The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus


Abstract
1. Introduction
2. Systemic Lupus Erythematosus
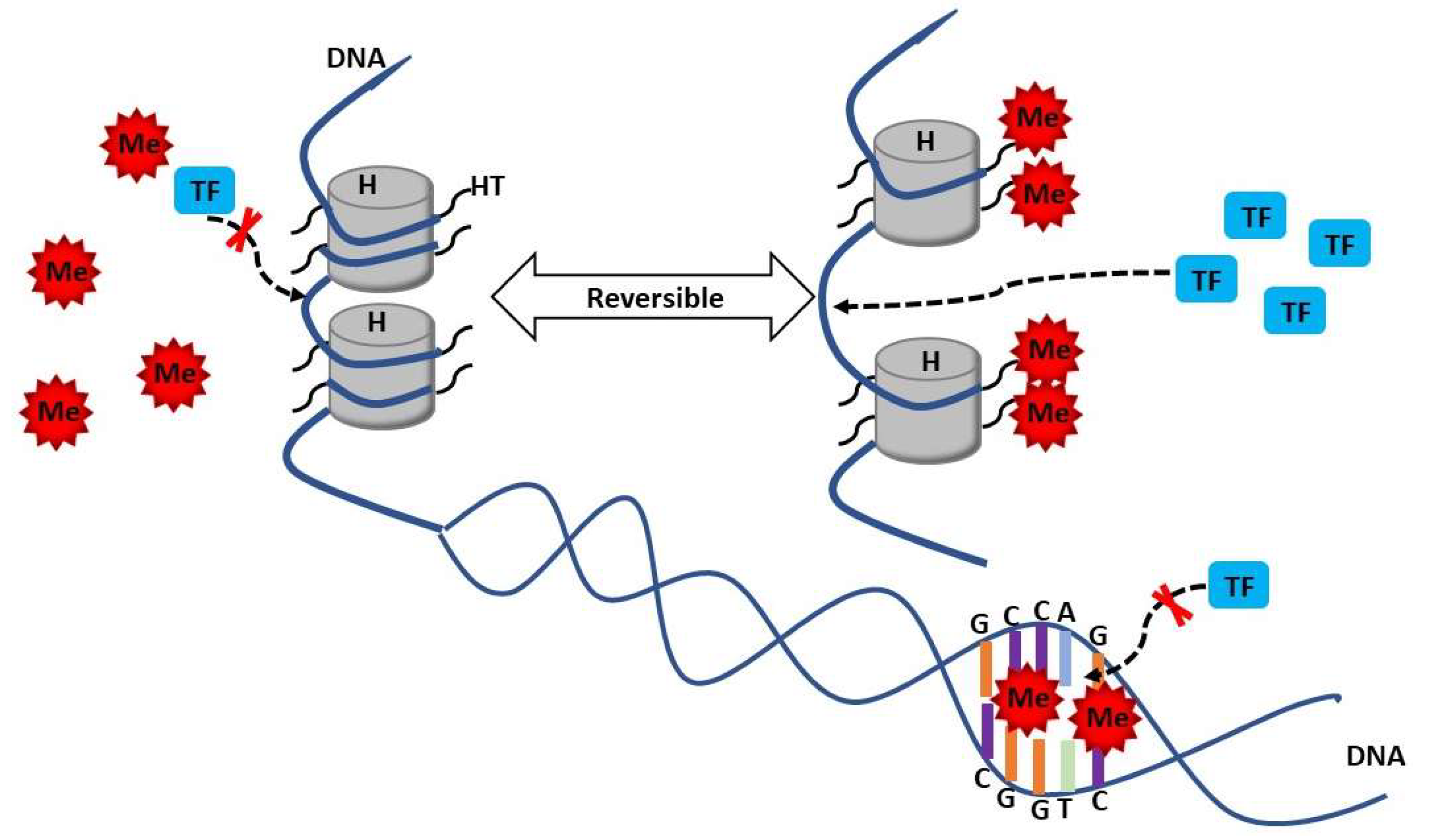
3. Methylation in SLE
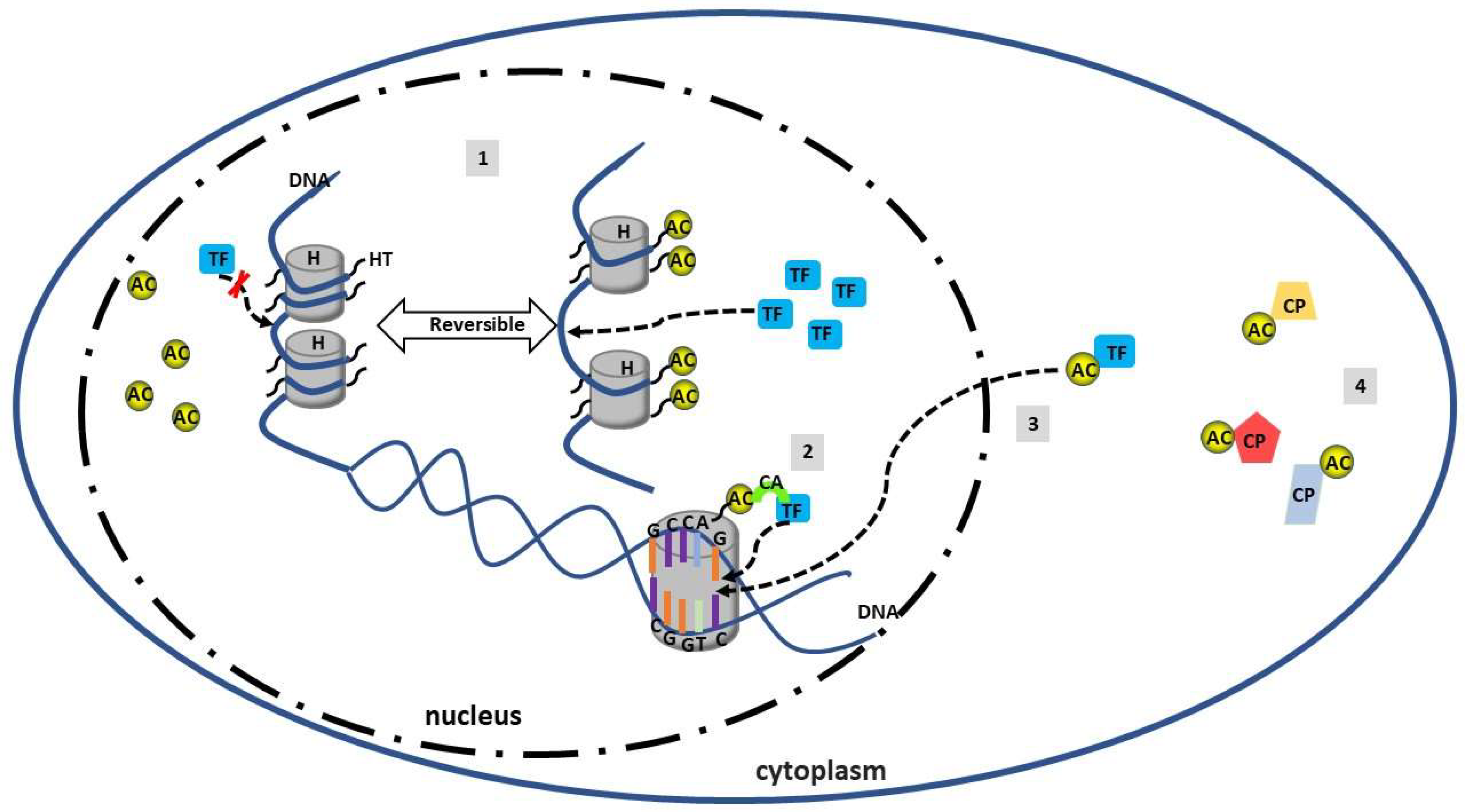
4. Acetylation in SLE
5. Metabolism and Epigenetic Crosstalk in Lupus
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SLE | Systemic lupus erythematosus |
| MHC | Major histocompatibility complex |
| HDACs | Histone deacetylase |
| HATs | Histone acetyltransferase |
| MEF2 | Myocyte enhancer factor 2 |
| TFN I/II | Type I/II interferons |
| TNF | Tumor necrosis factors |
| BLys | B-lymphocyte stimulators |
| LN | Lupus nephritis |
| SNPs | Single-nucleotide polymorphisms |
| TLR | Toll like receptor |
| SLEDAI | SLE disease activity index |
| TSA | Trichostatin |
| SAHA | Suberonylanilide hydroxamix acid |
| PBMC | Peripheral blood mononuclear cell |
| NZB/W | New Zealand Black/White |
References
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M. Epigenetics in SLE. Curr. Rheumatol. Rep. 2017, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.L.; Slansky, J.E. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012, 32, 1119–1136. [Google Scholar]
- Saito, Y.; Saito, H.; Liang, G.; Friedman, J.M. Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: A critical review. Clin. Rev. Allergy Immunol. 2014, 47, 128–135. [Google Scholar] [CrossRef]
- Jeffries, M.A. Epigenetic editing: How cutting-edge targeted epigenetic modification might provide novel avenues for autoimmune disease therapy. Clin. Immunol. 2018. [Google Scholar] [CrossRef]
- Jeffries, M.A.; Sawalha, A.H. Autoimmune disease in the epigenetic era: How has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev Clin. Immunol. 2015, 11, 45–58. [Google Scholar] [CrossRef]
- Regna, N.L.; Vieson, M.D.; Gojmerac, A.M.; Luo, X.M.; Caudell, D.L.; Reilly, C.M. HDAC expression and activity is upregulated in diseased lupus-prone mice. Int. Immunopharmacol. 2015, 29, 494–503. [Google Scholar] [CrossRef]
- Reilly, C.M.; Regna, N.; Mishra, N. HDAC inhibition in lupus models. Mol. Med. 2011, 17, 417–425. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Global H4 acetylation analysis by ChIP-chip in systemic lupus erythematosus monocytes. Genes Immun. 2010, 11, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Nititham, J.; Elboudwarej, E.; Quach, H.L.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Genome-Wide Assessment of Differential DNA Methylation Associated with Autoantibody Production in Systemic Lupus Erythematosus. PLoS ONE 2015, 10, e0129813. [Google Scholar] [CrossRef] [PubMed]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, A.H.; Jeffries, M.; Webb, R.; Lu, Q.; Gorelik, G.; Ray, D.; Osban, J.; Knowlton, N.; Johnson, K.; Richardson, B. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008, 9, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Wahren-Herlenius, M.; Dorner, T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013, 382, 819–831. [Google Scholar] [CrossRef]
- Deng, Y.; Tsao, B.P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat. Rev. Rheumatol. 2010, 6, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.H.; Cohen, P.L. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Xue, D.; Kagan, V. Programmed cell clearance: Molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem. Biophys. Res. Commun. 2010, 396, 7–10. [Google Scholar] [CrossRef]
- Jacob, C.; Lebrun-Julien, F.; Suter, U. How histone deacetylases control myelination. Mol. Neurobiol. 2011, 44, 303–312. [Google Scholar] [CrossRef]
- Mackay, M.; Oswald, M.; Sanchez-Guerrero, J.; Lichauco, J.; Aranow, C.; Kotkin, S.; Korsunsky, I.; Gregersen, P.K.; Diamond, B. Molecular signatures in systemic lupus erythematosus: Distinction between disease flare and infection. Lupus Sci. Med. 2016, 3, e000159. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, L.T.; Feldman, C.H.; Liu, J.; Alarcon, G.S.; Fischer, M.A.; Winkelmayer, W.C.; Costenbader, K.H. Prevalence, incidence, and demographics of systemic lupus erythematosus and lupus nephritis from 2000 to 2004 among children in the US Medicaid beneficiary population. Arthritis Rheum. 2012, 64, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Kwok, C.L.; Ho, L.Y.; Chan, P.T.; Yip, S.F. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheum. 2011, 63, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.; Leone, V.; Pilkington, C.; Tullus, K.; Rangaraj, S.; McDonagh, J.E.; Gardner-Medwin, J.; Wilkinson, N.; Riley, P.; Tizard, J.; et al. Disease activity, severity, and damage in the UK Juvenile-Onset Systemic Lupus Erythematosus Cohort. Arthritis Rheum. 2012, 64, 2356–2365. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Keen, C.L.; Shoenfeld, Y.; Gershwin, M.E. Surviving the butterfly and the wolf: Mortality trends in systemic lupus erythematosus. Autoimmun. Rev. 2004, 3, 423–453. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Habib, H.B.; Islam, M.; Ahmad, B.; Majid, S.; Saeed, W.; Shah, S.M.; Ahmad, A. Survival analysis and prognostic indicators of systemic lupus erythematosus in Pakistani patients. Lupus 2009, 18, 848–855. [Google Scholar] [CrossRef]
- Flower, C.; Hennis, A.J.; Hambleton, I.R.; Nicholson, G.D.; Liang, M.H.; Barbados National Lupus Registry Group. Systemic lupus erythematosus in an African Caribbean population: Incidence, clinical manifestations, and survival in the Barbados National Lupus Registry. Arthritis Care Res. 2012, 64, 1151–1158. [Google Scholar]
- Pons-Estel, G.J.; Serrano, R.; Plasin, M.A.; Espinosa, G.; Cervera, R. Epidemiology and management of refractory lupus nephritis. Autoimmun. Rev. 2011, 10, 655–663. [Google Scholar] [CrossRef]
- Baranowska-Daca, E.; Choi, Y.J.; Barrios, R.; Nassar, G.; Suki, W.N.; Truong, L.D. Nonlupus nephritides in patients with systemic lupus erythematosus: A comprehensive clinicopathologic study and review of the literature. Hum. Pathol. 2001, 32, 1125–1135. [Google Scholar] [CrossRef]
- Andrews, K.T.; Tran, T.N.; Wheatley, N.C.; Fairlie, D.P. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 2009, 9, 292–308. [Google Scholar] [CrossRef]
- Rullo, O.J.; Tsao, B.P. Recent insights into the genetic basis of systemic lupus erythematosus. Ann. Rheum. Dis. 2013, 72 (Suppl. 2), ii56–ii61. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Kammer, G.M. Molecular aberrations in human systemic lupus erythematosus. Mol. Med. Today 2000, 6, 418–424. [Google Scholar] [CrossRef]
- Harley, J.B.; Moser, K.L.; Gaffney, P.M.; Behrens, T.W. The genetics of human systemic lupus erythematosus. Curr. Opin. Immunol. 1998, 10, 690–696. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Zampeli, E.; Klinman, D.M.; Gershwin, M.E.; Moutsopoulos, H.M. A comprehensive evaluation for the treatment of lupus nephritis. J. Autoimmun. 2017, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Magrath, I.T.; Balow, J.E. Epstein-Barr virus induces normal B cell responses but defective suppressor T cell responses in patients with systemic lupus erythematosus. J. Immunol. 1983, 131, 1797–1801. [Google Scholar] [PubMed]
- Kang, I.; Quan, T.; Nolasco, H.; Park, S.H.; Hong, M.S.; Crouch, J.; Pamer, E.G.; Howe, J.G.; Craft, J. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J. Immunol. 2004, 172, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Costenbader, K.H.; Feskanich, D.; Stampfer, M.J.; Karlson, E.W. Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheum. 2007, 56, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B. Primer: Epigenetics of autoimmunity. Nat. Clin. Pract. Rheumatol. 2007, 3, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Shafi, G.; Aliya, N.; Jyothy, A. Histone modifications dictate specific biological readouts. J. Genet. Genom. 2009, 36, 75–88. [Google Scholar] [CrossRef]
- Kalden, J.R. Defective phagocytosis of apoptotic cells: Possible explanation for the induction of autoantibodies in SLE. Lupus 1997, 6, 326–327. [Google Scholar] [CrossRef]
- Gaipl, U.S.; Kuhn, A.; Sheriff, A.; Munoz, L.E.; Franz, S.; Voll, R.E.; Kalden, J.R.; Herrmann, M. Clearance of apoptotic cells in human SLE. Curr. Dir. Autoimmun. 2006, 9, 173–187. [Google Scholar]
- Qiao, B.; Wu, J.; Chu, Y.W.; Wang, Y.; Wang, D.P.; Wu, H.S.; Xiong, S.D. Induction of systemic lupus erythematosus-like syndrome in syngeneic mice by immunization with activated lymphocyte-derived DNA. Rheumatology 2005, 44, 1108–1114. [Google Scholar] [CrossRef]
- Wen, Z.K.; Xu, W.; Xu, L.; Cao, Q.H.; Wang, Y.; Chu, Y.W.; Xiong, S.D. DNA hypomethylation is crucial for apoptotic DNA to induce systemic lupus erythematosus-like autoimmune disease in SLE-non-susceptible mice. Rheumatology 2007, 46, 1796–1803. [Google Scholar] [CrossRef]
- Yasuda, K.; Richez, C.; Uccellini, M.B.; Richards, R.J.; Bonegio, R.G.; Akira, S.; Monestier, M.; Corley, R.B.; Viglianti, G.A.; Marshak-Rothstein, A.; et al. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J. Immunol. 2009, 183, 3109–3117. [Google Scholar] [CrossRef]
- Richardson, B.; Scheinbart, L.; Strahler, J.; Gross, L.; Hanash, S.; Johnson, M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990, 33, 1665–1673. [Google Scholar] [CrossRef]
- Liang, J.; Zhu, X.H.; Qin, H.H.; Lin, J.R.; Wang, D.Q.; Huang, L.; Luo, X.Q.; Xu, J.H. A correlation study on the effects of DNMT1 on methylation levels in CD4(+) T cells of SLE patients. Int. J. Clin. Exp. Med. 2015, 8, 19701–19708. [Google Scholar]
- Richardson, B.; Sawalha, A.H.; Ray, D.; Yung, R. Murine models of lupus induced by hypomethylated T cells (DNA hypomethylation and lupus.). Methods Mol. Biol. 2012, 900, 169–180. [Google Scholar] [PubMed]
- Lu, Q.; Kaplan, M.; Ray, D.; Ray, D.; Zacharek, S.; Gutsch, D.; Richardson, B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, Y.B.; Pirrotta, V. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef]
- Zhang, Q.; Long, H.; Liao, J.; Zhao, M.; Liang, G.; Wu, X.; Zhang, P.; Ding, S.; Luo, S.; Lu, Q. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J. Autoimmun. 2011, 37, 180–189. [Google Scholar] [CrossRef]
- Gray, S.G. Perspectives on epigenetic-based immune intervention for rheumatic diseases. Arthritis Res. Ther. 2013, 15, 207. [Google Scholar] [CrossRef]
- Tsou, P.S.; Coit, P.; Kilian, N.C.; Sawalha, A.H. EZH2 Modulates the DNA Methylome and Controls T Cell Adhesion Through Junctional Adhesion Molecule A in Lupus Patients. Arthritis Rheumatol. 2018, 70, 98–108. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, W.G. The changing face of HDAC inhibitor depsipeptide. Curr. Cancer Drug Targets 2009, 9, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef] [PubMed]
- Oelke, K.; Lu, Q.; Richardson, D.; Wu, A.; Deng, C.; Hanash, S.; Richardson, B. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum. 2004, 50, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Ulff-Moller, C.J.; Asmar, F.; Liu, Y.; Svendsen, A.J.; Busato, F.; Gronbaek, K.; Tost, J.; Jacobsen, S. Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Esteller, M.; Richardson, B.C. The epigenetic face of systemic lupus erythematosus. J. Immunol. 2006, 176, 7143–7147. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Briggs, S.D.; Allis, C.D. Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 2000, 12, 326–333. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Li, H.; Patel, D.J.; Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983–994. [Google Scholar] [CrossRef]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Kurdistani, S.K.; Grunstein, M. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 2003, 4, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 143–153. [Google Scholar] [CrossRef]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef]
- Passier, R.; Zeng, H.; Frey, N.; Naya, F.J.; Nicol, R.L.; McKinsey, T.A.; Overbeek, P.; Richardson, J.A.; Grant, S.R.; Olson, E.N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Investig. 2000, 105, 1395–1406. [Google Scholar] [CrossRef]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, H.; Jiao, J.; Xu, W. The structure and function of histone deacetylases: The target for anti-cancer therapy. Curr. Med. Chem. 2008, 15, 2840–2849. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.D.; Cai, R.; Bhatia, U.; Asselbergs, F.A.; Song, C.; Terry, R.; Trogani, N.; Widmer, R.; Atadja, P.; Cohen, D. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem. 2002, 277, 6656–6666. [Google Scholar] [CrossRef]
- Guardiola, A.R.; Yao, T.P. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. 2002, 277, 3350–3356. [Google Scholar] [CrossRef]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2008, 86, 537–543. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, J.; Zabidi-Hussin, Z.A.M.H.; Sasongko, T.H. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet. Mol. Biol. 2013, 36, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Dell’Aversana, C.; Benedetti, R.; Petraglia, F.; Carissimo, A.; Petrizzi, V.B.; D’Arco, A.M.; Abbondanza, C.; Nebbioso, A.; Altucci, L. HDAC2 deregulation in tumorigenesis is causally connected to repression of immune modulation and defense escape. Oncotarget 2015, 6, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.F.; Chen, Y.M.; Lin, C.Y.; Huang, H.L.; Yeh, H.; Chang, Y.T.; Huang, K.T.; Lin, M.C. Histone deacetylase 2 (HDAC2) attenuates lipopolysaccharide (LPS)-induced inflammation by regulating PAI-1 expression. J. Inflamm. 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Mielcarek, M.; Zielonka, D.; Carnemolla, A.; Marcinkowski, J.T.; Guidez, F. HDAC4 as a potential therapeutic target in neurodegenerative diseases: A summary of recent achievements. Front. Cell. Neurosci. 2015, 9, 42. [Google Scholar] [CrossRef]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Hohenhaus, D.M.; Kelly, G.M.; Kamal, N.A.; Gupta, P.; Labzin, L.I.; Schroder, K.; Garceau, V.; Barbero, S.; Iyer, A.; et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J. Biol. Chem. 2013, 288, 25362–25374. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Vieson, M.D.; Luo, X.M.; Chafin, C.B.; Puthiyaveetil, A.G.; Hammond, S.E.; Caudell, D.L.; Jarpe, M.B.; Reilly, C.M. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin. Immunol. 2016, 162, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Brown, D.R.; Olorenshaw, I.M.; Kammer, G.M. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc. Natl. Acad. Sci. USA 2001, 98, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Warke, V.G.; Fisher, C.U.; Tsokos, G.C. Effect of trichostatin A on human T cells resembles signaling abnormalities in T cells of patients with systemic lupus erythematosus: A new mechanism for TCR zeta chain deficiency and abnormal signaling. J. Cell. Biochem. 2002, 85, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.M.; Mishra, N.; Miller, J.M.; Joshi, D.; Ruiz, P.; Richon, V.M.; Marks, P.A.; Gilkeson, G.S. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J. Immunol. 2004, 173, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Chen, E.; Li, Y.; Zhu, X.; Zhang, T.; Zhu, X. Effects of Arsenic Trioxide on INF-gamma Gene Expression in MRL/lpr Mice and Human Lupus. Biol. Trace Elem. Res. 2018, 184, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Busby, S.A.; Shabanowitz, J.; Hunt, D.F.; Mishra, N. Resetting the epigenetic histone code in the MRL-lpr/lpr mouse model of lupus by histone deacetylase inhibition. J. Proteome Res. 2005, 4, 2032–2042. [Google Scholar] [CrossRef]
- Lu, Z.P.; Ju, Z.L.; Shi, G.Y.; Zhang, J.W.; Sun, J. Histone deacetylase inhibitor Trichostatin A reduces anti-DNA autoantibody production and represses IgH gene transcription. Biochem. Biophys. Res. Commun. 2005, 330, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.M.; Thomas, M.; Gogal, R., Jr.; Olgun, S.; Santo, A.; Sodhi, R.; Samy, E.T.; Peng, S.L.; Gilkeson, G.S.; Mishra, N. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J. Autoimmun. 2008, 31, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Waibel, M.; Christiansen, A.J.; Hibbs, M.L.; Shortt, J.; Jones, S.A.; Simpson, I.; Light, A.; O’Donnell, K.; Morand, E.F.; Tarlinton, D.M.; et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat. Commun. 2015, 6, 6838. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Long, H.; Zhao, M.; Yin, H.; Lu, Q. Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand. J. Rheumatol. 2009, 38, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.T.; Shi, L.; Maurer, K.; Song, L.; Zhang, Z.; Petri, M.; Sullivan, K.E. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics 2015, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sun, Y.; Gao, F.; Wu, X.; Tang, J.; Yin, H.; Luo, Y.; Richardson, B.; Lu, Q. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J. Autoimmun. 2010, 35, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Cao, Q.; Reilly, C.M.; Young, N.L.; Garcia, B.A.; Mishra, N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Chafin, C.B.; Hammond, S.E.; Puthiyaveetil, A.G.; Caudell, D.L.; Reilly, C.M. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 2014, 151, 29–42. [Google Scholar] [CrossRef] [PubMed]
- White, A.O.; Wood, M.A. Does stress remove the HDAC brakes for the formation and persistence of long-term memory? Neurobiol. Learn. Mem. 2014, 112, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liao, X.; Vieson, M.D.; Chen, M.; Scott, R.; Kazmierczak, J.; Luo, X.M.; Reilly, C.M. Selective HDAC6 inhibition decreases early stage of lupus nephritis by down-regulating both innate and adaptive immune responses. Clin. Exp. Immunol. 2018, 191, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Vieson, M.D.; Gojmerac, A.M.; Khan, D.; Dai, R.; van Duzer, J.H.; Mazitschek, R.; Caudell, D.L.; Liao, X.; Luo, X.M.; Reilly, C.M. Treatment with a selective histone deacetylase 6 inhibitor decreases lupus nephritis in NZB/W mice. Histol. Histopathol. 2017, 32, 1317–1332. [Google Scholar] [PubMed]
- Lennard Richard, M.L.; Nowling, T.K.; Brandon, D.; Watson, D.K.; Zhang, X.K. Fli-1 controls transcription from the MCP-1 gene promoter, which may provide a novel mechanism for chemokine and cytokine activation. Mol. Immunol. 2015, 63, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Karam, E.; Williams, S.; Watson, D.K.; Gilkeson, G.; Zhang, X.K. Fli-1 transcription factor affects glomerulonephritis development by regulating expression of monocyte chemoattractant protein-1 in endothelial cells in the kidney. Clin. Immunol. 2012, 145, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Zhang, X.K. The Friend leukaemia virus integration 1 (Fli-1) transcription factor affects lupus nephritis development by regulating inflammatory cell infiltration into the kidney. Clin. Exp. Immunol. 2014, 177, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Lennard Richard, M.L.; Brandon, D.; Lou, N.; Sato, S.; Caldwell, T.; Nowling, T.K.; Gilkeson, G.; Zhang, X.K. Acetylation impacts Fli-1-driven regulation of granulocyte colony stimulating factor. Eur. J. Immunol. 2016, 46, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tan, Y.; Peng, Q.; Huang, C.; Guo, Y.; Liang, G.; Zhu, B.; Huang, Y.; Liu, A.; Wang, Z.; et al. IL-6/STAT3 pathway induced deficiency of RFX1 contributes to Th17-dependent autoimmune diseases via epigenetic regulation. Nat. Commun. 2018, 9, 583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Long, H.; Chang, C.; Zhao, M.; Lu, Q. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: A comprehensive overview. Cell. Mol. Life Sci. 2018, 75, 3353–3369. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Perl, A. Metabolic control of T cell activation and death in SLE. Autoimmun. Rev. 2009, 8, 184–189. [Google Scholar] [CrossRef]
- Perl, A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. N. Y. Acad. Sci. 2015, 1346, 33–44. [Google Scholar] [CrossRef]
- Shah, D.; Sah, S.; Nath, S.K. Interaction between glutathione and apoptosis in systemic lupus erythematosus. Autoimmun. Rev. 2013, 12, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sivakumar, R.; Titov, A.A.; Choi, S.C.; Morel, L. Metabolic Factors that Contribute to Lupus Pathogenesis. Crit. Rev. Immunol. 2016, 36, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Li, D.; Li, X.; Ren, J.; Xu, J.; Ding, L.; Dou, H.; Hou, Y. mTOR inhibitor INK128 attenuates systemic lupus erythematosus by regulating inflammation-induced CD11b(+)Gr1(+) cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1865, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Perl, A. mTOR signaling: A central pathway to pathogenesis in systemic lupus erythematosus? Discov. Med. 2010, 9, 173–178. [Google Scholar] [PubMed]
- Li, X.; Zhang, X.; Pan, Y.; Shi, G.; Ren, J.; Fan, H.; Dou, H.; Hou, Y. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim. Biophys. Sin. 2018, 50, 888–896. [Google Scholar] [CrossRef]
- Oaks, Z.; Winans, T.; Huang, N.; Banki, K.; Perl, A. Activation of the Mechanistic Target of Rapamycin in SLE: Explosion of Evidence in the Last Five Years. Curr. Rheumatol. Rep. 2016, 18, 73. [Google Scholar] [CrossRef]
- Huang, N.; Perl, A. Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol. 2018, 39, 562–576. [Google Scholar] [CrossRef]
- Lai, Z.W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef]
- Boros, F.A.; Bohar, Z.; Vecsei, L. Genetic alterations affecting the genes encoding the enzymes of the kynurenine pathway and their association with human diseases. Mutat. Res. 2018, 776, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: The serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar] [PubMed]
- Perl, A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Perl, A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8-double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J. Immunol. 2014, 192, 4134–4144. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Zhong, L.; Mostoslavsky, R. The histone deacetylase SIRT6: At the crossroads between epigenetics, metabolism and disease. Curr. Top. Med. Chem. 2013, 13, 2991–3000. [Google Scholar] [CrossRef]
- Chen, H.; Fan, M.; Pfeffer, L.M.; Laribee, R.N. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012, 40, 6534–6546. [Google Scholar] [CrossRef] [PubMed]
- Birner, A.; Platzer, M.; Bengesser, S.A.; Dalkner, N.; Fellendorf, F.T.; Queissner, R.; Pilz, R.; Rauch, P.; Maget, A.; Hamm, C.; et al. Increased breakdown of kynurenine towards its neurotoxic branch in bipolar disorder. PLoS ONE 2017, 12, e0172699. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Grant, R. Effects of Kynurenine Pathway Inhibition on NAD Metabolism and Cell Viability in Human Primary Astrocytes and Neurons. Int. J. Tryptophan Res. 2011, 4, 29–37. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Histone Deacetylases (HDAC) Classification | Enzymatic Activity | Mechanism of Action | Location | Substrates | HDAC Inhibitor | Autoimmunity and Systemic Lupus Erythematosus (SLE) Involvement |
|---|---|---|---|---|---|---|
| Class I. | ||||||
| HDAC1 | Enhanced when incorporated into complexes | 1 class I catalytic domain | Nucleus | p53, RB, MyoD, NF-kB, DNMTI, DNMT3a, MBD2, Sp1, BRCA1, MeCP2, ATM, Smad7 [61,92] | Valproic acid, phenylbutyrate, MS-275, Romidepsin, Suberoylanilide Hydroxamic Acid [93] | Overexpression of HDAC1 increases the activity of the 3’-IgH enhancers. HDAC1 is recruited to the IgH enhancer region, and TSA treatment of B cells reduced the production of anti-DNA autoantibodies. |
| HDAC2 | Enhanced when incorporated into complexes | 1 class I catalytic domain | Nucleus | RB, NF-kB, BRCA1, DNMTI [61] | Valproic Acid, phenylbutyrate, Suberoylanilide Hydroxamic Acid, MS-275, Romidepsin [93,94,95] | Critical for transcriptional regulation, cell cycle progression and developmental processes. |
| HDAC3 | Enhanced when incorporated into complexes | 1 class I catalytic domain | Nucleus/Cytoplasm | RB, NF-kB, Smad7, Stat3, SRY [61] | Valproic Acid, Suberoylanilide Hydroxamic Acid, MS-275 [93,96] | HDAC3 gene expression is decreased in SLE monocytes, involved in macrophage polarization. |
| HDAC8 | Fully active in isolation | 1 class I catalytic domain | Nucleus | Not Reported | Suberoylanilide Hydroxamic Acid, Resveratrol, APHA, Curcumin [93,97] | Downregulate the expression of pro-inflammatory cytokines (TNF-alpha, TGF-beta, IL-1beta, and IL-6). |
| Class IIa. | ||||||
| HDAC4 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | GCMa, GATA-1, HP-1 [92,98,99] | Not reported | Role in pro-inflammatory gene expression. |
| HDAC5 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | GCMa, Smad7, HP-1 [92,100] | TSA [93] | HDAC5 mRNA expression is enhanced in inflammatory states. |
| HDAC7 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | PLAG1, PLAG2 [92,101] | Not reported | Promotes inflammatory responses in macrophages, regulates TLR responses in macrophages, regulates LPS signaling. |
| HDAC9 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | Not Reported | Suberoylanilide Hydroxamic Acid, MS-275 [93] | Regulates Foxp3-dependent suppression. Increase in Treg cells—decrease in suppressive activity. HDAC9 inhibition may benefit SLE patients as shown in MRL/lpr mice. |
| Class IIb. | ||||||
| HDAC6 | Acts on structural proteins | 2 class II catalytic domains with 1215 amino acids. SE14 repeats. BUZ is ZnF domain | Mainly cytoplasmic | Smad7, α-Tubulin, Hsp90 [61,102] | M344 [92,93,103] | HDAC6 is overexpressed in SLE—causes an increased B cell development and response. Inhibition causes reduced germinal center B cells, T follicular cells and IFN-gamma secreting cells. |
| HDAC10 | Not measurable | 2 class II catalytic domains | Nucleus/Cytoplasm | Not reported | Not reported | Overexpressed in B cells from the spleen. |
| Class IV. | ||||||
| HDAC11 | Regulates immune activation and immune tolerance | 1 class IV catalytic domain | Nucleus | Not reported | Not reported | Gene expression is decreased in SLE monocytes, negative transcriptional regulator |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Panther, E.; Liao, X.; Grammer, A.C.; Lipsky, P.E.; Reilly, C.M. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2018, 19, 4007. https://doi.org/10.3390/ijms19124007
Ren J, Panther E, Liao X, Grammer AC, Lipsky PE, Reilly CM. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2018; 19(12):4007. https://doi.org/10.3390/ijms19124007
Chicago/Turabian StyleRen, Jingjing, Eric Panther, Xiaofeng Liao, Amrie C. Grammer, Peter E. Lipsky, and Chris M. Reilly. 2018. "The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus" International Journal of Molecular Sciences 19, no. 12: 4007. https://doi.org/10.3390/ijms19124007
APA StyleRen, J., Panther, E., Liao, X., Grammer, A. C., Lipsky, P. E., & Reilly, C. M. (2018). The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. International Journal of Molecular Sciences, 19(12), 4007. https://doi.org/10.3390/ijms19124007