Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease?


 ,
,  ,
,  ,
,  ,
, 
 ,
, 
 , ,
, ,  ,
,  , add
Show full author list
, add
Show full author list
Abstract
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genetic Analysis
4.3. α-Galactosidase A Activity Assay
4.4. LysoGb3 Determination
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FD | Fabry Disease |
| α-Gal A | α-galactosidase A |
| GVUS | genetic variants of unknown significance |
References
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Kornreich, R.; Desnick, R.J.; Bishop, D.F. Nucleotide sequence of the human alpha-galactosidase A gene. Nucleic Acids Res. 1989, 17, 3301–3302. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. Alpha-galactosidase A deficiency: Fabry disease. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N. Engl. J. Med. 1995, 333, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Kodama, C.; Takenaka, T.; Tanaka, A.; Yasumoto, Y.; Yoshida, A.; Kanzaki, T.; Enriquez, A.L.; Eng, C.M.; Tanaka, H.; et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003, 64, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Lelis, A.; Mirocha, J.; Wilcox, W.R. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet. Med. 2007, 9, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Marchesoni, C.L.; Roa, N.; Pardal, A.M.; Neumann, P.; Cáceres, G.; Martínez, P.; Kisinovsky, I.; Bianchi, S.; Tarabuso, A.L.; Reisin, R.C. Misdiagnosis in Fabry disease. J. Pediatr. 2010, 156, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Lidove, O.; Kaminsky, P.; Hachulla, E.; Leguy-Seguin, V.; Lavigne, C.; Marie, I.; Maillot, F.; Serratrice, C.; Masseau, A.; Chérin, P.; et al. Fabry disease “The New Great Imposter”: Results of the French Observatoire in Internal Medicine Departments (FIMeD). Clin. Genet. 2012, 81, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.P.; Hornemann, T.; Gawinecka, J.; Theswet, E.; Hilz, M.J.; Kasper, D.C. Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients withFabry disease. Mol. Genet. MeTable 2018, 123, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Vieitez, I.; Souto-Rodriguez, O.; Fernandez-Mosquera, L.; San Millan, B.; Teijeira, S.; Fernandez-Martin, J.; Martinez-Sanchez, F.; Aldamiz-Echevarria, L.J.; Lopez-Rodriguez, M.; Navarro, C.; et al. Fabry disease in the Spanish population: Observational study with detection of 77 patients. Orphanet J. Rare Dis. 2018, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Colon, C.; Ortolano, S.; Melcon-Crespo, C.; Alvarez, J.V.; Lopez-Suarez, O.E.; Couce, M.L.; Fernández-Lorenzo, J.R. Newborn screening for Fabry disease in the north-west of Spain. Eur. J. Pediatr. 2017, 176, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Saito, O.; Kusano, E.; Akimoto, T.; Asano, Y.; Kitagawa, T.; Suzuki, K.; Ishige, N.; Akiba, T.; Saito, A.; Ishimura, E.; et al. Prevalence of Fabry disease in dialysis patients: Japan Fabry disease screening study (J-FAST). Clin. Exp. Nephrol. 2016, 20, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Namazova-Baranova, L.S.; Baranov, A.A.; Pushkov, A.A.; Savostyanov, K.V. Fabry disease in children: A federal screening programme in Russia. Eur. J. Pediatr. 2017, 176, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Webster, H.D. Effects of psychosine (galactosylsphingosine) on the survival and the fine structure of cultured Schwann cells. J. Neuropathol. Exp. Neurol. 1993, 52, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Schueler, U.H.; Kolter, T.; Kaneski, C.R.; Blusztajn, J.K.; Herkenham, M.; Sandhoff, K.; Brady, R.O. Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: A model system for assessing neuronal damage in Gaucher disease type 2 and 3. Neurobiol. Dis. 2003, 14, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M. Biomarkers for Fabry Disease. In Fabry Disease; Elstein, D., Altarescu, G., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 133–152. [Google Scholar]
- Rombach, S.M.; Dekker, N.; Bouwman, M.G.; Linthorst, G.E.; Zwinderman, A.H.; Wijburg, F.A.; Kuiper, S.; Vd Bergh Weerman, M.A.; Groener, J.E.; Poorthuis, B.J.; et al. Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochim. Biophys. Acta. 2010, 1802, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Rombach, S.M.; Twickler, T.B.; Aerts, J.M.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E. Vasculopathy in patients with Fabry disease: Current controversies and research directions. Mol. Genet. MeTable 2010, 99, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Van der Tol, L.; Smid, B.E.; Poorthuis, B.J.; Biegstraaten, M.; Deprez, R.H.; Linthorst, G.E.; Hollak, C.E. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.P.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.B. Diagnosis of sphingolipidoses: A new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. 2017, 55, 403–414. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
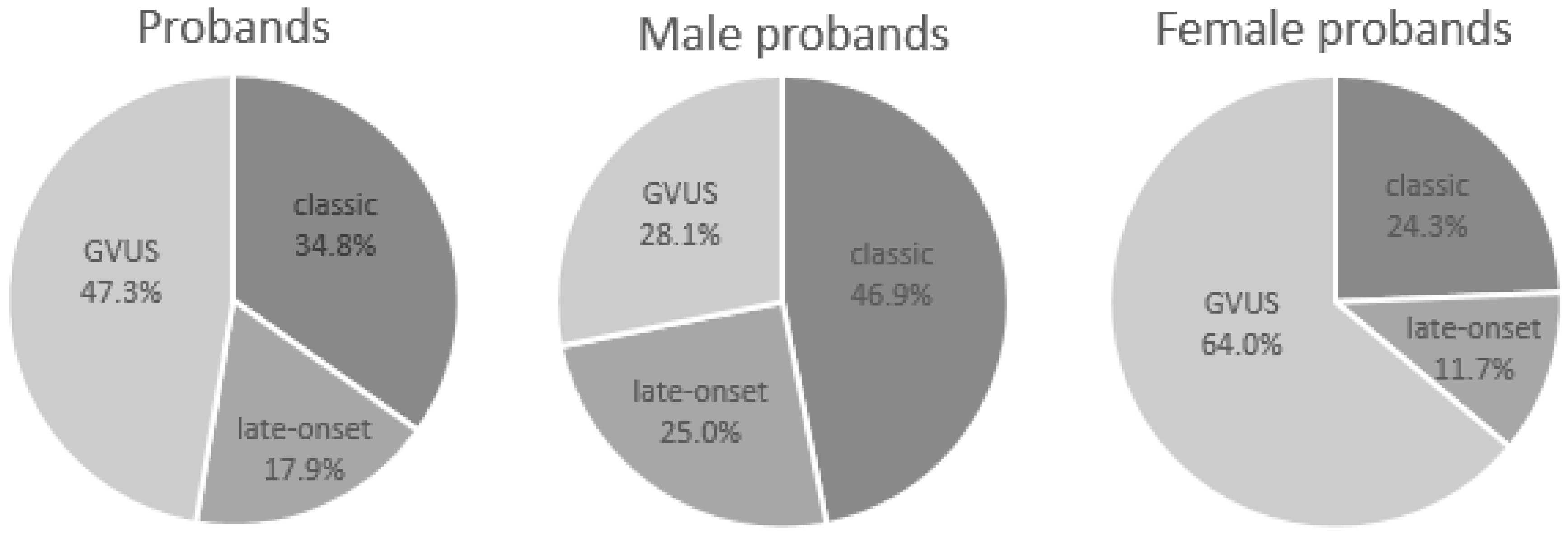
| Overall | Probands | Male Probands | Female Probands |
| Patients (%) | 207 | 96 (46.4%) | 111 (53.6%) |
| Average age [min-max] | 44.9 [0–85] | 42.3 [0–69] | 47.0 [4–85] |
| Average α-Gal A activity | 4.7 | 1.8 | 7.2 |
| Average LysoGb3 | 11.9 | 24.4 | 2.0 |
| Nephrology % | 37.2 | 38.6 | 36.1 |
| Neurology % | 23.7 | 13.5 | 32.4 |
| Cardiology % | 18.4 | 22.9 | 14.4 |
| Pediatrics % | 13.0 | 17.7 | 9.0 |
| Metabolic diseases % | 2.4 | 2.1 | 2.7 |
| Internal medicine % | 1.9 | − | 3.6 |
| Other % | 3.4 | 5.2 | 1.8 |
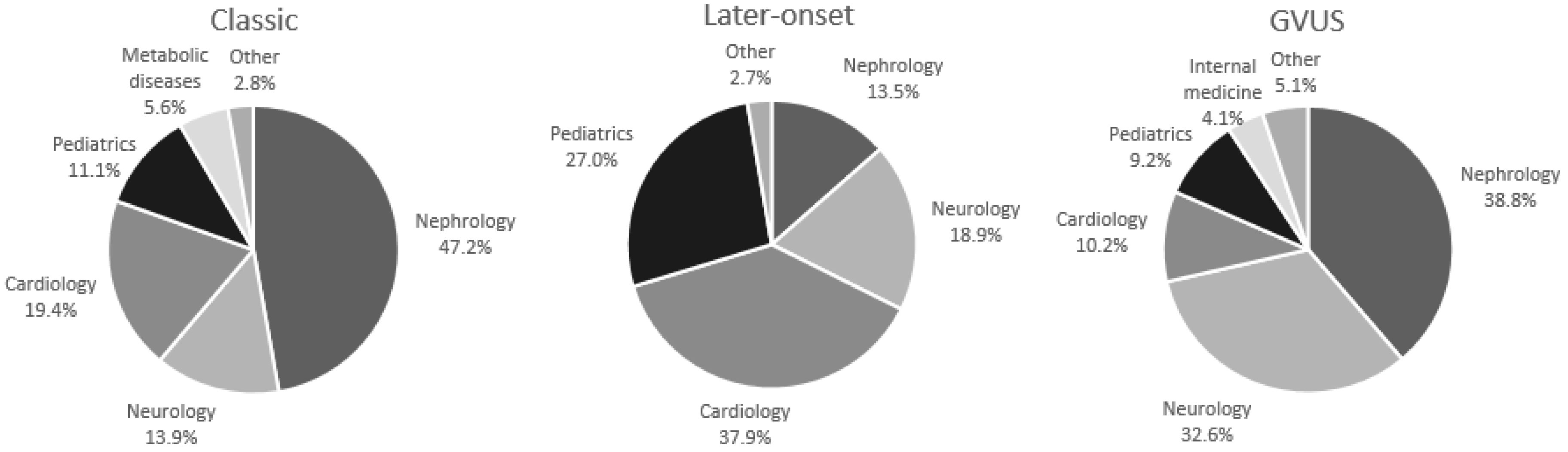
| Classic phenotype (%) | 72 (34.8%) | 45 (46.9%) | 27 (24.3%) |
| Late-onset (%) | 37 (17.9%) | 24 (25.0%) | 13 (11.7%) |
| GVUS (%) | 98 (47.3%) | 27 (28.1%) | 71 (64.0%) |
| Classic Phenotype | Probands | Male Probands | Female Probands |
| Patients (%) | 72 | 45 (62.5%) | 27 (37.5%) |
| Average age [min-max] | 45.8 [0–73] | 45.4 [0–57] | 46.5 [18–73] |
| Average α-Gal A activity | 1.8 | 0.1 | 4.7 |
| Average LysoGb3 | 27.9 | 43.3 | 4.8 |
| Nephrology % | 47.2 | 48.9 | 44.5 |
| Neurology % | 13.9 | 11.1 | 18.5 |
| Cardiology % | 19.4 | 20.0 | 18.5 |
| Pediatrics % | 11.1 | 13.4 | 7.4 |
| Metabolic diseases % | 5.6 | 2.2 | 11.1 |
| Internal medicine % | − | − | − |
| Other % | 2.8 | 4.4 | − |
| Late-Onset | Probands | Male Probands | Female Probands |
| Patients (%) | 37 | 24 (64.9%) | 13 (35.1%) |
| Average age [min-max] | 45.0 [0–78] | 44.7 [0–69] | 45.5 [14–78] |
| Average α-Gal A activity | 2.1 | 0.6 | 4.5 |
| Average LysoGb3 | 4.7 | 5.5 | 3.1 |
| Nephrology % | 13.5 | 20.8 | − |
| Neurology % | 18.9 | 4.2 | 46.1 |
| Cardiology % | 37.9 | 41.6 | 30.8 |
| Pediatrics % | 27.0 | 29.2 | 23.1 |
| Metabolic diseases % | − | − | − |
| Internal medicine % | − | − | − |
| Other % | 2.7 | 4.2 | − |
| GVUS | Probands | Male Probands | Female Probands |
| Patients (%) | 98 | 27 (27.6%) | 71 (72.4%) |
| Average age [min-max] | 44.2 [1–85] | 34.0 [1–58] | 47.3 [4–85] |
| Average α-Gal A activity | 8.2 | 6.2 | 9.0 |
| Average LysoGb3 | 0.9 | 0.8 | 0.9 |
| Nephrology % | 38.8 | 37.1 | 39.4 |
| Neurology % | 32.6 | 25.9 | 35.2 |
| Cardiology % | 10.2 | 11.1 | 9.9 |
| Pediatrics % | 9.2 | 14.8 | 7.1 |
| Metabolic diseases % | 1.0 | 3.7 | − |
| Internal medicine % | 4.1 | − | 5.6 |
| Other % | 4.1 | 7.4 | 2.8 |
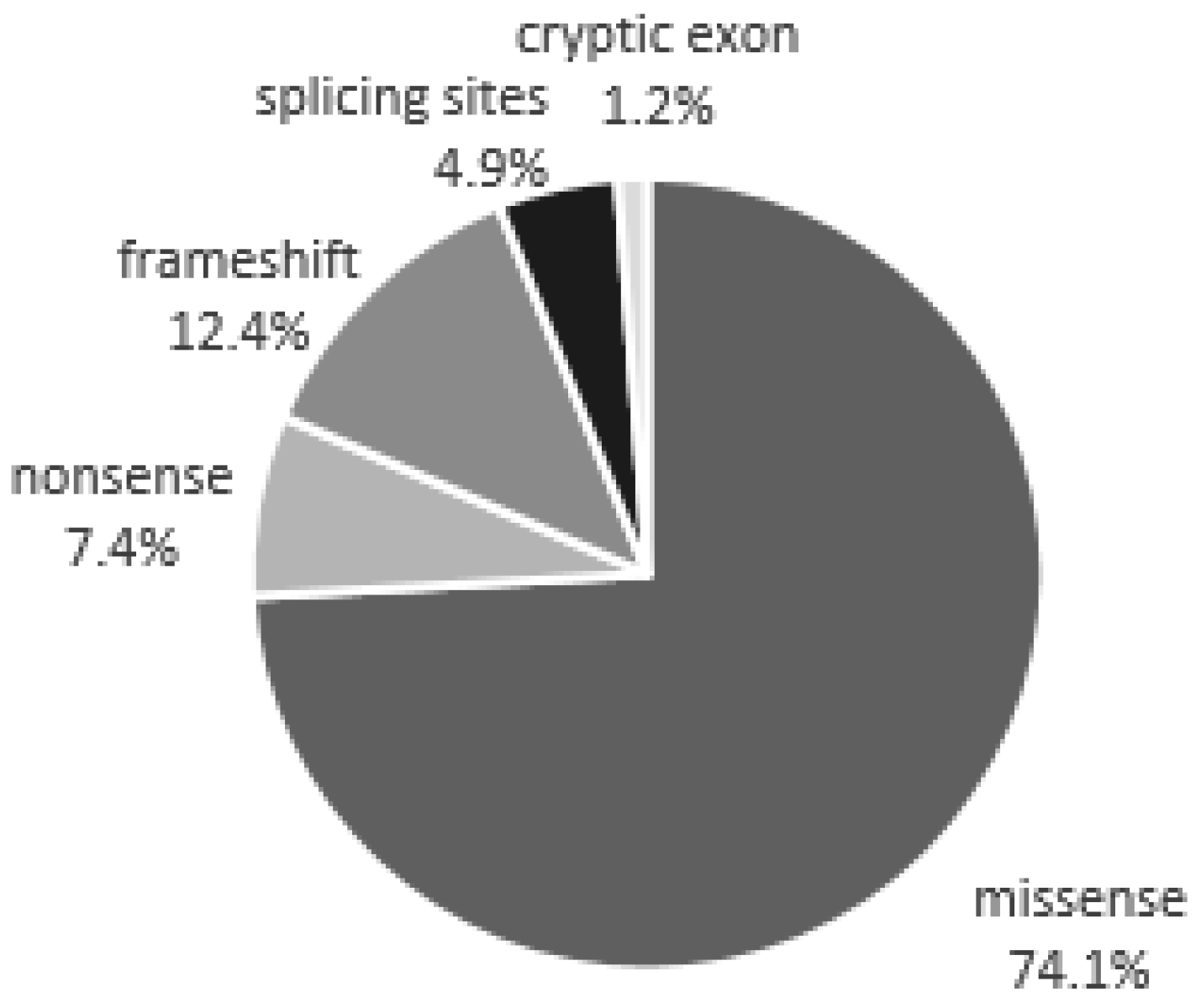
| Mutations Detected in Patients with a Classic Phenotype | |||
| Missense | Nonsense | Frameshift | Splicing sites |
| p.M1T | p.Q57X | c.124_125delTA | IVS2−2>G |
| p.L54F | p.W162X | c.428dupC | IVS3+1G>A |
| p.E59K | p.R220X | c.583insGAATA | IVS4+5G>T |
| p.R112C | p.R227X | c.614delC | IVS5+1G>T |
| p.G116A | p.E341X | c.618del10 + c.629insA | |
| p.Y152H | p.C382X | c.666delC | |
| p.A156V | c.718_719delAA | ||
| p.D165H | c.846_847delTC | ||
| p.D170N | c.946delG | ||
| p.C172Y | c.1163delTCC | ||
| p.G183A | |||
| p.G183S | |||
| p.Y184N | |||
| p.Y222D | |||
| p.C223Y | |||
| p.R227L | |||
| p.W236S | |||
| p.L243F | |||
| p.N249K | |||
| p.G261C | |||
| p.P265L | |||
| p.I270T | |||
| p.L275H | |||
| p.Q279H | |||
| p.Q279K | |||
| p.A288D | |||
| p.M290T | |||
| p.R301G | |||
| p.R342Q | |||
| p.I354K | |||
| p.R356G | |||
| p.R356Q | |||
| p.R356W | |||
| p.E358G | |||
| p.G361R | |||
| p.R363H | |||
| p.G373S | |||
| p.T412P | |||
| Mutations detected in patients with late-onset | |||
| Missense | Cryptic exon | ||
| p.L19P p.V22G p.D33Y | IVS4+919G>A | ||
| p.M51I | |||
| p.I91T | |||
| p.R112H | |||
| p.F113L | |||
| p.K130E | |||
| P.I253T | |||
| p.N215S | |||
| p.T246I | |||
| p.M290L | |||
| p.R301Q | |||
| p.G395A | |||
| p.V413A | |||
| Genetic Variants of Unknown Significance | |||
| Missense | |||
| p.N5D | |||
| p.R118C | |||
| p.R118H | |||
| p.S126G | |||
| p.A143T | |||
| p.D313Y | |||
| p.T385A | |||
| Overall | Patients | Male Patients | Female Patients |
| Patients (%) | 471 | 185 (39.3%) | 286 (60.7%) |
| Average age [min-max] | 40.2 [0–85] | 40.1 [0–73] | 40.2 [1–85] |
| Average α-Gal A activity | 4.1 | 1.9 | 5.6 |
| Average LysoGb3 | 9.7 | 19.8 | 2.6 |
| Classic Phenotype | Patients | Male Patients | Female Patients |
| Patients (%) | 172 | 78 (45.3%) | 94 (54.7%) |
| Average age [min-max] | 38.2 [0–85] | 39.3 [0–63] | 37.3 [6–85] |
| Average α-Gal A activity | 2.2 | 0.1 | 3.9 |
| Average LysoGb3 | 20.3 | 38.0 | 4.6 |
| Late-Onset | Patients | Male Patients | Female Patients |
| Patients (%) | 111 | 47 (42.3%) | 64 (57.7%) |
| Average age [min-max] | 40.2 [0–78] | 45.5 [0–73] | 36.2 [6–78] |
| Average α-Gal A activity | 2.4 | 0.5 | 3.7 |
| Average LysoGb3 | 3.8 | 5.0 | 2.1 |
| GVUS | Patients | Male Patients | Female Patients |
| Patients (%) | 188 | 60 (31.9%) | 128 (68.1%) |
| Average age [min-max] | 41.9 [1–85] | 36.7 [1–77] | 44.2 [1–85] |
| Average α-Gal A activity | 7.2 | 5.5 | 8.1 |
| Average LysoGb3 | 0.9 | 0.8 | 0.9 |
| n. Mutations | % Mutations | n. Probands | % Probands | n. Positives | % Positives | |
|---|---|---|---|---|---|---|
| Classic | 58 | 71.6% | 72 | 34.8% | 172 | 36.5% |
| Late-onset | 16 | 19.8% | 37 | 17.9% | 111 | 23.6% |
| GVUS | 7 | 8.6% | 98 | 47.3% | 188 | 39.9% |
| Overall | 81 | 207 | 471 |
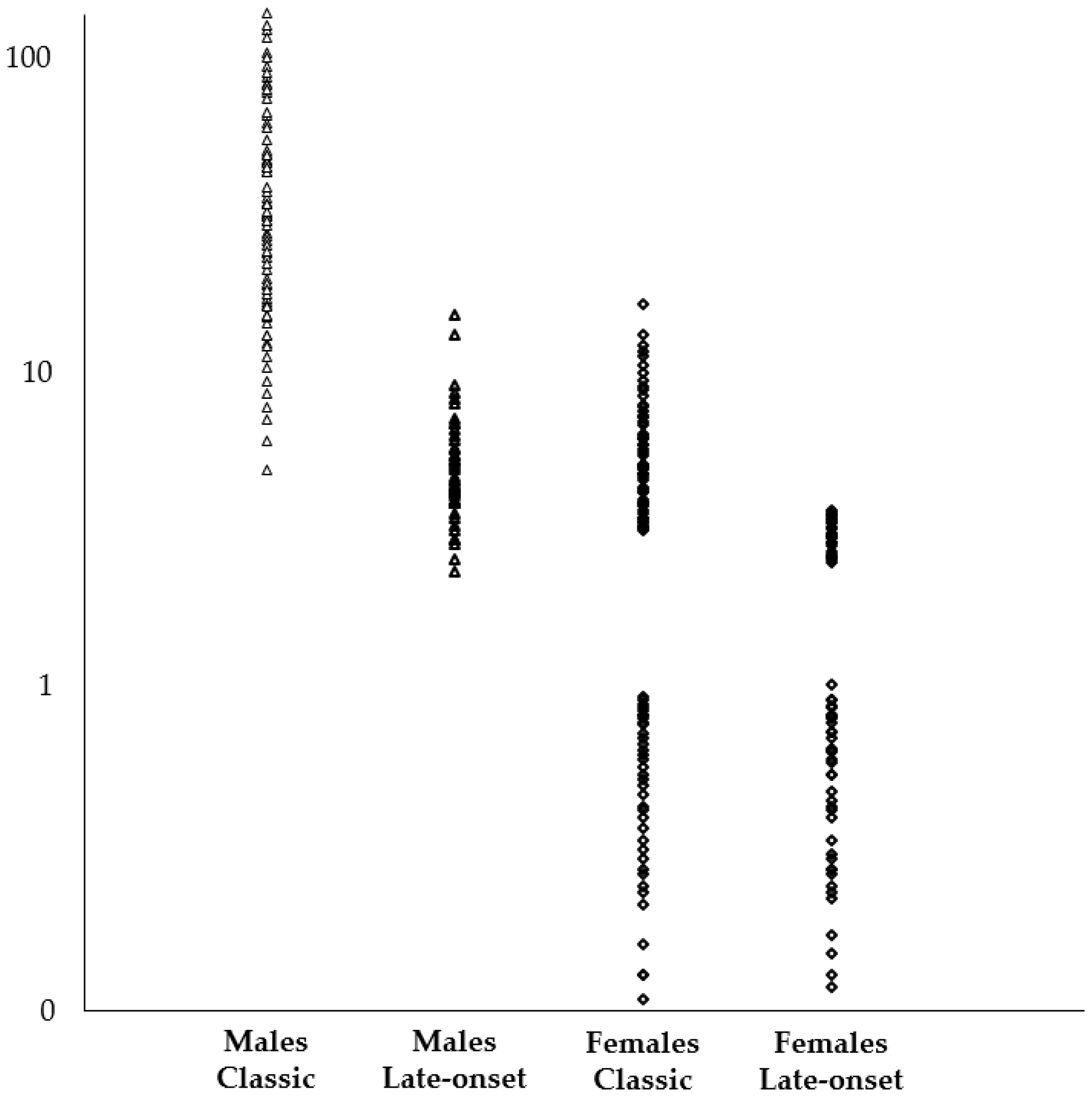
| α-Gal A Activity | % Patients with LysoGb3 in Blood | Average LysoGb3 [min-max] | ||
|---|---|---|---|---|
| Classic Phenotype | Males | Null or almost null | 100% | 38.0 nmol/L [4.8–137.2] |
| Females | Variable | 56.8% | 7.2 nmol/L [3.1–16.2] | |
| Late-onset | Males | Residual | 100% | 5.0 nmol/L [2.3–15] |
| Females | Variable | 40.1% | 3.2 nmol/L [2.5–3.6] | |
| GVUS | Males | Normal | 0 | − |
| Females | Normal | 0 | − | |
| Healthy controls | Males | Normal | 0 | − |
| Females | Normal | 0 | − | |
| Mutation | Symptomatology | α-Gal A Activity | LysoGb3 in Blood |
|---|---|---|---|
| Responsible for Classic Phenotype | Systemic, severe | Null or almost null | Yes (high) |
| Responsible for Late-Onset Phenotype | Single organ and/or late-onset | Residual | Yes (low) |
| GVUS | Mild or absent | Normal | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. https://doi.org/10.3390/ijms19123726
Duro G, Zizzo C, Cammarata G, Burlina A, Burlina A, Polo G, Scalia S, Oliveri R, Sciarrino S, Francofonte D, et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? International Journal of Molecular Sciences. 2018; 19(12):3726. https://doi.org/10.3390/ijms19123726
Chicago/Turabian StyleDuro, Giovanni, Carmela Zizzo, Giuseppe Cammarata, Alessandro Burlina, Alberto Burlina, Giulia Polo, Simone Scalia, Roberta Oliveri, Serafina Sciarrino, Daniele Francofonte, and et al. 2018. "Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease?" International Journal of Molecular Sciences 19, no. 12: 3726. https://doi.org/10.3390/ijms19123726
APA StyleDuro, G., Zizzo, C., Cammarata, G., Burlina, A., Burlina, A., Polo, G., Scalia, S., Oliveri, R., Sciarrino, S., Francofonte, D., Alessandro, R., Pisani, A., Palladino, G., Napoletano, R., Tenuta, M., Masarone, D., Limongelli, G., Riccio, E., Frustaci, A., ... Colomba, P. (2018). Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? International Journal of Molecular Sciences, 19(12), 3726. https://doi.org/10.3390/ijms19123726