NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma



Abstract
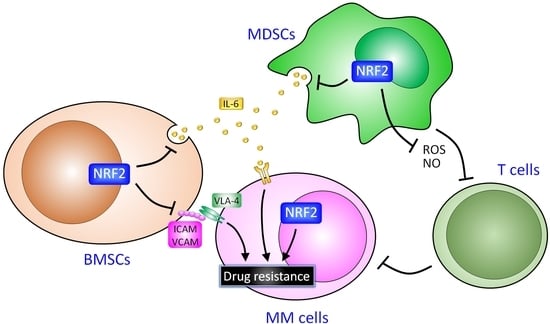
{kind=link}
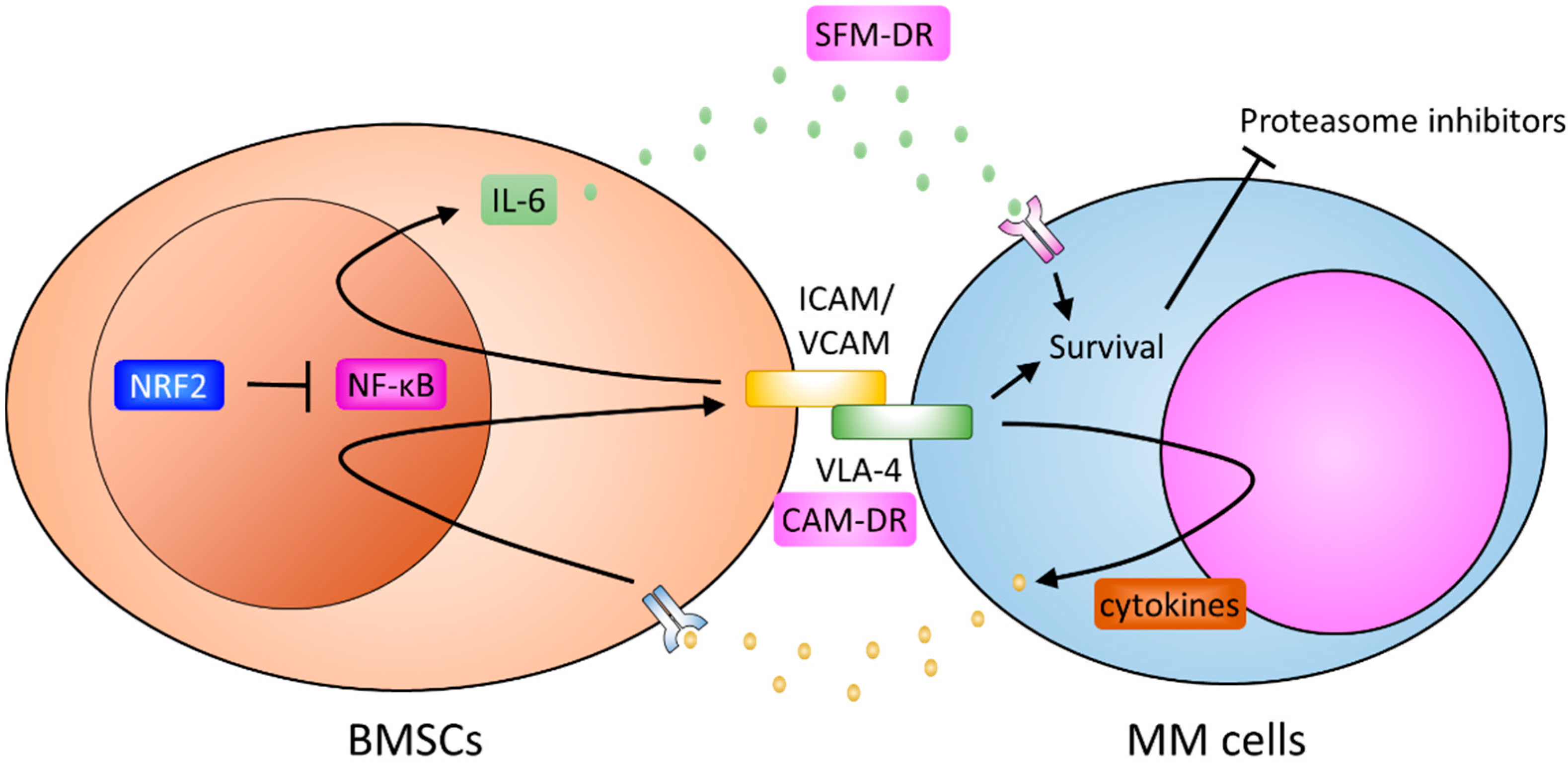{kind=link}
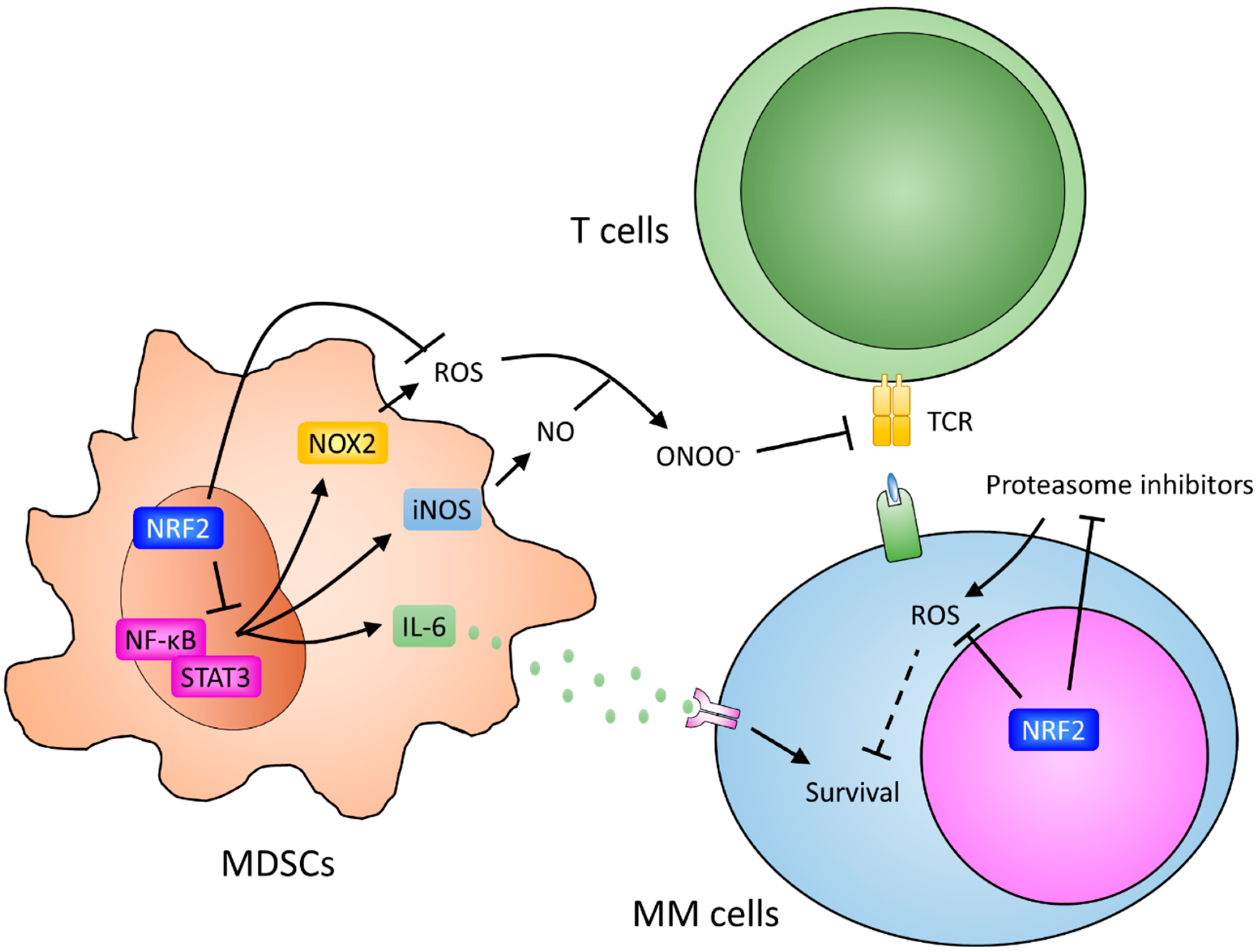{kind=link}
1. Introduction
2. Dual Roles of Nuclear Factor Erythroid-Derived-2-Like 2 (NRF2) in Cancer Development and Therapy
3. Resistance from the Bone Marrow Microenvironment
4. Soluble Factors of Drug Resistance
5. Cell Adhesion Mediated Resistance
6. Bone Marrow Stromal Cells and Resistance
7. Myeloid-Derived Suppressor Cells and Resistance
8. Potential Roles of NRF2 in Cells in the Marrow Microenvironment
9. The Role of NRF2 in MM Cells
10. Interaction of Bone Marrow Microenvironment and MicroRNA
11. Other Environmental Factors for the Resistance
12. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MM | multiple myeloma |
| PIs | proteasome inhibitors |
| IMiDs | immunomodulatory drugs |
| CD | cluster of differentiation |
| SLAMF7 | signal lymphocytic activation molecule F7 |
| BCMA | B cell maturation agents |
| CAR-T | chimeric antigen receptor T |
| MGUS | monoclonal gammopathy of undetermined significance |
| ROS | reactive oxygen species |
| NRF2 | nuclear factor erytheroid-derived-2-like 2 |
| Maf | musculoaponeurotic fibrosarcoma |
| NF-κB | nuclear factor-κB |
| MDSCs | myeloid-derived suppressor cells |
| BMSCs | bone marrow stromal cells |
| SFM-DR | soluble factor-mediated drug resistance |
| CAM-DR | cell-adhesion mediated drug resistance |
| JAK/STAT | Janus kinase/Signal transducer and activator of transcription |
| IL-6 | interleukin-6 |
| VEGF | vascular endothelial growth factor |
| IGF-1 | insulin-like growth factor-1 |
| PI3K | phosphatidylinositol-4,5-bisphosphate 3-kinase |
| HGF | hepatocyte growth factor |
| SDF-1 | stroma-derived factor-1 |
| EGF | epithelial growth factor |
| EGFR | epidermal growth factor receptor |
| TJP1 | tight junction protein 1 |
| TNF-B | tumor necrosis factor-B |
| VLA-4 | Very Late Antigen-4 |
| SDC1 | Syndecan-1 |
| VCAM-1 | vascular Cell Adhesion Molecule-1 |
| LFA-1 | lymphocyte Function-Associated Antigen-1 |
| MUC-1 | Mucin-1 antigen |
| ICAM-1 | intercellular Adhesion Molecule-1 |
| HA | hyaluronic acid |
| RANKL | receptor activator of nuclear kappa B ligand |
| HSP-70 | heat shock protein-70 |
| TNF-α | tumor necrosis factor alpha |
| MIP-1α | macrophage inflammatory protein 1 alpha |
| OPN | osteopontin |
| CXCL12 | c-x-c motif chemokine 12 |
| PSGL1 | P-selectin glycoprotein ligand-1 |
| Erk1/2 | extracellular signal–regulated kinase 1/2 |
| APRIL | proliferation-inducing ligand |
| Tregs | regulatory T cells |
| G-MDSCs | granulocytic MDSCs |
| M-MDSCs | monocytic MDSCs |
| ARG1 | arginase 1 |
| NO | nitric oxide |
| iNOS | inducible NOS |
| NOX | NADPH oxidase |
| TCR | T cell receptor |
| O2− | superoxide ion |
| ONOO− | peroxynitrite |
| CCL2 | C-C motif chemokine ligand 2 |
| IFN | interferon |
| CSCs | cancer stem cells |
| Keap1 | Kelch-like ECH-associated protein 1 |
| HO-1 | heme oxygenase-1 |
| NQO-1 | NAD(P)H:quinone oxidoreductase-1 |
| SOD | superoxide dismutase |
| GCLC | glutamate cysteine ligase |
| GST | glutathione S-transferase |
| AREs | antioxidant response elements |
| HDAC3 | histone deacetylase 3 |
| CBP | CREB-binding protein |
| POMP | proteasome maturation protein |
| PTEN | phosphatase and tensin homolog |
| ER | endoplasmic reticulum |
| IRE1α | kinase/endoribonuclease inositol requiring enzyme 1α |
| PERK | protein kinase RNA-like ER kinase |
| IRE1 | inositol-requiring enzyme 1 |
| ATF6 | activating transcription factor 6 |
| HIF-1α | hypoxia-inducible factor-1α |
| FGF-2 | fibroblast growth factor-2 |
| UPR | unfolded protein response |
| LPS | lipopolysaccharide |
| TLRs | Toll-like receptors |
References
- Kristinsson, S.Y.; Landgren, O.; Dickman, P.W.; Derolf, A.R.; Bjorkholm, M. Patterns of survival in multiple myeloma: A population-based study of patients diagnosed in Sweden from 1973 to 2003. J. Clin. Oncol. 2007, 25, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Rollig, C.; Knop, S.; Bornhauser, M. Multiple myeloma. Lancet 2015, 385, 2197–2208. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Ailawadhi, S.; Advani, P.; Yang, D.; Ghosh, R.; Swaika, A.; Roy, V.; Foran, J.; Colon-Otero, G.; Chanan-Khan, A. Impact of access to NCI- and NCCN-designated cancer centers on outcomes for multiple myeloma patients: A SEER registry analysis. Cancer 2016, 122, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Ailawadhi, S.; Jacobus, S.; Sexton, R.; Stewart, A.K.; Dispenzieri, A.; Hussein, M.A.; Zonder, J.A.; Crowley, J.; Hoering, A.; Barlogie, B.; et al. Disease and outcome disparities in multiple myeloma: Exploring the role of race/ethnicity in the Cooperative Group clinical trials. Blood Cancer J. 2018, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Dingli, D.; Lacy, M.Q.; Dispenzieri, A.; Hayman, S.R.; Buadi, F.K.; Rajkumar, S.V.; Litzow, M.R.; Gertz, M.A. Outcome after autologous stem cell transplantation for multiple myeloma in patients with preceding plasma cell disorders. Br. J. Haematol. 2008, 141, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, J.S.; Touzeau, C.; Moreau, P. Triplet combinations in relapsed/refractory myeloma: Update on recent phase 3 trials. Expert Rev. Hematol. 2017, 10, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Richardson, P.G.; Moreau, P.; Anderson, K.C. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat. Rev. Clin. Oncol. 2015, 12, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Kawano, Y.; Podar, K. Targeting the Bone Marrow Microenvironment. Cancer Treat. Res. 2016, 169, 63–102. [Google Scholar] [PubMed]
- Shank, B.R.; Brown, V.T.; Schwartz, R.N. Multiple myeloma maintenance therapy: A review of the pharmacologic treatment. J. Oncol. Pharm. Pract. 2015, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S. Treatment of MM: Upcoming Novel Therapies. Cancer Treat. Res. 2016, 169, 195–205. [Google Scholar] [PubMed]
- Lonial, S.; Boise, L.H.; Kaufman, J. How I treat high-risk myeloma. Blood 2015, 126, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wasch, R.; Giaccone, L.; Auner, H.W.; Caers, J.; Gramatzki, M.; van de Donk, N.; Oliva, S.; et al. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica 2018, 103, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Schoester, M.; de Leeuw, K. Clinical modulation of multidrug resistance in multiple myeloma: Effect of cyclosporine on resistant tumor cells. J. Clin. Oncol. 1994, 12, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of myeloma. Annu. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bailey, R.J.; Ahmann, G.J.; Rajkumar, S.V.; Hoyer, J.D.; Lust, J.A.; Kyle, R.A.; Gertz, M.A.; Greipp, P.R.; Dewald, G.W. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood 2002, 100, 1417–1424. [Google Scholar] [PubMed]
- Farrell, M.L.; Reagan, M.R. Soluble and Cell-Cell-Mediated Drivers of Proteasome Inhibitor Resistance in Multiple Myeloma. Front. Endocrinol. 2018, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ria, R.; Semeraro, F.; Merchionne, F.; Coluccia, M.; Boccarelli, A.; Scavelli, C.; Nico, B.; Gernone, A.; Battelli, F.; et al. Endothelial cells in the bone marrow of patients with multiple myeloma. Blood 2003, 102, 3340–3348. [Google Scholar] [CrossRef] [PubMed]
- Alimbetov, D.; Askarova, S.; Umbayev, B.; Davis, T.; Kipling, D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1690. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kitadate, A.; Abe, F.; Saitoh, H.; Michishita, Y.; Hatano, Y.; Kawabata, Y.; Kitabayashi, A.; Teshima, K.; Kume, M.; et al. Hypoxia-inducible microRNA-210 regulates the DIMT1-IRF4 oncogenic axis in multiple myeloma. Cancer Sci. 2017, 108, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Shen, H.; Xu, L.; Feng, J.; Tang, H.; Zhang, N.; Chen, X.; Gao, G. Monoclonal Antibodies versus Histone Deacetylase Inhibitors in Combination with Bortezomib or Lenalidomide plus Dexamethasone for the Treatment of Relapsed or Refractory Multiple Myeloma: An Indirect-Comparison Meta-Analysis of Randomized Controlled Trials. J. Immunol. Res. 2018, 2018, 7646913. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Molecular pathogenesis of multiple myeloma. Int. J. Clin. Oncol. 2015, 20, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Ubels, J.; Sonneveld, P.; van Beers, E.H.; Broijl, A.; van Vliet, M.H.; de Ridder, J. Predicting treatment benefit in multiple myeloma through simulation of alternative treatment effects. Nat. Commun. 2018, 9, 2943. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Bahlis, N.J. Targeting of adhesion molecules as a therapeutic strategy in multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 776–796. [Google Scholar] [CrossRef] [PubMed]
- Willenbacher, W.; Seeber, A.; Steiner, N.; Willenbacher, E.; Gatalica, Z.; Swensen, J.; Kimbrough, J.; Vranic, S. Towards Molecular Profiling in Multiple Myeloma: A Literature Review and Early Indications of Its Efficacy for Informing Treatment Strategies. Int. J. Mol. Sci. 2018, 19, 2087. [Google Scholar] [CrossRef] [PubMed]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.; Suzuki, T.; Yamamoto, M. Roles Nrf2 Plays in Myeloid Cells and Related Disorders. Oxid. Med. Cell. Longev. 2013, 2013, 529219. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Kensler, T.W. Nrf2 signaling: An adaptive response pathway for protection against environmental toxic insults. Mutat. Res. 2008, 659, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; She, D.; Sakashita, S.; Zhu, C.Q.; Pintilie, M.; Shepherd, F.A.; Tsao, M.S. NRF2 Pathway Activation and Adjuvant Chemotherapy Benefit in Lung Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Chang, J.T.; Wu, D.W.; Huang, C.C.; Lee, H. Corrigendum to “Cytoplasmic localization of Nrf2 promotes colorectal cancer with more aggressive tumors via upregulation of PSMD4” [Free Radic. Biol. Med. 2016, 95, 121–132]. Free. Radic. Biol. Med. 2017, 104, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Namani, A.; Matiur Rahaman, M.; Chen, M.; Tang, X. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.F.; Yao, J.; Gao, S.G.; Wang, X.S.; Peng, X.Q.; Yang, Y.T.; Feng, X.S. Nrf2 overexpression predicts prognosis and 5-FU resistance in gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5231–5235. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.A.; Niegisch, G.; Albrecht, P.; van Koeveringe, G.; Romano, A.; Albers, P.; Schulz, W.A.; Hoffmann, M.J. Various Mechanisms Involve the Nuclear Factor (Erythroid-Derived 2)-Like (NRF2) to Achieve Cytoprotection in Long-Term Cisplatin-Treated Urothelial Carcinoma Cell Lines. Int. J. Mol. Sci. 2017, 18, 1680. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Choi, B.H.; Kwak, M.K. Activation of NRF2 by p62 and proteasome reduction in sphere-forming breast carcinoma cells. Oncotarget 2015, 6, 8167–8184. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-H.; Kwak, M.-K. Shadows of NRF2 in cancer: Resistance to chemotherapy. Curr. Opin. Toxicol. 2016, 1, 20–28. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Braggio, E.; Kortum, K.M.; Stewart, A.K. SnapShot: Multiple Myeloma. Cancer Cell 2015, 28, 678.e1. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget 2017, 8, 20394–20409. [Google Scholar] [CrossRef] [PubMed]
- Shay, G.; Hazlehurst, L.; Lynch, C.C. Dissecting the multiple myeloma-bone microenvironment reveals new therapeutic opportunities. J. Mol. Med. 2016, 94, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Sobieraj-Teague, M.; Kuss, B.J.; Pitson, S.M. Resistance to proteasome inhibitors and other targeted therapies in myeloma. Br. J. Haematol. 2018, 182, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Carrino, M.; Semenzato, G.; Piazza, F. Old and Young Actors Playing Novel Roles in the Drama of Multiple Myeloma Bone Marrow Microenvironment Dependent Drug Resistance. Int. J. Mol. Sci. 2018, 19, 1512. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.R.; Jaiswal, R.; Brown, R.D.; Luk, F.; Bebawy, M. Multiple myeloma and persistence of drug resistance in the age of novel drugs (Review). Int. J. Oncol. 2016, 49, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Lin, S.F. Mechanisms of Drug Resistance in Relapse and Refractory Multiple Myeloma. BioMed Res. Int. 2015, 2015, 341430. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Dalton, W.S. Environmental-mediated drug resistance: A target for multiple myeloma therapy. Expert Rev. Hematol. 2009, 2, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Matthes, T.; Manfroi, B.; Zeller, A.; Dunand-Sauthier, I.; Bogen, B.; Huard, B. Autocrine amplification of immature myeloid cells by IL-6 in multiple myeloma-infiltrated bone marrow. Leukemia 2015, 29, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: Implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009, 69, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, M.A.; Cusmai, A.; Iodice, G.; Dammacco, F. Autocrine interleukin-6 production and highly malignant multiple myeloma: Relation with resistance to drug-induced apoptosis. Blood 2001, 97, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Chen, Q.; Kuhn, D.J.; Small, G.W.; Hunsucker, S.A.; Strader, J.S.; Corringham, R.E.; Zaki, M.H.; Nemeth, J.A.; Orlowski, R.Z. Inhibition of interleukin-6 signaling with CNTO 328 enhances the activity of bortezomib in preclinical models of multiple myeloma. Clin. Cancer Res. 2007, 13, 6469–6478. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ria, R.; Ribatti, D.; Semeraro, F.; Djonov, V.; Di Raimondo, F.; Dammacco, F. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica 2003, 88, 176–185. [Google Scholar] [PubMed]
- Gupta, D.; Treon, S.P.; Shima, Y.; Hideshima, T.; Podar, K.; Tai, Y.T.; Lin, B.; Lentzsch, S.; Davies, F.E.; Chauhan, D.; et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia 2001, 15, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Zhang, L.; An, G.; Sui, W.; Yu, Z.; Zou, D.; Xu, Y.; Chang, H.; Qiu, L. Suppressing miRNA-15a/-16 expression by interleukin-6 enhances drug-resistance in myeloma cells. J. Hematol. Oncol. 2011, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.J.; Berkova, Z.; Jones, R.J.; Woessner, R.; Bjorklund, C.C.; Ma, W.; Davis, R.E.; Lin, P.; Wang, H.; Madden, T.L.; et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012, 120, 3260–3270. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, L.; Johnsen, H.E.; Maes, K.; Menu, E.; Van Valckenborgh, E.; Overgaard, M.T.; Nyegaard, M.; Conover, C.A.; Vanderkerken, K.; De Bruyne, E. The insulin-like growth factor system in multiple myeloma: Diagnostic and therapeutic potential. Oncotarget 2016, 7, 48732–48752. [Google Scholar] [CrossRef] [PubMed]
- Chiron, D.; Maiga, S.; Surget, S.; Descamps, G.; Gomez-Bougie, P.; Traore, S.; Robillard, N.; Moreau, P.; Le Gouill, S.; Bataille, R.; et al. Autocrine insulin-like growth factor 1 and stem cell factor but not interleukin 6 support self-renewal of human myeloma cells. Blood Cancer J. 2013, 3, e120. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.U.; Baykov, V.; Ro, T.B.; Brabrand, S.; Waage, A.; Sundan, A.; Borset, M. Human myeloma cells adhere to fibronectin in response to hepatocyte growth factor. Haematologica 2005, 90, 479–488. [Google Scholar] [PubMed]
- Zhang, X.D.; Baladandayuthapani, V.; Lin, H.; Mulligan, G.; Li, B.; Esseltine, D.W.; Qi, L.; Xu, J.; Hunziker, W.; Barlogie, B.; et al. Tight Junction Protein 1 Modulates Proteasome Capacity and Proteasome Inhibitor Sensitivity in Multiple Myeloma via EGFR/JAK1/STAT3 Signaling. Cancer Cell 2016, 29, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009, 23, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Yoon, S.S.; Harrison, S.J.; Morris, S.R.; Smith, D.A.; Brigandi, R.A.; Gauvin, J.; Kumar, R.; Opalinska, J.B.; Chen, C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 2014, 124, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef] [PubMed]
- Baljevic, M.; Zaman, S.; Baladandayuthapani, V.; Lin, Y.H.; de Partovi, C.M.; Berkova, Z.; Amini, B.; Thomas, S.K.; Shah, J.J.; Weber, D.M.; et al. Phase II study of the c-MET inhibitor tivantinib (ARQ 197) in patients with relapsed or relapsed/refractory multiple myeloma. Ann. Hematol. 2017, 96, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin. Cancer Res. 2008, 14, 2519–2526. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Dalton, W.S. Cell adhesion is a key determinant in de novo multidrug resistance (MDR): New targets for the prevention of acquired MDR. Mol. Cancer Ther. 2001, 1, 69–78. [Google Scholar] [PubMed]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Damiano, J.S.; Buyuksal, I.; Pledger, W.J.; Dalton, W.S. Adhesion to fibronectin via β1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 2000, 19, 4319–4327. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.Z. Adhesion molecules—The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Koyama, D.; Mukai, H.Y.; Furukawa, Y. Suitable drug combination with bortezomib for multiple myeloma under stroma-free conditions and in contact with fibronectin or bone marrow stromal cells. Int. J. Hematol. 2014, 99, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Morsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for cell adhesion-mediated drug resistance of multiple myeloma cells in vivo. Int. J. Biol. Mark. 2006, 21, 218–222. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Enkemann, S.A.; Beam, C.A.; Argilagos, R.F.; Painter, J.; Shain, K.H.; Saporta, S.; Boulware, D.; Moscinski, L.; Alsina, M.; et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res. 2003, 63, 7900–7906. [Google Scholar] [PubMed]
- Nefedova, Y.; Cheng, P.; Alsina, M.; Dalton, W.S.; Gabrilovich, D.I. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood 2004, 103, 3503–3510. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [PubMed]
- Ayala, F.; Dewar, R.; Kieran, M.; Kalluri, R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia 2009, 23, 2233–2241. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Z.; Yao, C. Survivin is upregulated in myeloma cell lines cocultured with mesenchymal stem cells. Leuk. Res. 2010, 34, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, C.C.; Baladandayuthapani, V.; Lin, H.Y.; Jones, R.J.; Kuiatse, I.; Wang, H.; Yang, J.; Shah, J.J.; Thomas, S.K.; Wang, M.; et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: Therapeutic implications. Leukemia 2014, 28, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Gibbons Johnson, R.M.; Dong, H. Functional Expression of Programmed Death-Ligand 1 (B7-H1) by Immune Cells and Tumor Cells. Front. Immunol. 2017, 8, 961. [Google Scholar] [CrossRef] [PubMed]
- Nimmanapalli, R.; Gerbino, E.; Dalton, W.S.; Gandhi, V.; Alsina, M. HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br. J. Haematol. 2008, 142, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Herlihy, S.E.; Lin, C.; Nefedova, Y. Bone marrow myeloid cells in regulation of multiple myeloma progression. Cancer Immunol. Immunother. 2017, 66, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Karsdal, M.A.; Martin, T.J. Osteoclast-derived coupling factors in bone remodeling. Calcif. Tissue Int. 2014, 94, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zhu, K.; Qiu, L.; Li, S.; Niu, H.; Hao, M.; Yang, S.; Zhao, Z.; Lai, Y.; Anderson, J.L.; et al. Critical role of AKT protein in myeloma-induced osteoclast formation and osteolysis. J. Biol. Chem. 2013, 288, 30399–30410. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Choi, S.J.; Kurihara, N.; Koide, M.; Oba, Y.; Roodman, G.D. Macrophage inflammatory protein-1alpha is an osteoclastogenic factor in myeloma that is independent of receptor activator of nuclear factor κB ligand. Blood 2001, 97, 3349–3353. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Hiura, K.; Wilde, J.; Shioyasono, A.; Moriyama, K.; Hashimoto, T.; Kido, S.; Oshima, T.; Shibata, H.; Ozaki, S.; et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: A vicious cycle between bone destruction and myeloma expansion. Blood 2004, 104, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Bitner, H.; Leiba, M.; Gutwein, O.; Koren-Michowitz, M.; Ostrovsky, O.; Abraham, M.; Wald, H.; Galun, E.; Peled, A.; et al. Multiple myeloma cells recruit tumor-supportive macrophages through the CXCR4/CXCL12 axis and promote their polarization toward the M2 phenotype. Oncotarget 2014, 5, 11283–11296. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cai, Z.; Wang, S.; Zhang, X.; Qian, J.; Hong, S.; Li, H.; Wang, M.; Yang, J.; Yi, Q. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood 2009, 114, 3625–3628. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Campbell, R.A.; Chang, Y.; Li, M.; Wang, C.S.; Li, J.; Sanchez, E.; Share, M.; Steinberg, J.; Berenson, A.; et al. Pleiotrophin produced by multiple myeloma induces transdifferentiation of monocytes into vascular endothelial cells: A novel mechanism of tumor-induced vasculogenesis. Blood 2009, 113, 1992–2002. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, J.; Qian, J.; Qiu, P.; Hanabuchi, S.; Lu, Y.; Wang, Z.; Liu, Z.; Li, H.; He, J.; et al. PSGL-1/selectin and ICAM-1/CD18 interactions are involved in macrophage-induced drug resistance in myeloma. Leukemia 2013, 27, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, C.Y.; Wu, S.S.; Liang, Q.H.; Yuan, L.Q.; Liao, E.Y. Novel adipokines and bone metabolism. Int. J. Endocrinol. 2013, 2013, 895045. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Vazquez Rodriguez, G.; Abrahamsson, A.; Jensen, L.D.E.; Dabrosin, C. Adipocytes Promote Early Steps of Breast Cancer Cell Dissemination via Interleukin-8. Front. Immunol. 2018, 9, 1767. [Google Scholar] [CrossRef] [PubMed]
- Caers, J.; Deleu, S.; Belaid, Z.; De Raeve, H.; Van Valckenborgh, E.; De Bruyne, E.; Defresne, M.P.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 2007, 21, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Falank, C.; Fairfield, H.; Reagan, M.R. Reflections on Cancer in the Bone Marrow: Adverse Roles of Adipocytes. Curr. Mol. Biol. Rep. 2017, 3, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Vicennati, V.; Vottero, A.; Friedman, C.; Papanicolaou, D.A. Hormonal regulation of interleukin-6 production in human adipocytes. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.A.; Oberheu, K.; Polusani, S.R.; Ortega, V.; Velagaleti, G.V.; Oyajobi, B.O. PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia 2014, 28, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- El Arfani, C.; De Veirman, K.; Maes, K.; De Bruyne, E.; Menu, E. Metabolic Features of Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 1200. [Google Scholar] [CrossRef] [PubMed]
- Westhrin, M.; Moen, S.H.; Kristensen, I.B.; Buene, G.; Mylin, A.K.; Turesson, I.; Abildgaard, N.; Waage, A.; Standal, T. Chemerin is elevated in multiple myeloma patients and is expressed by stromal cells and pre-adipocytes. Biomark. Res. 2018, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Gerace, D.; Allegra, A.G.; Vaddinelli, D.; Bianco, O.; Musolino, C. The adipose organ and multiple myeloma: Impact of adipokines on tumor growth and potential sites for therapeutic intervention. Eur. J. Intern. Med. 2018, 53, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.V.; Edwards, C.M. Adipokines, adiposity, and bone marrow adipocytes: Dangerous accomplices in multiple myeloma. J. Cell. Physiol. 2018, 233, 9159–9166. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Pope, B.; Murray, A.; Esdale, W.; Sze, D.M.; Gibson, J.; Ho, P.J.; Hart, D.; Joshua, D. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood 2001, 98, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.; Fernandes, S.; Melinkeri, S.; Kale, V.; Limaye, L. Compromised functionality of monocyte-derived dendritic cells in multiple myeloma patients may limit their use in cancer immunotherapy. Sci. Rep. 2018, 8, 5705. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Gunther, A.; Klausz, K.; Staudinger, M.; Peipp, M.; Penas, E.M.; Rose-John, S.; Wijdenes, J.; Gramatzki, M. Due to interleukin-6 type cytokine redundancy only glycoprotein 130 receptor blockade efficiently inhibits myeloma growth. Haematologica 2017, 102, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Ratta, M.; Fagnoni, F.; Curti, A.; Vescovini, R.; Sansoni, P.; Oliviero, B.; Fogli, M.; Ferri, E.; Della Cuna, G.R.; Tura, S.; et al. Dendritic cells are functionally defective in multiple myeloma: The role of interleukin-6. Blood 2002, 100, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, A.; Hutchinson, A.; Dhodapkar, K.; Mazumder, A.; Vesole, D.; Angitapalli, R.; Jagannath, S.; Dhodapkar, M.V. Enhancement of clonogenicity of human multiple myeloma by dendritic cells. J. Exp. Med. 2006, 203, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Winter, O.; Hartig, C.; Siebels, S.; Szyska, M.; Tiburzy, B.; Meng, L.; Kulkarni, U.; Fahnrich, A.; Bommert, K.; et al. Eosinophils and megakaryocytes support the early growth of murine MOPC315 myeloma cells in their bone marrow niches. PLoS ONE 2014, 9, e109018. [Google Scholar] [CrossRef] [PubMed]
- Duwe, A.K.; Singhal, S.K. The immunoregulatory role of bone marrow. I. Suppression of the induction of antibody responses to T-dependent and T-independent antigens by cells in the bone marrow. Cell. Immunol. 1979, 43, 362–371. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425, author reply 426. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed]
- Hestdal, K.; Ruscetti, F.W.; Ihle, J.N.; Jacobsen, S.E.; Dubois, C.M.; Kopp, W.C.; Longo, D.L.; Keller, J.R. Characterization and regulation of RB6-8C5 antigen expression on murine bone marrow cells. J. Immunol. 1991, 147, 22–28. [Google Scholar] [PubMed]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.B.; Raveney, B.J.; Munder, M. Monocyte dependent regulation of autoimmune inflammation. Curr. Mol. Med. 2009, 9, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Damuzzo, V.; Pinton, L.; Desantis, G.; Solito, S.; Marigo, I.; Bronte, V.; Mandruzzato, S. Complexity and challenges in defining myeloid-derived suppressor cells. Cytom. Part B Clin. Cytom. 2015, 88, 77–91. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Van Ginderachter, J.A.; Lub, S.; De Beule, N.; Thielemans, K.; Bautmans, I.; Oyajobi, B.O.; De Bruyne, E.; Menu, E.; Lemaire, M.; et al. Multiple myeloma induces Mcl-1 expression and survival of myeloid-derived suppressor cells. Oncotarget 2015, 6, 10532–10547. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.R.; Martner, A.; Pisklakova, A.; Condamine, T.; Chase, T.; Vogl, T.; Roth, J.; Gabrilovich, D.; Nefedova, Y. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J. Immunol. 2013, 190, 3815–3823. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, J.; Liyadipitiya, T.; Brown, R.; Yang, S.; Suen, H.; Woodland, N.; Nassif, N.; Hart, D.; Fromm, P.; Weatherburn, C.; et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk. Lymphoma 2014, 55, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Pyzer, A.R.; Cole, L.; Rosenblatt, J.; Avigan, D.E. Myeloid-derived suppressor cells as effectors of immune suppression in cancer. Int. J. Cancer 2016, 139, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Ibanez-Vea, M.; Zuazo, M.; Gato, M.; Arasanz, H.; Fernandez-Hinojal, G.; Escors, D.; Kochan, G. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Current Knowledge and Future Perspectives. Arch. Immunol. Ther. Exp. 2018, 66, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T cell receptor expression and antigen-specific T cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Matsui, S.; Park, J.M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med. 2003, 198, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Vickers, S.M.; MacMillan-Crow, L.A.; Green, M.; Ellis, C.; Thompson, J.A. Association of increased immunostaining for inducible nitric oxide synthase and nitrotyrosine with fibroblast growth factor transformation in pancreatic cancer. Arch. Surg. 1999, 134, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.V.; Martins, A.R.; de Nucci, G.; Murad, F.; Soares, F.A. Expression of nitric oxide synthase isoforms and nitrotyrosine immunoreactivity by B cell non-Hodgkin’s lymphomas and multiple myeloma. Histopathology 2001, 39, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 2005, 201, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Mundy-Bosse, B.L.; Lesinski, G.B.; Jaime-Ramirez, A.C.; Benninger, K.; Khan, M.; Kuppusamy, P.; Guenterberg, K.; Kondadasula, S.V.; Chaudhury, A.R.; La Perle, K.M.; et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011, 71, 5101–5110. [Google Scholar] [CrossRef] [PubMed]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar] [PubMed]
- Szuster-Ciesielska, A.; Hryciuk-Umer, E.; Stepulak, A.; Kupisz, K.; Kandefer-Szerszen, M. Reactive oxygen species production by blood neutrophils of patients with laryngeal carcinoma and antioxidative enzyme activity in their blood. Acta Oncol. 2004, 43, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer 2014, 134, 2853–2864. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.G.; Borutaite, V.; Brown, G.C. Superoxide dismutase and hydrogen peroxide cause rapid nitric oxide breakdown, peroxynitrite production and subsequent cell death. Biochim. Biophys. Acta 1999, 1454, 275–288. [Google Scholar] [CrossRef]
- Segal, B.H.; Veys, P.; Malech, H.; Cowan, M.J. Chronic granulomatous disease: Lessons from a rare disorder. Biol. Blood Marrow Transplant. 2011, 17, S123–S131. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Megiel, C.; Russell, S.M.; Bingham, B.; Arger, N.; Woo, T.; Epstein, A.L. Functional characterization of human Cd33+ and Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J. Transl. Med. 2011, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Chen, E.I. Cancer stem cells, tumor dormancy, and metastasis. Front. Endocrinol. 2012, 3, 125. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.; Zhang, X.G.; Jourdan, M.; Boiron, J.M.; Portier, M.; Lu, Z.Y.; Wijdenes, J.; Brochier, J.; Bataille, R. Interleukin-6 is the central tumor growth factor in vitro and in vivo in multiple myeloma. Eur. Cytokine Netw. 1990, 1, 193–201. [Google Scholar] [PubMed]
- Bataille, R.; Jourdan, M.; Zhang, X.G.; Klein, B. Serum levels of interleukin 6, a potent myeloma cell growth factor, as a reflect of disease severity in plasma cell dyscrasias. J. Clin. Investig. 1989, 84, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Lee, O.Y.; Shon, S.Y.; Nam, O.; Ryu, P.M.; Seo, M.W.; Lee, D.S. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013, 15, R79. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.L.; Zhang, J.; Dong, L.W.; Fu, W.J.; Du, J.; Shi, H.G.; Jiang, H.; Ye, F.; Xi, H.; Zhang, C.Y.; et al. URI regulates tumorigenicity and chemotherapeutic resistance of multiple myeloma by modulating IL-6 transcription. Cell Death Dis. 2014, 5, e1126. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; De Beule, N.; Maes, K.; Menu, E.; De Bruyne, E.; De Raeve, H.; Fostier, K.; Moreaux, J.; Kassambara, A.; Hose, D.; et al. Extracellular S100A9 Protein in Bone Marrow Supports Multiple Myeloma Survival by Stimulating Angiogenesis and Cytokine Secretion. Cancer Immunol. Res. 2017, 5, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, I.R.; Condamine, T.; Lin, C.; Herlihy, S.E.; Garfall, A.; Vogl, D.T.; Gabrilovich, D.I.; Nefedova, Y. Bone marrow PMN-MDSCs and neutrophils are functionally similar in protection of multiple myeloma from chemotherapy. Cancer Lett. 2016, 371, 117–124. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Van Ginderachter, J.A.; Van Riet, I.; De Beule, N.; Lub, S.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Van Valckenborgh, E. Myeloid Derived Suppressor Cell Mediated AMPK Activation Regulates Multiple Myeloma Cell Survival. Blood 2014, 124, 2009. [Google Scholar]
- Binsfeld, M.; Muller, J.; Lamour, V.; De Veirman, K.; De Raeve, H.; Bellahcene, A.; Van Valckenborgh, E.; Baron, F.; Beguin, Y.; Caers, J.; et al. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 2016, 7, 37931–37943. [Google Scholar] [CrossRef] [PubMed]
- Buelna-Chontal, M.; Zazueta, C. Redox activation of Nrf2 & NF-κB: A double end sword? Cell Signal. 2013, 25, 2548–2557. [Google Scholar] [PubMed]
- Liu, G.H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, F.; Kang, L.; Wang, Z.; Wang, Y. Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating Nrf2/Bach1 equilibrium. BMC Pulm. Med. 2017, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Huang, Q.; Chen, J.; Wang, M.; Pan, H.; Wang, R.; Zhou, H.; Zhou, Z.; Liu, J.; Yang, F.; et al. Calycosin suppresses expression of pro-inflammatory cytokines via the activation of p62/Nrf2-linked heme oxygenase 1 in rheumatoid arthritis synovial fibroblasts. Pharmacol. Res. 2016, 113, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Kang, B.; Suh, H.J.; Choi, H.-S. Red ginseng-derived saponin fraction suppresses the obesity-induced inflammatory responses via Nrf2-HO-1 pathway in adipocyte-macrophage co-culture system. Biomed. Pharmacother. 2018, 108, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.M.; Suh, K.S.; Kim, Y.J.; Hong, S.M.; Park, S.Y.; Chon, S. Glabridin Alleviates the Toxic Effects of Methylglyoxal on Osteoblastic MC3T3-E1 Cells by Increasing Expression of the Glyoxalase System and Nrf2/HO-1 Signaling and Protecting Mitochondrial Function. J. Agric. Food Chem. 2016, 64, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Lai, Y.S.; Tsai, H.J.; Kuo, C.C.; Yen, B.L.; Yeh, S.P.; Sun, H.S.; Hung, W.C. The oncometabolite R-2-hydroxyglutarate activates NF-κB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. 2016, 6, 32428. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Guo, Y.; Yu, J.; Qi, J.; Shi, W.; Wu, X.; Ni, H.; Ju, S. miRNA-202 in bone marrow stromal cells affects the growth and adhesion of multiple myeloma cells by regulating B cell-activating factor. Clin. Exp. Med. 2016, 16, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin prevents reperfusion injury following ischemic stroke in rats via inhibition of NFκB, ICAM-1, MMP-9 and caspase-3 expression. Mol. Med. Rep. 2017, 16, 4710–4720. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Rajesh, M.; Zhang, J.; Zhou, S.; Wang, S.; Sun, J.; Tan, Y.; Zheng, Y.; Cai, L. Protection by dimethyl fumarate against diabetic cardiomyopathy in type 1 diabetic mice likely via activation of nuclear factor erythroid-2 related factor 2. Toxicol. Lett. 2018, 287, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Yu, Z.; Zheng, Y.; Wang, L.; Qin, X.; Cheng, G.; Ci, X. Isovitexin Exerts Anti-Inflammatory and Anti-Oxidant Activities on Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting MAPK and NF-κB and Activating HO-1/Nrf2 Pathways. Int. J. Biol. Sci. 2016, 12, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.W.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994, 269, 4705–4708. [Google Scholar] [PubMed]
- Goldring, C.E.; Narayanan, R.; Lagadec, P.; Jeannin, J.F. Transcriptional inhibition of the inducible nitric oxide synthase gene by competitive binding of NF-κB/Rel proteins. Biochem. Biophys. Res. Commun. 1995, 209, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Ye, X.; Malik, A.B. In vivo inhibition of nuclear factor-κB activation prevents inducible nitric oxide synthase expression and systemic hypotension in a rat model of septic shock. J. Immunol. 1997, 159, 3976–3983. [Google Scholar] [PubMed]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M. Regulation of NADPH oxidase subunit p22(phox) by NF-kB in human aortic smooth muscle cells. Arch. Physiol. Biochem. 2007, 113, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Greifenberg, V.; Ribechini, E.; Rossner, S.; Lutz, M.B. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur. J. Immunol. 2009, 39, 2865–2876. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Poe, S.L.; Oriss, T.B.; Krishnamoorthy, N.; Yarlagadda, M.; Wenzel, S.E.; Billiar, T.R.; Ray, A.; Ray, P. TLR4/MyD88-induced CD11b+Gr-1intF4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. 2010, 3, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bi, Y.; Han, F.; Lu, Y.; Wang, J.; Zhang, Z.; Liu, G. Myeloid-derived suppressor cells in immunity and autoimmunity. Expert Rev. Clin. Immunol. 2015, 11, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000, 25, 496–502. [Google Scholar] [CrossRef]
- Wojciak, J.M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009, 28, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T., Jr.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Youn, J.I.; Weber, H.; Iclozan, C.; Lu, L.; Cotter, M.J.; Meyer, C.; Becerra, C.R.; Fishman, M.; Antonia, S.; et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Yu Sun, L.Z.; Shafat, M.S.; Bowles, K.M.; Rushworth, S.A. Dual Activation of NRF2 in Multiple Myeloma and Bone Marrow Mesenchymal Stromal Cells Regulates Chemotherapy Resistance. Blood 2016, 128, 3287. [Google Scholar]
- Barrera, L.N.; Rushworth, S.A.; Bowles, K.M.; MacEwan, D.J. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle 2012, 11, 2248–2252. [Google Scholar] [CrossRef] [PubMed]
- Riz, I.; Hawley, T.S.; Marsal, J.W.; Hawley, R.G. Noncanonical SQSTM1/p62-Nrf2 pathway activation mediates proteasome inhibitor resistance in multiple myeloma cells via redox, metabolic and translational reprogramming. Oncotarget 2016, 7, 66360–66385. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, H.; Orlowski, R.Z. Proteasome Maturation Protein (POMP) is Associated with Proteasome Inhibitor Resistance in Myeloma, and Its Suppression Enhances The Activity of Bortezomib and Carfilzomib. Blood 2013, 122, 280. [Google Scholar]
- Li, B.; Fu, J.; Chen, P.; Ge, X.; Li, Y.; Kuiatse, I.; Wang, H.; Wang, H.; Zhang, X.; Orlowski, R.Z. The Nuclear Factor (Erythroid-derived 2)-like 2 and Proteasome Maturation Protein Axis Mediate Bortezomib Resistance in Multiple Myeloma. J. Biol. Chem. 2015, 290, 29854–29868. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Tuckett, A.Z.; Fennell, M.; Garippa, R.; Zakrzewski, J.L. Repurposing of the CDK inhibitor PHA-767491 as a NRF2 inhibitor drug candidate for cancer therapy via redox modulation. Investig. New Drugs 2018, 36, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Tian, W.; La Clair, J.J.; Tan, A.C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Wakabayashi, N.; Kensler, T.W. Keap1/Nrf2 pathway in the frontiers of cancer and non-cancer cell metabolism. Biochem. Soc. Trans. 2015, 43, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Otsuki, A.; Keleku-Lukwete, N.; Yamamoto, M. Overview of redox regulation by Keap1–Nrf2 system in toxicology and cancer. Curr. Opin. Toxicol. 2016, 1, 29–36. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Yang, Y.-X.; Zhe, H.; He, Z.-X.; Zhou, S.-F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Dev. Ther. 2014, 8, 2075–2088. [Google Scholar]
- Zhu, B.; Ju, S.; Chu, H.; Shen, X.; Zhang, Y.; Luo, X.; Cong, H. The potential function of microRNAs as biomarkers and therapeutic targets in multiple myeloma. Oncol. Lett. 2018, 15, 6094–6106. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Rukov, J.L.; Wilentzik, R.; Jaffe, I.; Vinther, J.; Shomron, N. Pharmaco-miR: Linking microRNAs and drug effects. Brief. Bioinform. 2014, 15, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Rastgoo, N.; Abdi, J.; Hou, J.; Chang, H. Role of epigenetics-microRNA axis in drug resistance of multiple myeloma. J. Hematol. Oncol. 2017, 10, 121. [Google Scholar] [CrossRef] [PubMed]
- Leone, E.; Morelli, E.; Di Martino, M.T.; Amodio, N.; Foresta, U.; Gulla, A.; Rossi, M.; Neri, A.; Giordano, A.; Munshi, N.C.; et al. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin. Cancer Res. 2013, 19, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Yao, Y.; Wang, Y.; Liu, B.; Wu, W.; Chen, J.; Su, F.; Yao, H.; Song, E. Up-regulation of miR-21 mediates resistance to trastuzumab therapy for breast cancer. J. Biol. Chem. 2011, 286, 19127–19137. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Dubaybo, H.; Ali, S.; Goncalves, P.; Kollepara, S.L.; Sethi, S.; Philip, P.A.; Li, Y. Down-regulation of miR-221 inhibits proliferation of pancreatic cancer cells through up-regulation of PTEN, p27(kip1), p57(kip2), and PUMA. Am. J. Cancer Res. 2013, 3, 465–477. [Google Scholar] [PubMed]
- Wang, X.; Li, C.; Ju, S.; Wang, Y.; Wang, H.; Zhong, R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk. Lymphoma 2011, 52, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-J.; Yu, J.; Li, J.-Y.; Liu, Y.-T.; Zhong, R.-Q. Circulating microRNA expression is associated with genetic subtype and survival of multiple myeloma. Med. Oncol. 2012, 29, 2402–2408. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-J.; Chu, Z.-B.; Hu, Y.; Lin, J.; Wang, Z.; Jiang, M.; Chen, M.; Wang, X.; Kang, Y.; Zhou, Y.; et al. Targeting the miR-221-222/PUMA/BAK/BAX Pathway Abrogates Dexamethasone Resistance in Multiple Myeloma. Cancer Res. 2015, 75, 4384–4397. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.-D.; Zhang, H.; Zhang, P.; Zheng, Y.-S.; Zhang, X.-J.; Han, B.-W.; Luo, X.-Q.; Xu, L.; Zhou, H.; Qu, L.-H.; et al. Down-regulated miR-331-5p and miR-27a are associated with chemotherapy resistance and relapse in leukaemia. J. Cell. Mol. Med. 2011, 15, 2164–2175. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Jian, H.; Chang, H. Role of micro-RNAs in drug resistance of multiple myeloma. Oncotarget 2016, 7, 60723–60735. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.K.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.; et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 2006, 1, e116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shu, L.; Kong, A.N. MicroRNAs: New players in cancer prevention targeting Nrf2, oxidative stress and inflammatory pathways. Curr. Pharmacol. Rep. 2015, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.M.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J.; Rushworth, S.A. NRF2-driven miR-125B1 and miR-29B1 transcriptional regulation controls a novel anti-apoptotic miRNA regulatory network for AML survival. Cell Death Differ. 2015, 22, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Vincenz, L.; Jager, R.; O’Dwyer, M.; Samali, A. Endoplasmic reticulum stress and the unfolded protein response: Targeting the Achilles heel of multiple myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef] [PubMed]
- White-Gilbertson, S.; Hua, Y.; Liu, B. The role of endoplasmic reticulum stress in maintaining and targeting multiple myeloma: A double-edged sword of adaptation and apoptosis. Front. Genet. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Mutis, T.; Garssen, J.; Redegeld, F. Characterization of the Toll-like receptor expression profile in human multiple myeloma cells. PLoS ONE 2013, 8, e60671. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Garssen, J.; Redegeld, F. Toll-like receptors in human multiple myeloma: New insight into inflammation-related pathogenesis. Curr. Mol. Med. 2014, 14, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenet. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Kizaki, M.; Tabayashi, T. The Role of Intracellular Signaling Pathways in the Pathogenesis of Multiple Myeloma and Novel Therapeutic Approaches. J. Clin. Exp. Hematop. 2016, 56, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. The Multiple Myeloma Drug Pipeline-2018: A Review of Small Molecules and Their Therapeutic Targets. Clin. Lymphoma Myeloma Leuk. 2018, 18, 611–627. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yen, C.-H.; Hsiao, H.-H. NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 3503. https://doi.org/10.3390/ijms19113503
Yen C-H, Hsiao H-H. NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma. International Journal of Molecular Sciences. 2018; 19(11):3503. https://doi.org/10.3390/ijms19113503
Chicago/Turabian StyleYen, Chia-Hung, and Hui-Hua Hsiao. 2018. "NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma" International Journal of Molecular Sciences 19, no. 11: 3503. https://doi.org/10.3390/ijms19113503
APA StyleYen, C.-H., & Hsiao, H.-H. (2018). NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma. International Journal of Molecular Sciences, 19(11), 3503. https://doi.org/10.3390/ijms19113503