Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Cholesterol
2. Cholesterol Metabolism
2.1. Cholesterol Synthesis
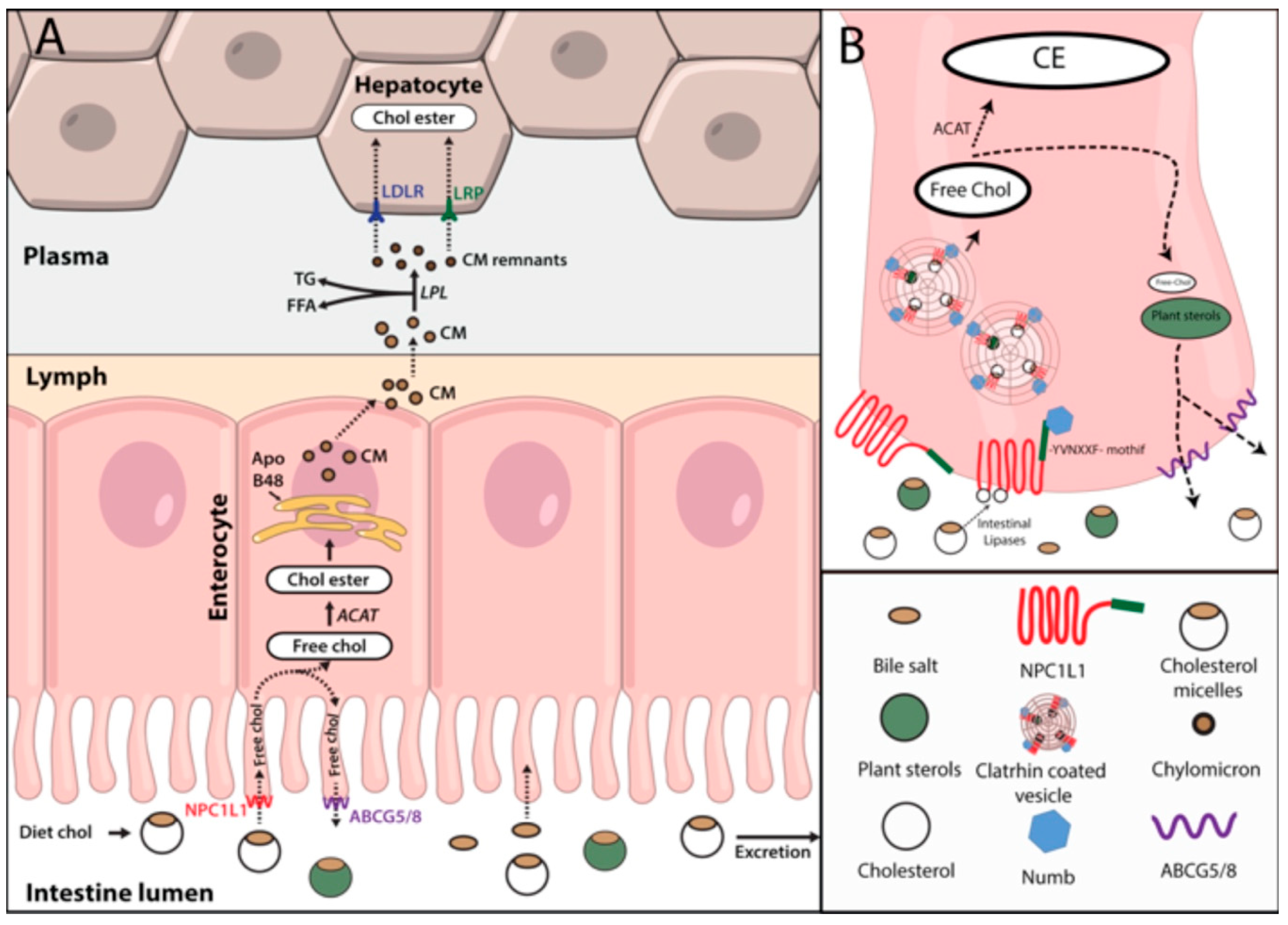
2.2. Cholesterol Absorption
2.3. Hepatic Cholesterol Efflux
2.4. Cholesterol Influx
Cholesterol Influx Regulation
2.5. Reverse Cholesterol Transport (RCT)
2.6. Bile Acid Excretion
3. Familial Hypercholesterolemia
3.1. Genetics of FH
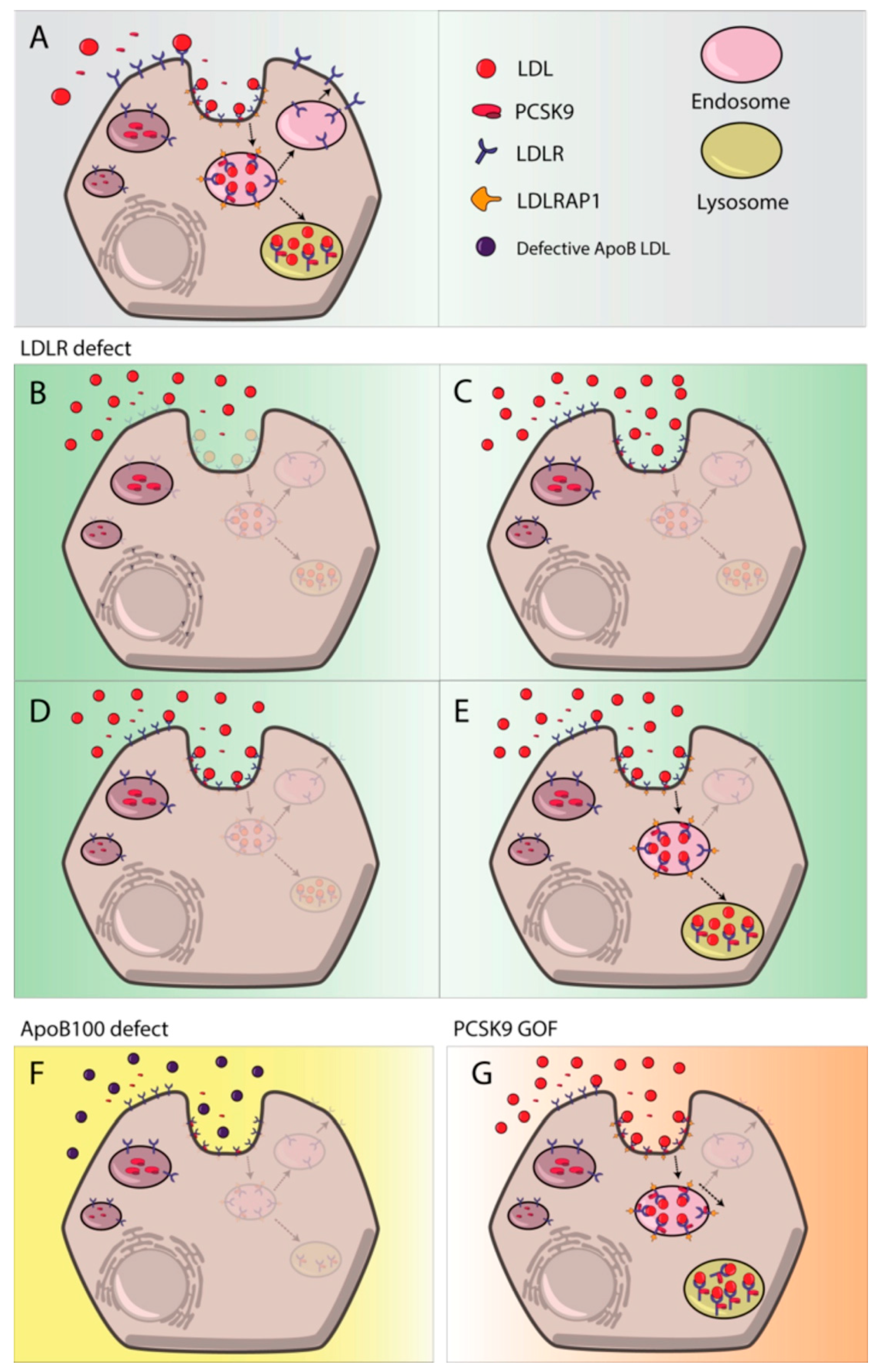
3.1.1. LDLR
3.1.2. APOB
3.1.3. PCSK9
3.1.4. LDLRAP1
3.2. Second Generation FH
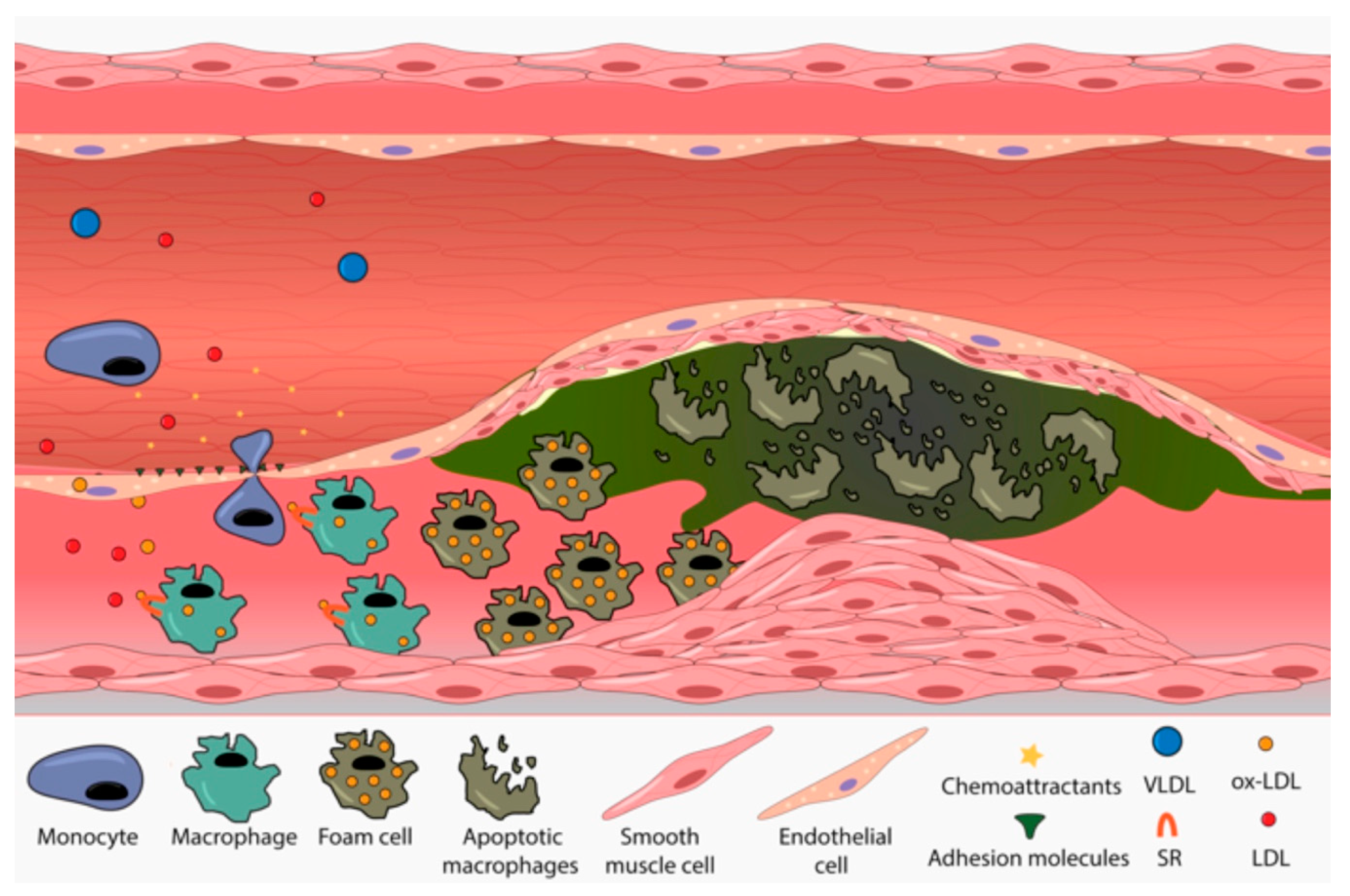
3.3. FH Implication in Cardiovascular Disease
3.4. FH Diagnosis
3.5. Evolution of Lipid Lowering Therapies
3.5.1. Statins
3.5.2. Niacin
3.5.3. Bile Acid Sequestrants
3.5.4. Ezetimibe
3.5.5. Human Monoclonal Anti-PCSK9 Antibodies
3.5.6. Other Treatments
3.6. Nutraceuticals in FH
3.7. Current FH Situation
3.8. Functional Studies as a Complement to Genetic Testing
Funding
Acknowledgments
Conflicts of Interest
References
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-Resolution Crystal Structure of an Engineered Human 2-Adrenergic G Protein-Coupled Receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Grouleff, J.; Irudayam, S.J.; Skeby, K.K.; Schiøtt, B. The influence of cholesterol on membrane protein structure, function, and dynamics studied by molecular dynamics simulations. BBA Biomembr. 2015, 1848, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Liu, J.; Song, B.-L.; Li, B.-L.; Chang, C.C.Y.; Chang, T.-Y. Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J. Steroid Biochem. Mol. Biol. 2015, 151, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Y.; Goldstein, J.L.; Brown, M.S.; Radhakrishnan, A. Cholesterol-induced conformational changes in the sterolsensing domain of the Scap protein suggest feedback mechanism to control cholesterol synthesis. J. Biol. Chem. 2017, 292, 8729–8737. [Google Scholar] [CrossRef] [PubMed]
- Theesfeld, C.L.; Pourmand, D.; Davis, T.; Garza, R.M.; Hampton, R.Y. The sterol-sensing domain (SSD) directly mediates signal-regulated endoplasmic reticulum-associated degradation (ERAD) of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase isozyme Hmg2. J. Biol. Chem. 2011, 286, 26298–26307. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Baier, C.J.; Evans, L.S.; Williamson, P.T.F.; Fantini, J.; Barrantes, F.J. Relevance of CARC and CRAC Cholesterol-Recognition Motifs in the Nicotinic Acetylcholine Receptor and Other Membrane-Bound Receptors. Curr. Top. Membr. 2017, 80, 3–23. [Google Scholar] [PubMed]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Midzak, A.; Papadopoulos, V. Binding domain-driven intracellular trafficking of sterols for synthesis of steroid hormones, bile acids and oxysterols. Traffic 2014, 15, 895–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J. Vitamin D and Cardiovascular Disease. Annu. Rev. Med. 2016, 67, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Goldstein, J. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. Sterol molecule: Structure, biosynthesis, and function. Steroids 1992, 57, 378–383. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar] [PubMed]
- Goedeke, L.; Fernández-Hernando, C. Regulation of cholesterol homeostasis. Cell. Mol. Life Sci. 2012, 69, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Fears, R. The contribution of the cholesterol biosynthetic pathway to intermediary metabolism and cell function. Biochem. J. 1981, 199, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.M. Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 2014, 25, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Betters, J.L.; Yu, L. NPC1L1 and cholesterol transport. FEBS Lett. 2010, 584, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Li, P.S.; Fu, Z.-Y.; Zhang, Y.-Y.; Zhang, J.-H.; Xu, C.-Q.; Ma, Y.-T.; Li, B.L.; Song, B.-L. The clathrin adaptor Numb regulates intestinal cholesterol absorption through dynamic interaction with NPC1L1. Nat. Med. 2014, 20, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E. Sitosterolemia: A review and update of pathophysiology, clinical spectrum, diagnosis, and management. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Marcel, Y.L.; Innerarity, T.L.; Spilman, C.; Mahley, R.W.; Protter, A.A.; Milne, R.W. Mapping of human apolipoprotein B antigenic determinants. Arterioscler. Thromb. Vasc. Biol. 1987, 7, 166–175. [Google Scholar] [CrossRef]
- Julve, J.; Martín-Campos, J.M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Chylomicrons: Advances in biology, pathology, laboratory testing, and therapeutics. Clin. Chim. Acta 2016, 455, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.B. Mechanisms of chylomicron uptake into lacteals. Ann. N. Y. Acad. Sci. 2010, 1207, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Miller, N.E. Lymphatic transport of high-density lipoproteins and chylomicrons. J. Clin. Investig. 2014, 124, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, G. Role of lipoprotein lipase in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Dallinga-Thie, G.M.; Franssen, R.; Mooij, H.L.; Visser, M.E.; Hassing, H.C.; Peelman, F.; Kastelein, J.J.P.; Péterfy, M.; Nieuwdorp, M. The metabolism of triglyceride-rich lipoproteins revisited: New players, new insight. Atherosclerosis 2010, 211, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Heeren, J. Apolipoprotein E Recycling: Implications for Dyslipidemia and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 26, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Frazier-Wood, A.C.; Kabagambe, E.K.; Wojczynski, M.K.; Borecki, I.B.; Tiwari, H.K.; Smith, C.E.; Ordovas, J.M.; Arnett, D.K. The association between LRP-1 variants and chylomicron uptake after a high fat meal. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R.; Naoumova, R.P.; Watts, G.F. Role of cholesterol in regulating apolipoprotein B secretion by the liver. J. Lipid Res. 1996, 37, 439–447. [Google Scholar] [PubMed]
- Ganesan, L.P.; Mates, J.M.; Cheplowitz, A.M.; Avila, C.L.; Zimmerer, J.M.; Yao, Z.; Maiseyeu, A.; Rajaram, M.V.S.; Robinson, J.M.; Anderson, C.L. Scavenger receptor B1, the HDL receptor, is expressed abundantly in liver sinusoidal endothelial cells. Sci. Rep. 2016, 6, 20646. [Google Scholar] [CrossRef] [PubMed]
- Oude Elferink, R.P.; Groen, A. Mechanisms of biliary lipid secretion and their role in lipid homeostasis. Semin. Liver Dis. 2000, 20, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Pavanello, C.; Bertolini, S. Microsomal transfer protein (MTP) inhibition—A novel approach to the treatment of homozygous hypercholesterolemia. Ann. Med. 2014, 46, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, S.O.; Borén, J. Apolipoprotein B secretory regulation by degradation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L.; Zhang, H.; Fan, J.; Zhang, F.; Yu, M.; Shi, L.; Yang, L.; Lam, S.M.; Wang, H.; et al. Mea6 controls VLDL transport through the coordinated regulation of COPII assembly. Cell Res. 2016, 26, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Hossain, T.; Riad, A.; Siddiqi, S.; Parthasarathy, S.; Siddiqi, S.A. Mature VLDL triggers the biogenesis of a distinct vesicle from the trans-Golgi network for its export to the plasma membrane. Biochem. J. 2014, 459, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Doonan, L.M.; Fisher, E.A.; Brodsky, J.L. Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol-lowering drugs? BBA Mol. Cell Biol. Lipids 2018, 1863, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Siddiqi, S.A. Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Adewole, O.L.; Brunt, E.M.; Abumrad, N.A. CD36 deletion reduces VLDL secretion, modulates liver prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. J. Lipid Res. 2013, 54, 2988–2997. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Benito-Vicente, A.; Stef, M.; Ostolaza, H.; Palacios, L.; Martin, C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis 2015, 238, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Soufi, M.; Rust, S.; Walter, M.; Schaefer, J.R. A combined LDL receptor/LDL receptor adaptor protein 1 mutation as the cause for severe familial hypercholesterolemia. Gene 2013, 521, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž.; Reiner, Ž.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Ćorić, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency—An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Higuero, J.A.; Benito-Vicente, A.; Etxebarria, A.; Milicua, J.C.G.; Ostolaza, H.; Arrondo, J.L.R.; Martín, C. Structural changes induced by acidic pH in human apolipoprotein B-100. Sci. Rep. 2016, 6, 36324. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Benito-Vicente, A.; Palacios, L.; Stef, M.; Cenarro, A.; Civeira, F.; Ostolaza, H.; Martin, C. Functional characterization and classification of frequent low-density lipoprotein receptor variants. Hum. Mutat. 2015, 36, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-H.; Jiang, N.; Yao, P.-B.; Zheng, X.-L.; Cayabyab, F.S.; Tang, C.-K. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin. Chim. Acta 2014, 429, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reue, K.; Fong, L.G.; Young, S.G.; Tontonoz, P. Feedback Regulation of Cholesterol Uptake by the LXR-IDOL-LDLR Axis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2541–2546. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D. PCSK9: An enigmatic protease. BBA Mol. Cell Biol. Lipids 2008, 1781, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR Regulates Cholesterol Uptake Through Idol-Dependent Ubiquitination of the LDL Receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Wierød, L.; Cameron, J.; Strøm, T.B.; Leren, T.P. Studies of the autoinhibitory segment comprising residues 31–60 of the prodomain of PCSK9: Possible implications for the mechanism underlying gain-of-function mutations. Mol. Genet. Metab. 2016, 9, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Luna Saavedra, Y.G.; Zhang, J.; Seidah, N.G. PCSK9 Prosegment Chimera as Novel Inhibitors of LDLR Degradation. PLoS ONE 2013, 8, e72113. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.I.; Swedberg, J.E.; Withka, J.M.; Rosengren, K.J.; Akcan, M.; Clayton, D.J.; Daly, D.L.; Cheneval, O.; Borzilleri, K.A.; Griffor, M.; et al. Design and Synthesis of Truncated EGF-A Peptides that Restore LDL-R Recycling in the Presence of PCSK9 In Vitro. Chem. Biol. 2014, 21, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Adijiang, A.; Mah, M.; Zhang, D. Characterization of the role of EGF-A of low density lipoprotein receptor in PCSK9 binding. J. Lipid Res. 2013, 54, 3345–3357. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Mangelsdorf, D.J. Liver X Receptor Signaling Pathways in Cardiovascular Disease. Mol. Endocrinol. 2003, 17, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mao, Y.; Narimatsu, Y.; Ye, Z.; Tian, W.; Goth, C.K.; Lira-Navarrete, E.; Pedersen, N.B.; Benito-Vicente, A.; Martin, C.; et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J. Biol. Chem. 2018, 293, 7408–7422. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Wagschal, A.; Fernández-Hernando, C.; Näär, A.M. miRNA regulation of LDL-cholesterol metabolism. BBA Mol. Cell Biol. Lipids 2016, 1861, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Wagschal, A.; Najafi-Shoushtari, S.H.; Wang, L.; Goedeke, L.; Sinha, S.; deLemos, A.S.; Black, J.C.; Ramírez, C.M.; Li, Y.; Tewhey, R.; et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015, 21, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cordoba, S.L.; Salido-Guadarrama, I.; Rodriguez-Dorantes, M.; Hidalgo-Miranda, A. miRNA biogenesis: Biological impact in the development of cancer. Cancer Biol. Ther. 2014, 15, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Rotllan, N.; Ramírez, C.M.; Aranda, J.F.; Canfrán-Duque, A.; Araldi, E.; Fernández-Hernando, A.; Langhi, C.; Cabo, R.D.; Baldán, Á.; et al. miR-27b inhibits LDLR and ABCA1 expression but does not influence plasma and hepatic lipid levels in mice. Atherosclerosis 2015, 243, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; Khosroheidari, M.; Eddy, E.; Done, S.C. MicroRNA-27a decreases the level and efficiency of the LDL receptor and contributes to the dysregulation of cholesterol homeostasis. Atherosclerosis 2015, 242, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Rotllan, N.; Canfrán-Duque, A.; Aranda, J.F.; Ramírez, C.M.; Araldi, E.; Lin, C.-S.; Anderson, N.N.; Wagschal, A.; Cabo, R.d.; et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Chroni, A.; Tietge, U.J.; Zanotti, I.; Escolà-Gil, J.C.; Bernini, F. Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2015; Volume 224, pp. 181–206. [Google Scholar]
- Wang, S.; Smith, J.D. ABCA1 and nascent HDL biogenesis. BioFactors 2014, 40, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Westerterp, M.; Koetsveld, J.; Fernández-Hernando, C.; Yvan-Charvet, L.; Wang, N.; Sessa, W.C.; Tall, A.R. ATP-binding cassette transporter G1 and high-density lipoprotein promote endothelial NO synthesis through a decrease in the interaction of caveolin-1 and endothelial NO synthase. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Gillard, B.K.; Rosales, C.; Xu, B., Jr.; Gotto, A.M.; Pownall, H.J. Rethinking reverse cholesterol transport and dysfunctional high-density lipoproteins. J. Clin. Lipidol. 2018, 12, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Hao, L.; Xie, H.; Hao, C.; Liu, C.; Li, W.; Xiong, X.; Zhao, D. The therapeutic potential of CETP inhibitors: A patent review. Expert Opin. Ther. Pat. 2018, 28, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Dikkers, A.; Tietge, U.J.F. Biliary cholesterol secretion: More than a simple ABC. World J. Gastroenterol. 2010, 16, 5936–5945. [Google Scholar] [PubMed]
- Halilbasic, E.; Claudel, T.; Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 2013, 58, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Norlin, M.; Wikvall, K. Enzymes in the Conversion of Cholesterol into Bile Acids. Curr. Mol. Med. 2007, 7, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Qian, K.; Jiang, N.; Zheng, X.L.; Cayabyab, F.S.; Tang, C.K. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clin. Chim. Acta 2014, 428, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Bonamassa, B.; Moschetta, A. Atherosclerosis: Lessons from LXR and the intestine. Trends Endocrinol. Metab. 2013, 24, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Gordo-Gilart, R.; Andueza, S.; Hierro, L.; Jara, P.; Alvarez, L. Functional Rescue of Trafficking-Impaired ABCB4 Mutants by Chemical Chaperones. PLoS ONE 2016, 11, e0150098. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.E. Balancing cholesterol synthesis and absorption in the gastrointestinal tract. J. Clin. Lipidol. 2008, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Akram, A.; Kondapally Seshasai, S.R.; Cole, D.; Watts, G.F.; Hovingh, G.K.; Kastelein, J.J.; Mata, P.; Raal, F.J.; Santos, R.D.; et al. Pooling and expanding registries of familial hypercholesterolaemia to assess gaps in care and improve disease management and outcomes: Rationale and design of the global EAS Familial Hypercholesterolaemia Studies Collaboration. Atheroscler. Suppl. 2016, 22, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Cenarro, A.; Etxebarria, A.; de Castro-Orós, I.; Stef, M.; Bea, A.M.; Palacios, L.; Mateo-Gallego, R.; Benito-Vicente, A.; Ostolaza, H.; Tejedor, T.; et al. The p.Leu167del mutation in APOE gene causes autosomal dominant hypercholesterolemia by down-regulation of LDL receptor expression in hepatocytes. J. Clin. Endocrinol. Metab. 2016, 101, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.; Zelcer, N.; Kastelein, J.J.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.; Stein, E.; Shendure, J.; Hobbs, H.H.; Cohen, J.C. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum. Mol. Genet. 2010, 19, 4313–4318. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Prim. 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Iacocca, M.A.; Hegele, R.A. Role of DNA copy number variation in dyslipidemias. Curr. Opin. Lipidol. 2018, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Palacios, L.; Stef, M.; Tejedor, D.; Uribe, K.B.; Oleaga, A.; Irigoyen, L.; Torres, B.; Ostolaza, H.; Martin, C. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum. Mutat. 2012, 33, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.K.M.; Musa, F.R.; Bell, C.; Walker, S.W. LDLR gene synonymous mutation c.1813C > T results in mRNA splicing variation in a kindred with familial hypercholesterolaemia. Ann. Clin. Biochem. 2015, 52, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Holla, Ø.L.; Kulseth, M.A.; Berge, K.E.; Leren, T.P.; Ranheim, T. Nonsense-mediated decay of human LDL receptor mRNA. Scand. J. Clin. Lab. Investig. 2009, 69, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Benito-Vicente, A.; Medeiros, A.M.; Reeves, K.; Martin, C.; Bourbon, M. Further evidence of novel APOB mutations as a cause of Familial Hypercholesterolaemia. Atherosclerosis 2018. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Etxebarria, A.; Soutar, A.K.; Martin, C.; Bourbon, M. Novel functional APOB mutations outside LDL-binding region causing familial hypercholesterolaemia. Hum. Mol. Genet. 2014, 23, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Dron, J.S.; Hegele, R.A. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr. Opin. Lipidol. 2017, 28, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.A.; Berge, K.E.; Leren, T.P. The unique role of proprotein convertase subtilisin/kexin 9 in cholesterol homeostasis. J. Intern. Med. 2009, 266, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Eng. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Soutar, A.K.; Naoumova, R.P. Mechanisms of disease: Genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Quagliarini, F.; Quagliarini, F.; Vallvé, J.C.; Campagna, F.; Alvaro, A.; Fuentes-Jimenez, F.J.; Sirinian, M.I.; Meloni, F.; Masana, L.; Arca, M. Autosomal recessive hypercholesterolemia in Spanish kindred due to a large deletion in the ARH gene. Mol. Genet. Metab. 2007, 92, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Nomura, A.; Yamagishi, M.; Kawashiri, M.A. First case of sitosterolemia caused by double heterozygous mutations in ABCG5 and ABCG8 genes. J. Clin. Lipidol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, L.; Chen, P.P.; Wu, Y.; Su, P.Y.; Wang, L.Y. A case of sitosterolemia misdiagnosed as familial hypercholesterolemia: A 4-year follow-up. J. Clin. Lipidol. 2018, 12, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Muso, E. Beneficial effect of LDL-apheresis in refractory nephrotic syndrome. Clin. Exp. Nephrol. 2014, 18, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Zaritsky, J.J.; Fornoni, A.; Smoyer, W.E. Dyslipidaemia in nephrotic syndrome: Mechanisms and treatment. Nat. Rev. Nephrol. 2017, 14, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Chrostek, L.; Supronowicz, L.; Panasiuk, A.; Cylwik, B.; Gruszewska, E.; Flisiak, R. The effect of the severity of liver cirrhosis on the level of lipids and lipoproteins. Clin. Exp. Med. 2014, 14, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.V.; Elisaf, M.S.; Liberopoulos, E.N. Effects of thyroid dysfunction on lipid profile. Open Cardiovasc. Med. J. 2011, 5, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Nemes, K.; Åberg, F.; Gylling, H.; Isoniemi, H. Cholesterol metabolism in cholestatic liver disease and liver transplantation: From molecular mechanisms to clinical implications. World J. Hepatol. 2016, 8, 924. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics-2015 update: A report from the American Heart Association. Circulation 2015, 131, 434–441. [Google Scholar] [CrossRef]
- Schwenke, D.C.; Carew, T.E. Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis 1989, 9, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Vascular adhesion molecules in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Freeman, M.W. Scavenger receptors in atherosclerosis: Beyond lipid uptake. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Changing concepts of atherogenesis. J. Intern. Med. 2000, 247, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Böhm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef]
- Bench, T.J.; Jeremias, A.; Brown, D.L. Matrix metalloproteinase inhibition with tetracyclines for the treatment of coronary artery disease. Pharmacol. Res. 2011, 64, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Scientific Steering Committe on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 1991, 303, 893–896. [Google Scholar] [CrossRef]
- Williams, R.R.; Hunt, S.C.; Schumacher, M.C.; Hegele, R.A.; Leppert, M.F.; Ludwig, E.H.; Hopkins, P.N. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am. J. Cardiol. 1993, 72, 171–176. [Google Scholar] [CrossRef]
- Ito, M.K.; Watts, G.F. Challenges in the Diagnosis and Treatment of Homozygous Familial Hypercholesterolemia. Drugs 2015, 75, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Gagné, C.; Gaudet, D.; Bruckert, E. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation 2002, 105, 2469–2475. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Santos, R.D.; Blom, D.J.; Marais, A.D.; Charng, M.J.; Cromwell, W.C.; Lachmann, R.H.; Gaudet, D.; Tan, J.L.; Chasan-Taber, S.; et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 998–1006. [Google Scholar] [CrossRef]
- ENDO, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Marais, A.D.; Raal, F.J.; Stein, E.A.; Rader, D.J.; Blasetto, J.; Palmer, M.; Wilpshaar, W. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis 2008, 197, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Walter, M.F.; Day, C.A.; Jacob, R.F. Intermolecular differences of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors contribute to distinct pharmacologic and pleiotropic actions. Am. J. Cardiol. 2005, 96, 11F–23F. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Germershausen, J.I.; Hunt, V.M.; Bostedor, R.G.; Bailey, P.J.; Karkas, J.D.; Alberts, A.W. Tissue selectivity of the cholesterol-lowering agents lovastatin, simvastatin and pravastatin in rats in vivo. Biochem. Biophys. Res. Commun. 1989, 158, 667–675. [Google Scholar] [CrossRef]
- McKenney, J.M. Pharmacologic characteristics of statins. Clin. Cardiol. 2003, 26, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Preiss, D.; Murray, H.M. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Rev. Port. Cardiol. 2010, 29, 1077–1078. [Google Scholar] [CrossRef]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. Adverse effects of statin therapy: Perception vs. the evidence—Focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Heart J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef] [PubMed]
- Gille, A.; Bodor, E.T.; Ahmed, K.; Offermanns, S. Nicotinic Acid: Pharmacological Effects and Mechanisms of Action. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Julius, U. History of lipidology and lipoprotein apheresis. Atheroscler 2017, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Robidoux, J. Dyslipidemia management update. Curr. Opin. Pharmacol. 2017, 33, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Pizzoferrato, M.; Ponziani, F.R.; Gasbarrini, G.; Gasbarrini, A. Use and indications of cholestyramine and bile acid sequestrants. Int. Emerg. Med. 2013, 8, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H. A systematic review of bile acid sequestrant therapy in children with familial hypercholesterolemia. J. Clin. Lipidol. 2011, 5, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W. Niemann-Pick C1 Like 1 Protein Is Critical for Intestinal Cholesterol Absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Temel, R.E.; Tang, W.; Ma, Y.; Rudel, L.L.; Willingham, M.C.; Ioannou, Y.A.; Davies, J.P.; Nilsson, L.M.; Yu, L. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J. Clin. Investig. 2007, 117, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Hegele, R.A.; Fazio, S.; Cannon, C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018, 72, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Huijgen, R.; Ray, K.; Persons, J.; Kastelein, J.J.; Pencina, M.J. Determining When to Add Nonstatin Therapy. J. Am. Coll. Cardiol. 2016, 68, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Zodda, D.; Giammona, R.; Schifilliti, S. Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs. Pharmacy 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Polychronopoulos, G.; Tziomalos, K. Novel treatment options for the management of heterozygous familial hypercholesterolemia. Expert Rev. Clin. Pharmacol. 2017, 10, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R.; Catapano, A.; Saheb, S.; Atassi-Dumont, M.; Barbir, M.; Eriksson, M.; Paulweber, B.; Sijbrands, E.; Stalenhoef, A.F.; Parhofer, K.G. Severe hypercholesterolaemia: Therapeutic goals and eligibility criteria for LDL apheresis in Europe. Curr. Opin. Lipidol. 2010, 21, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Julius, U. Current Role of Lipoprotein Apheresis in the Treatment of High-Risk Patients. J. Cardiovasc. Dev. Dis. 2018, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Reiner, Z.; De Backer, G.; Graham, I.; Taskinen, M.R.; Wiklund, O.; Agewall, S.; Alegria, E.; Chapman, M.; Durrington, P.; et al. ESC/EAS Guidelines for the management of dyslipidaemias the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur. Heart J. 2011, 217, 3–46. [Google Scholar]
- Gylling, H.; Plat, J.; Turley, S.; Ginsberg, H.N.; Ellegård, L.; Jessup, W.; Jones, P.J.; Lütjohann, D.; Maerz, W.; Masana, L.; et al. Plant sterols and plant stanols in the management of dyslipidaemia and prevention of cardiovascular disease. Atherosclerosis 2014, 232, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Shishikura, Y.; Khokhar, S.; Murray, B.S. Effects of tea polyphenols on emulsification of olive oil in a small intestine model system. J. Agric. Food Chem. 2006, 54, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.; Ranheim, T.; Kulseth, M.A.; Leren, T.P.; Berge, K.E. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis 2008, 201, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, B.; Park, S.W.; Lee, H.S.; Chen, W.; Liu, J. Hepatocyte Nuclear Factor 1α Plays a Critical Role in PCSK9 Gene Transcription and Regulation by the Natural Hypocholesterolemic Compound Berberine. J. Biol. Chem. 2009, 284, 28885–28895. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, J.; Uang, Y.; Lin, C. Improved dissolution rate and oral bioavailability of lovastatin in red yeast rice products. Int. J. Pharm. 2013, 444, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Steering, S.; Register, B. Mortality in treated heterozygous familial hypercholesterolaemia: Implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis 1999, 142, 105–112. [Google Scholar]
- Palacios, L.; Grandoso, L.; Cuevas, N.; Olano-Martín, E.; Martinez, A.; Tejedor, D.; Stef, M. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis 2012, 221, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Civeira, F.; Ros, E.; Jarauta, E.; Plana, N.; Zambon, D.; Puzo, J.; Martinez de Esteban, J.P.; Ferrando, J.; Zabala, S.; Almagro, F.; et al. Comparison of Genetic versus Clinical Diagnosis in Familial Hypercholesterolemia. Am. J. Cardiol. 2008, 102, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Starr, B.; Hadfield, S.G.; Hutten, B.A.; Lansberg, P.J.; Leren, T.P.; Damgaard, D.; Neil, H.A.; Humphries, S.E. Development of sensitive and specific age- and gender- specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin. Chem. Lab. Med. 2008, 46, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Alves, A.C.; Etxebarria, A.; Medeiros, A.M.; Martin, C.; Bourbon, M. The importance of an integrated analysis of clinical, molecular, and functional data for the genetic diagnosis of familial hypercholesterolemia. Genet. Med. 2015, 17, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Huijgen, R.; Kindt, I.; Defesche, J.C.; Kastelein, J.J.P. Cardiovascular risk in relation to functionality of sequence variants in the gene coding for the low-density lipoprotein receptor: A study among 29,365 individuals tested for 64 specific low-density lipoprotein-receptor sequence variants. Eur. Heart J. 2012, 33, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Strøm, T.B.; Tveten, K.; Leren, T.P. PCSK9 acts as a chaperone for the LDL receptor in the endoplasmic reticulum. Biochem. J. 2014, 457, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants. Int. J. Mol. Sci. 2018, 19, 1676. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. https://doi.org/10.3390/ijms19113426
Benito-Vicente A, Uribe KB, Jebari S, Galicia-Garcia U, Ostolaza H, Martin C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. International Journal of Molecular Sciences. 2018; 19(11):3426. https://doi.org/10.3390/ijms19113426
Chicago/Turabian StyleBenito-Vicente, Asier, Kepa B. Uribe, Shifa Jebari, Unai Galicia-Garcia, Helena Ostolaza, and Cesar Martin. 2018. "Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease" International Journal of Molecular Sciences 19, no. 11: 3426. https://doi.org/10.3390/ijms19113426
APA StyleBenito-Vicente, A., Uribe, K. B., Jebari, S., Galicia-Garcia, U., Ostolaza, H., & Martin, C. (2018). Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. International Journal of Molecular Sciences, 19(11), 3426. https://doi.org/10.3390/ijms19113426