an experimental model of alcohol-induced steatohepatitis. Alcohol. Clin. Exp. Res. 2011, 35, 2139–2151. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Mato, J.M.; Vance, D.; Kaplowitz, N.; Fernandez-Checa, J.C. Acid sphingomyelinase-ceramide system in steatohepatitis: A novel target regulating multiple pathways. J. Hepatol. 2015, 62, 219–233. [Google Scholar] [CrossRef]
- Bearer, C.F.; Bailey, S.M.; Hoek, J.B. Advancing alcohol biomarkers research. Alcohol. Clin. Exp. Res. 2010, 34, 941–945. [Google Scholar] [CrossRef]
- Gonzalo, P.; Radenne, S.; Gonzalo, S. Biomarkers of chronic alcohol misuse. Curr. Biomark. Find. 2014, 4, 9–22. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Lai, X.; Ross, R.A.; Yu, Z.; Modlik, E.; Westerhold, C.; Heathers, L.; Paul, R.; O’Connor, S.; Crabb, D.W.; et al. Novel serum biomarkers for detection of excessive alcohol use. Alcohol. Clin. Exp. Res. 2015, 39, 556–565. [Google Scholar] [CrossRef]
- Batra, A.; Muller, C.A.; Mann, K.; Heinz, A. Alcohol dependence and harmful use of alcohol. Dtsch. Arzteblatt Int. 2016, 113, 301–310. [Google Scholar] [CrossRef]
- Heinz, A.; Deserno, L.; Zimmermann, U.S.; Smolka, M.N.; Beck, A.; Schlagenhauf, F. Targeted intervention: Computational approaches to elucidate and predict relapse in alcoholism. NeuroImage 2017, 151, 33–44. [Google Scholar] [CrossRef]
- Lenz, B.; Mühle, C.; Braun, B.; Weinland, C.; Bouna-Pyrrou, P.; Behrens, J.; Kubis, S.; Mikolaiczik, K.; Muschler, M.R.; Saigali, S.; et al. Prenatal and adult androgen activities in alcohol dependence. Acta Psychiatr. Scand. 2017, 136, 96–107. [Google Scholar] [CrossRef]
- Spargo, E. The acute effects of alcohol on plasma creatine kinase (CK) activity in the rat. J. Neurol. Sci. 1984, 63, 307–316. [Google Scholar] [CrossRef]
- Gupta, A.; Gupta, C.; Khurana, S. Evaluation of total creatine kinase levels in a spectrum of neuro-psychiatric disorders in a tertiary neurosciences centre. Int. J. Med. Public Health 2015, 5, 362–366. [Google Scholar] [CrossRef]
- Segal, M.; Avital, A.; Rusakov, A.; Sandbank, S.; Weizman, A. Serum creatine kinase activity differentiates alcohol syndromes of dependence, withdrawal and delirium tremens. Eur. Neuropsychopharmacol. 2009, 19, 92–96. [Google Scholar] [CrossRef]
- Tabara, Y.; Arai, H.; Hirao, Y.; Takahashi, Y.; Setoh, K.; Kawaguchi, T.; Kosugi, S.; Ito, Y.; Nakayama, T.; Matsuda, F.; et al. The causal effects of alcohol on lipoprotein subfraction and triglyceride levels using a Mendelian randomization analysis: The Nagahama study. Atherosclerosis 2017, 257, 22–28. [Google Scholar] [CrossRef]
- Vu, K.N.; Ballantyne, C.M.; Hoogeveen, R.C.; Nambi, V.; Volcik, K.A.; Boerwinkle, E.; Morrison, A.C. Causal role of alcohol consumption in an improved lipid profile: The Atherosclerosis Risk in Communities (ARIC) Study. PLoS ONE 2016, 11, e0148765. [Google Scholar] [CrossRef]
- Sillanaukee, P.; Koivula, T.; Jokela, H.; Myllyharju, H.; Seppa, K. Relationship of alcohol consumption to changes in HDL-subfractions. Eur. J. Clin. Investig. 1993, 23, 486–491. [Google Scholar] [CrossRef]
- Mühle, C.; Huttner, H.B.; Walter, S.; Reichel, M.; Canneva, F.; Lewczuk, P.; Gulbins, E.; Kornhuber, J. Characterization of acid sphingomyelinase activity in human cerebrospinal fluid. PLoS ONE 2013, 8, e62912. [Google Scholar] [CrossRef]
- Deevska, G.M.; Sunkara, M.; Morris, A.J.; Nikolova-Karakashian, M.N. Characterization of secretory sphingomyelinase activity, lipoprotein sphingolipid content and LDL aggregation in ldlr−/− mice fed on a high-fat diet. Biosci. Rep. 2012, 32, 479–490. [Google Scholar] [CrossRef]
- Nikolova-Karakashian, M. Alcoholic and non-alcoholic fatty liver disease: Focus on ceramide. Adv. Biol. Regul. 2018, 70, 40–50. [Google Scholar] [CrossRef]
- Reichel, M.; Richter-Schmidinger, T.; Mühle, C.; Rhein, C.; Alexopoulos, P.; Schwab, S.G.; Gulbins, E.; Kornhuber, J. The common acid sphingomyelinase polymorphism p.G508R is associated with self-reported allergy. Cell. Physiol. Biochem. 2014, 34, 82–91. [Google Scholar] [CrossRef]
- Rhein, C.; Mühle, C.; Kornhuber, J.; Reichel, M. Alleged detrimental mutations in the SMPD1 gene in patients with Niemann-Pick disease. Int. J. Mol. Sci. 2015, 16, 13649–13652. [Google Scholar] [CrossRef]
- Rhein, C.; Reichel, M.; Mühle, C.; Rotter, A.; Schwab, S.G.; Kornhuber, J. Secretion of acid sphingomyelinase is affected by its polymorphic signal peptide. Cell. Physiol. Biochem. 2014, 34, 1385–1401. [Google Scholar] [CrossRef]
- Reagan, J.W., Jr.; Hubbert, M.L.; Shelness, G.S. Posttranslational regulation of acid sphingomyelinase in Niemann-Pick type C1 fibroblasts and free cholesterol-enriched Chinese hamster ovary cells. J. Boil. Chem. 2000, 275, 38104–38110. [Google Scholar] [CrossRef]
- Zuo, L.; Wang, K.; Zhang, X.Y.; Krystal, J.H.; Li, C.S.; Zhang, F.; Zhang, H.; Luo, X. NKAIN1-SERINC2 is a functional, replicable and genome-wide significant risk gene region specific for alcohol dependence in subjects of European descent. Drug Alcohol Depend. 2013, 129, 254–264. [Google Scholar] [CrossRef]
- Zuo, L.J.; Wang, K.S.; Zhang, X.Y.; Li, C.S.R.; Zhang, F.Y.; Wang, X.P.; Chen, W.A.; Gao, G.M.; Zhang, H.P.; Krystal, J.H.; et al. Rare SERINC2 variants are specific for alcohol dependence in individuals of European descent. Pharmacogenet. Genom. 2013, 23, 395–402. [Google Scholar] [CrossRef]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Boil. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef]
- Rhein, C.; Tripal, P.; Seebahn, A.; Konrad, A.; Kramer, M.; Nagel, C.; Kemper, J.; Bode, J.; Mühle, C.; Gulbins, E.; et al. Functional implications of novel human acid sphingomyelinase splice variants. PLoS ONE 2012, 7, e35467. [Google Scholar] [CrossRef]
- Rhein, C.; Reichel, M.; Kramer, M.; Rotter, A.; Lenz, B.; Mühle, C.; Gulbins, E.; Kornhuber, J. Alternative splicing of SMPD1 coding for acid sphingomyelinase in major depression. J. Affect. Disord. 2017, 209, 10–15. [Google Scholar] [CrossRef]
- Kornhuber, J.; Kornhuber, H.H.; Backhaus, B.; Kornhuber, A.; Kaiserauer, C.; Wanner, W. The normal values of gamma-glutamyltransferase are falsely defined up to now: On the diagnosis of hypertension, obesity and diabetes with reference to “normal” consumption of alcohol. Versicherungsmedizin 1989, 41, 78–81. [Google Scholar]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuno, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef]
- Gegenhuber, B.; Weinland, C.; Kornhuber, J.; Mühle, C.; Lenz, B. OPRM1 A118G and serum beta-endorphin interact with sex and digit ratio (2D:4D) to influence risk and course of alcohol dependence. Eur. Neuropsychopharmacol. 2018. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Ozelius, L.J.; Bar-Shira, A.; Saunders-Pullman, R.; Mirelman, A.; Kornreich, R.; Gana-Weisz, M.; Raymond, D.; Rozenkrantz, L.; Deik, A.; et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013, 80, 1606–1610. [Google Scholar] [CrossRef]
- Dagan, E.; Schlesinger, I.; Ayoub, M.; Mory, A.; Nassar, M.; Kurolap, A.; Peretz-Aharon, J.; Gershoni-Baruch, R. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Park. Relat. Disord. 2015, 21, 1067–1071. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Liu, C.; Marioni, R.E.; Hedman, A.K.; Pfeiffer, L.; Tsai, P.C.; Reynolds, L.M.; Just, A.C.; Duan, Q.; Boer, C.G.; Tanaka, T.; et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 2018, 23, 422–433. [Google Scholar] [CrossRef]
- Heymann, H.M.; Gardner, A.M.; Gross, E.R. Aldehyde-induced DNA and protein adducts as biomarker tools for alcohol use disorder. Trends Mol. Med. 2018, 24, 144–155. [Google Scholar] [CrossRef]
- Logan, C.; Asadi, H.; Kok, H.K.; Looby, S.T.; Brennan, P.; O’Hare, A.; Thornton, J. Neuroimaging of chronic alcohol misuse. J. Med. Imaging Radiat. Oncol. 2017, 61, 435–440. [Google Scholar] [CrossRef]
- Zou, Y.; Murray, D.E.; Durazzo, T.C.; Schmidt, T.P.; Murray, T.A.; Meyerhoff, D.J. White matter microstructural correlates of relapse in alcohol dependence. Psychiatry Res. Neuroimaging 2018, 281, 92–100. [Google Scholar] [CrossRef]
- Tu, W.; Chu, C.; Li, S.; Liangpunsakul, S. Development and validation of a composite score for excessive alcohol use screening. J. Investig. Med. 2016, 64, 1006–1011. [Google Scholar] [CrossRef]
- Weinland, C.; Mühle, C.; Kornhuber, J.; Lenz, B. Body mass index and craving predict 24-month hospital readmissions of alcohol-dependent in-patients following withdrawal. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Weinland, C.; Braun, B.; Mühle, C.; Kornhuber, J.; Lenz, B. Cloninger type 2 score and Lesch typology predict hospital readmission of female and male alcohol-dependent inpatients during a 24-month follow-up. Alcohol. Clin. Exp. Res. 2017, 41, 1760–1767. [Google Scholar] [CrossRef]
- Stuppaeck, C.H.; Barnas, C.; Falk, M.; Guenther, V.; Hummer, M.; Oberbauer, H.; Pycha, R.; Whitworth, A.B.; Fleischhacker, W.W. Assessment of the alcohol withdrawal syndrome—Validity and reliability of the translated and modified Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-A). Addiction 1994, 89, 1287–1292. [Google Scholar] [CrossRef]
- Rumpf, H.-J.; Hapke, U.; John, U. Deutsche Version des CAGE Fragebogens (CAGE-G). In Elektronisches Handbuch zu Erhebungsinstrumenten im Suchtbereich (EHES); Version 3.00; Glöckner-Rist, A.F., Rist, H.K., Eds.; Zentrum für Umfragen, Methoden und Analysen: Mannheim, Germany, 2003. [Google Scholar]
- Rumpf, H.-J.; Meyer, C.; Hapke, U.; John, U. Deutsche Version des Alcohol Use Disorders Identification Test (AUDIT-G-L). In Elektronisches Handbuch zu Erhebungsinstrumenten im Suchtbereich (EHES); Version 3.00; Glöckner-Rist, A.F., Rist, H.K., Eds.; Zentrum für Umfragen, Methoden und Analysen: Mannheim, Germany, 2003. [Google Scholar]
- Mühle, C.; Kornhuber, J. Assay to measure sphingomyelinase and ceramidase activities efficiently and safely. J. Chromatogr. A 2017, 1481, 137–144. [Google Scholar] [CrossRef]
- Lenz, B.; Müller, C.P.; Stoessel, C.; Sperling, W.; Biermann, T.; Hillemacher, T.; Bleich, S.; Kornhuber, J. Sex hormone activity in alcohol addiction: Integrating organizational and activational effects. Prog. Neurobiol. 2012, 96, 136–163. [Google Scholar] [CrossRef]
- Clayton, J.A.; Collins, F.S. Policy: NIH to balance sex in cell and animal studies. Nature 2014, 509, 282–283. [Google Scholar] [CrossRef]
- Gulbins, E.; Palmada, M.; Reichel, M.; Lüth, A.; Böhmer, C.; Amato, D.; Müller, C.P.; Tischbirek, C.H.; Groemer, T.W.; Tabatabai, G.; et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat. Med. 2013, 19, 934–938. [Google Scholar] [CrossRef]
- Kornhuber, J.; Muehlbacher, M.; Trapp, S.; Pechmann, S.; Friedl, A.; Reichel, M.; Mühle, C.; Terfloth, L.; Groemer, T.W.; Spitzer, G.M.; et al. Identification of novel functional inhibitors of acid sphingomyelinase. PLoS ONE 2011, 6, e23852. [Google Scholar] [CrossRef]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Mühle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): A novel pharmacological group of drugs with broad clinical applications. Cell. Physiol. Biochem. 2010, 26, 9–20. [Google Scholar] [CrossRef]
- Babenko, N.A.; Semenova Ya, A. Sphingolipid turnover in the hippocampus and cognitive dysfunction in alcoholized rats: correction with the help of alimentary n-3 fatty acids. Neurophysiology 2010, 42, 206–212. [Google Scholar] [CrossRef]
- Collins, M.A. Alcohol abuse and docosahexaenoic acid: Effects on cerebral circulation and neurosurvival. Brain Circ. 2015, 1, 63–68. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Alcohol-Dependent Patients | Healthy Control Subjects | p Patients vs. Controls | p ♂ vs. ♀ | ||||
|---|---|---|---|---|---|---|---|---|
| ♂ | ♀ | ♂ | ♀ | ♂ | ♀ | Patients | Controls | |
| n | 113 | 87 | 133 | 107 | - | - | - | - |
| Age (years) | 48 (40–53) | 48 (42–55) | 48 (38–56) | 49 (39–55) | 0.794 | 0.772 | 0.308 | 0.762 |
| BMI (kg/m2) b | 24.9 (22.1–27.8) | 24.4 (22.1–29.3) | 27.7 (24.9–29.5) | 25.0 (21.7–28.6) | <0.001 | 0.961 | 0.963 | <0.001 |
| Active smokers (%) b | 77.9 | 76.9 | 21.8 | 18.7 | <0.001 | <0.001 | <0.001 | <0.001 |
| Age at onset of alcohol dependence (years) c | 30 (24–39) | 35 (28–42) | - | - | - | - | 0.015 | - |
| Previous withdrawal treatments (n) c | 6 (2–12) | 5 (2–11) | - | - | - | - | 0.892 | - |
| Alcohol concentration at admission (‰) b | 1.7 (0.5–2.4) | 1.2 (0.1–1.8) | - | - | - | - | 0.020 | - |
| CIWA-Ar total score c | 18 (15–23) | 16 (14–21) | - | - | - | - | 0.163 | - |
| CAGE score | - | - | 0 (0–1) | 0 (0–0) | - | - | - | 0.024 |
| AUDIT score b | - | - | 4 (3–6) | 3 (2–4) | - | - | - | <0.001 |
| Days until first alcohol-related readmission | 285 (57–730) | 625 (90–730) | - | - | - | - | 0.047 | - |
| Number of alcohol-related readmissions | 2 (0–4) | 1 (0–3) | - | - | - | - | 0.021 | - |
| S-ASM activity during recruitment (fmol/h/µL serum) | 224 (168–318) | 202 (157–256) | 150 (112–194) | 134 (97–181) | <0.001 | <0.001 | 0.049 | 0.100 |
| S-ASM activity during follow-up (fmol/h/µL serum) b | 214 (142–299) | 183 (159–251) | - | - | - | - | 0.175 | - |
| GGT (U/L) | 109 (50–275) | 57 (34–230) | 28 (21–41) | 17 (14–25) | <0.001 | <0.001 | 0.022 | <0.001 |
| ALT (U/L) | 48 (28–84) | 28 (20–50) | 30 (23–39) | 18 (14–24) | <0.001 | <0.001 | <0.001 | <0.001 |
| AST (U/L) | 51 (36–91) | 37 (28–67) | 29 (25–34) | 23 (20–26) | <0.001 | <0.001 | 0.002 | <0.001 |
| CDT (nephelometry, %) a | 2.8 (1.9–4.0) | 1.9 (1.6–2.5) | 1.5 (1.3–1.7) | 1.5 (1.3–1.6) | <0.001 | <0.001 | <0.001 | 0.486 |
| MCV (fl) a | 93 (90–96) | 95 (91–97) | 88 (85–91) | 88 (85–91) | <0.001 | <0.001 | 0.166 | 0.734 |
| Homocysteine (µmol) | 15 (12–23) | 15 (11–21) | 12 (10–14) | 10 (9–12) | <0.001 | <0.001 | 0.161 | <0.001 |
| CK (U/L) | 132 (81–220) | 89 (72–141) | 151 (112–213) | 92 (75–110) | 0.058 | 0.422 | 0.006 | <0.001 |
| Leukocytes (per nL) a | 7.2 (5.5–8.4) | 6.9 (5.2–8.6) | 5.8 (5.0–7.2) | 5.8 (4.7–6.7) | <0.001 | <0.001 | 0.341 | 0.173 |
| Thrombocytes (per nL) a | 198 (145–254) | 218 (181–266) | 230 (196–257) | 251 (214–287) | <0.001 | <0.001 | 0.021 | <0.001 |
| Triglycerides (mg/dL) | 165 (96–236) | 135 (95–221) | 135 (98–192) | 117 (80–165) | 0.244 | 0.012 | 0.479 | 0.008 |
| Total cholesterol (mg/dL) | 214 (185–252) | 216 (180–255) | 210 (189–239) | 223 (191–252) | 0.833 | 0.530 | 0.926 | 0.106 |
| High-density lipoprotein cholesterol (mg/dL) | 61 (51–80) | 68 (54–85) | 48 (43–57) | 62 (51–74) | <0.001 | 0.015 | 0.123 | <0.001 |
| Low-density lipoprotein cholesterol (mg/dL) a | 130 (96–160) | 122 (98–157) | 143 (125–171) | 146 (122–166) | <0.001 | <0.001 | 0.750 | 0.676 |
| Parameter | Patients | Controls | Patients | Controls | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ♂ | ♀ | ♂ | ♀ | |||||||||
| Rho | p | Rho | p | Rho | p | Rho | p | Rho | p | Rho | p | |
| GGT | 0.373 | 5.6 × 10−8 | 0.254 | 7.0 × 10−5 | 0.327 | 4.4 × 10−4 | 0.378 | 3.1 × 10−4 | 0.217 | 0.012 | 0.307 | 0.001 |
| ALT | 0.373 | 5.4 × 10−8 | 0.170 | 0.008 | 0.327 | 4.0 × 10−4 | 0.368 | 4.6 × 10−4 | 0.124 | 0.154 | 0.177 | 0.068 |
| AST | 0.499 | 5.4 × 10−14 | 0.122 | 0.060 | 0.465 | 2.2 × 10−7 | 0.524 | 1.9 × 10−7 | 0.092 | 0.291 | 0.093 | 0.340 |
| CDT | 0.378 | 3.4 × 10−8 | −0.062 | 0.341 | 0.311 | 7.9 × 10−4 | 0.400 | 1.2 × 10−4 | 0.030 | 0.732 | −0.162 | 0.095 |
| MCV | 0.172 | 0.016 | 0.139 | 0.032 | 0.154 | 0.104 | 0.248 | 0.022 | 0.170 | 0.050 | 0.085 | 0.388 |
| Homocysteine | 0.304 | 1.1 × 10−5 | 0.142 | 0.028 | 0.369 | 5.7 × 10−5 | 0.201 | 0.062 | 0.123 | 0.158 | 0.092 | 0.347 |
| CK | 0.218 | 0.002 | 0.008 | 0.902 | 0.167 | 0.076 | 0.228 | 0.033 | 0.015 | 0.868 | −0.114 | 0.243 |
| Leukocytes | −0.152 | 0.033 | 0.145 | 0.025 | −0.248 | 0.008 | −0.059 | 0.591 | 0.093 | 0.287 | 0.209 | 0.032 |
| Thrombocytes | −0.292 | 3.0 × 10−5 | 0.015 | 0.812 | −0.229 | 0.015 | −0.365 | 6.0 × 10−4 | 0.030 | 0.732 | 0.070 | 0.475 |
| Triglycerides | −0.214 | 0.002 | 0.126 | 0.052 | −0.233 | 0.013 | −0.188 | 0.082 | −0.004 | 0.965 | 0.252 | 0.009 |
| Cholesterol | 0.055 | 0.435 | 0.027 | 0.679 | 0.097 | 0.308 | 0.022 | 0.838 | 0.034 | 0.698 | 0.058 | 0.551 |
| HDL cholesterol | 0.229 | 0.001 | 0.004 | 0.954 | 0.272 | 0.004 | 0.215 | 0.045 | 0.139 | 0.112 | −0.086 | 0.379 |
| LDL cholesterol | −0.050 | 0.485 | 0.017 | 0.797 | −0.035 | 0.715 | −0.072 | 0.509 | −0.034 | 0.700 | 0.081 | 0.405 |
| Total Cohort | ♂ | ♀ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Y | Sens | Spec | AUC | Y | Sens | Spec | AUC | Y | Sens | Spec | |
| S-ASM (fmol/h/µL) | 0.771 | 151 | 0.850 | 0.554 | 0.787 | 224 | 0.504 | 0.910 | 0.753 | 151 | 0.816 | 0.607 |
| CDT (%) | 0.868 | 1.72 | 0.760 | 0.849 | 0.890 | 1.77 | 0.796 | 0.841 | 0.848 | 1.71 | 0.690 | 0.888 |
| GGT (U/L) | 0.853 | 39.5 | 0.740 | 0.817 | 0.843 | 44.0 | 0.779 | 0.774 | 0.886 | 33.5 | 0.770 | 0.869 |
| MCV (fl) | 0.799 | 90.1 | 0.787 | 0.695 | 0.778 | 90.0 | 0.777 | 0.684 | 0.824 | 91.8 | 0.729 | 0.830 |
| ALT (U/L) | 0.687 | 47.5 | 0.410 | 0.904 | 0.687 | 47.5 | 0.522 | 0.857 | 0.725 | 22.5 | 0.621 | 0.738 |
| AST (U/L) | 0.806 | 33.5 | 0.700 | 0.821 | 0.816 | 35.5 | 0.752 | 0.812 | 0.837 | 26.5 | 0.782 | 0.776 |
| Hcy (µmol) | 0.758 | 13.2 | 0.650 | 0.750 | 0.742 | 13.0 | 0.717 | 0.669 | 0.783 | 12.2 | 0.667 | 0.785 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mühle, C.; Weinland, C.; Gulbins, E.; Lenz, B.; Kornhuber, J. Peripheral Acid Sphingomyelinase Activity Is Associated with Biomarkers and Phenotypes of Alcohol Use and Dependence in Patients and Healthy Controls. Int. J. Mol. Sci. 2018, 19, 4028. https://doi.org/10.3390/ijms19124028
Mühle C, Weinland C, Gulbins E, Lenz B, Kornhuber J. Peripheral Acid Sphingomyelinase Activity Is Associated with Biomarkers and Phenotypes of Alcohol Use and Dependence in Patients and Healthy Controls. International Journal of Molecular Sciences. 2018; 19(12):4028. https://doi.org/10.3390/ijms19124028
Chicago/Turabian StyleMühle, Christiane, Christian Weinland, Erich Gulbins, Bernd Lenz, and Johannes Kornhuber. 2018. "Peripheral Acid Sphingomyelinase Activity Is Associated with Biomarkers and Phenotypes of Alcohol Use and Dependence in Patients and Healthy Controls" International Journal of Molecular Sciences 19, no. 12: 4028. https://doi.org/10.3390/ijms19124028
APA StyleMühle, C., Weinland, C., Gulbins, E., Lenz, B., & Kornhuber, J. (2018). Peripheral Acid Sphingomyelinase Activity Is Associated with Biomarkers and Phenotypes of Alcohol Use and Dependence in Patients and Healthy Controls. International Journal of Molecular Sciences, 19(12), 4028. https://doi.org/10.3390/ijms19124028