Efficiency of All-Trans Retinoic Acid on Gastric Cancer: A Narrative Literature Review


Abstract
1. Introduction
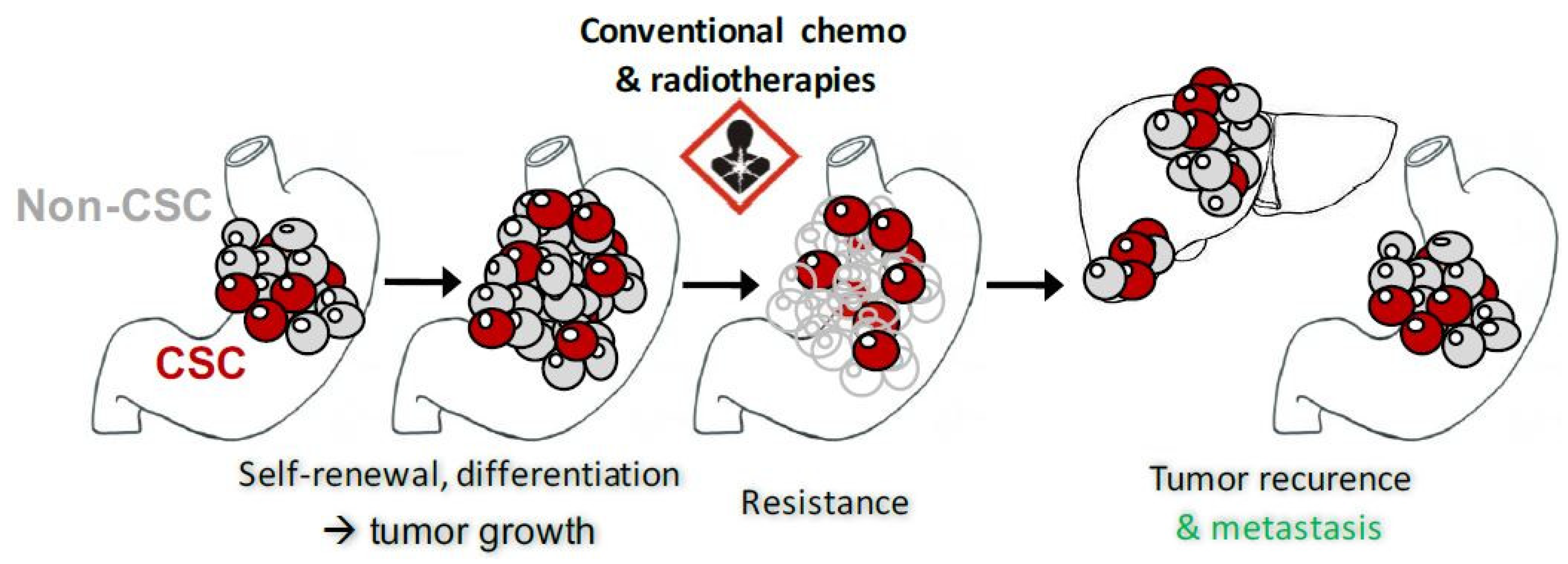
2. GCSC and Biomarkers
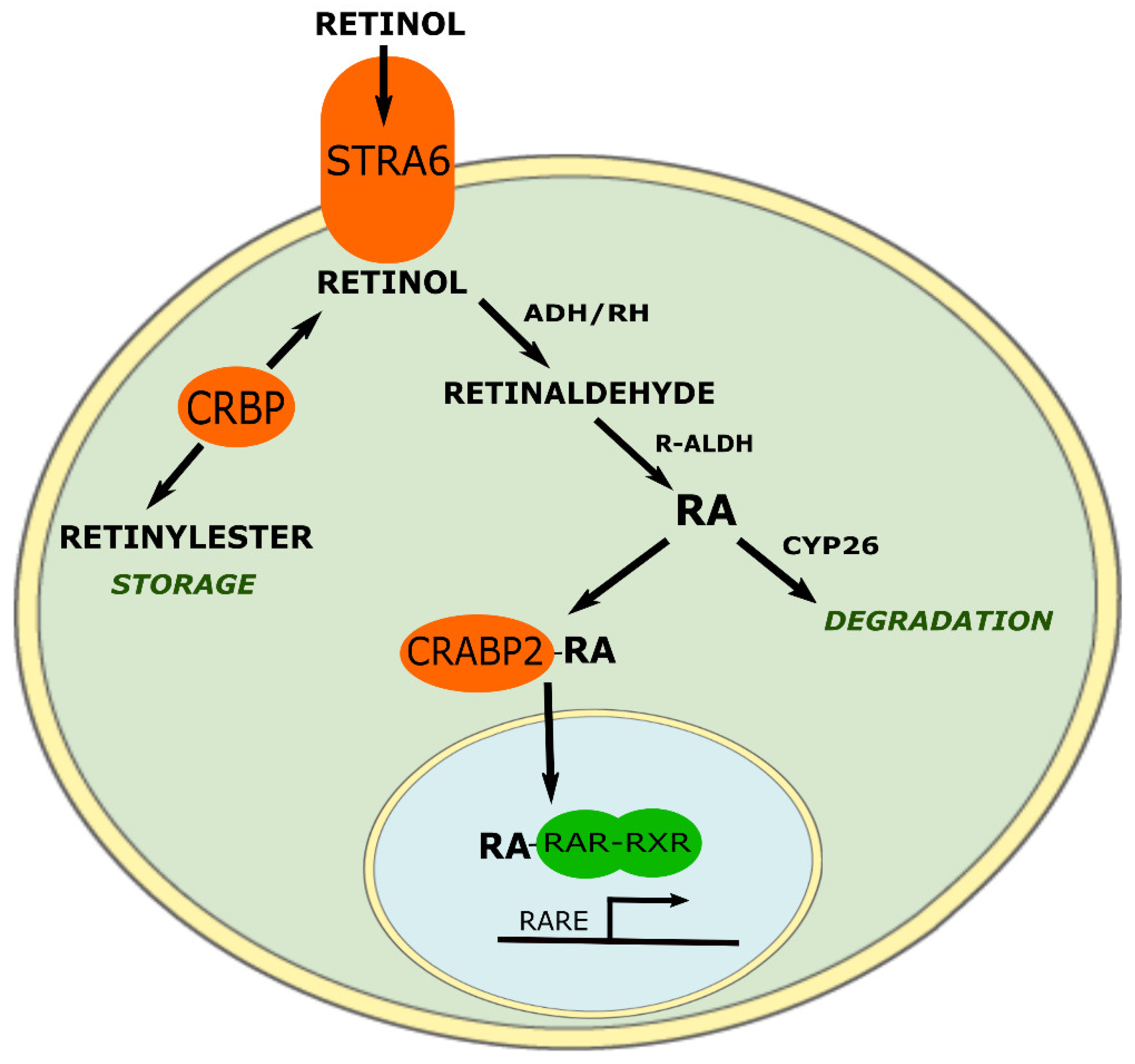
3. ATRA Historic and Targets
3.1. Retinoic Acid Receptor α (RARα)
3.2. Retinoic Acid Receptor β (RARβ)
3.3. Retinoic X Receptors (RXR)
3.4. RAR and RXR’s Expression Impact on GC Prognosis
4. ATRA on GC Models
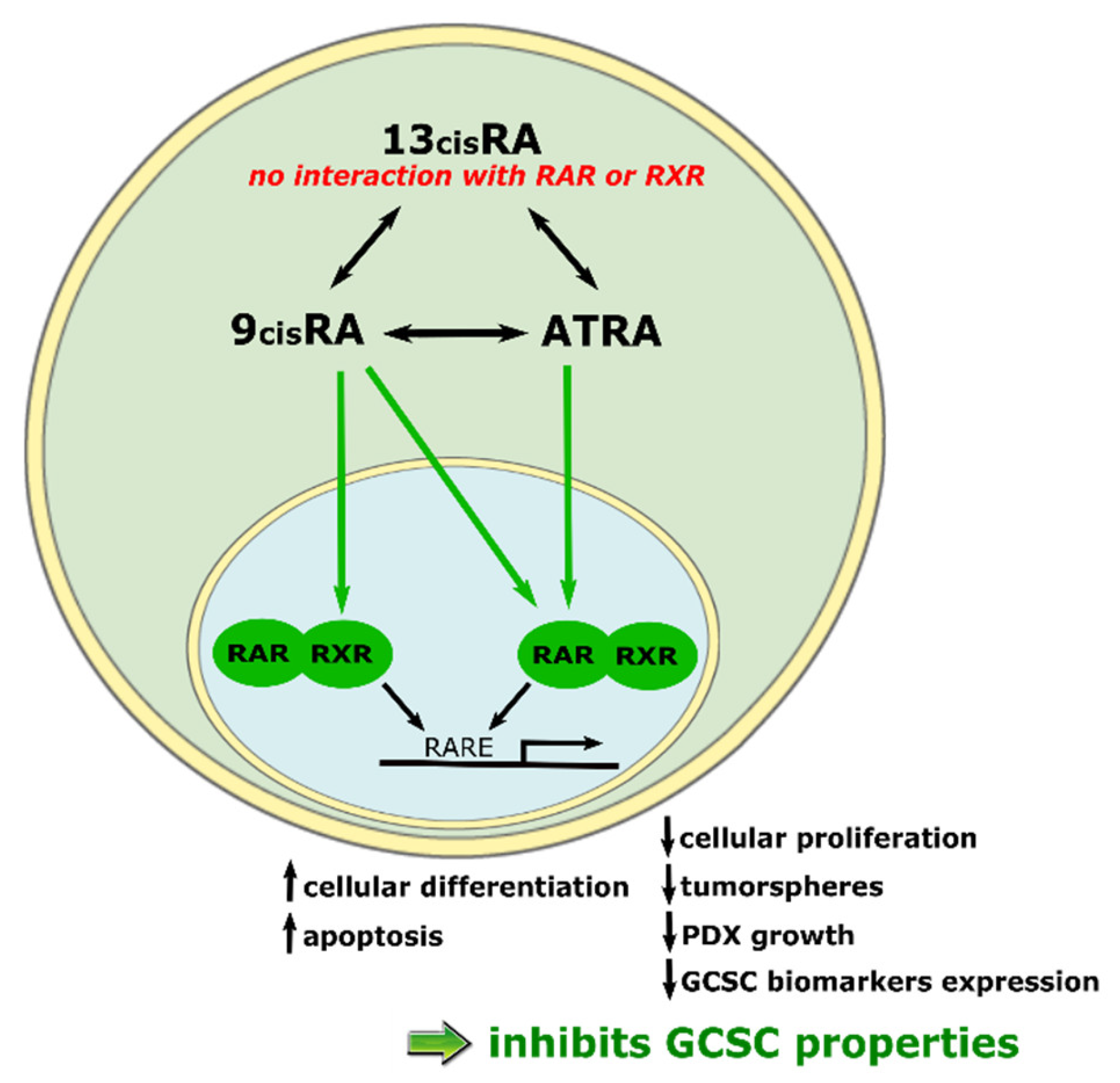
4.1. ATRA’s Mechanism of Action for Cell Cycle Blocking and Differentiation Initiation
4.2. ATRA’s Mechanism of Action for Apoptosis Initiation
4.3. ATRA’s Mechanism of Action for CSC Properties Inhibition
4.4. ATRA’s Anticancer Effect on Patients with GC
4.5. ATRA’s Anticancer Effect on Non-Epithelial GCs
4.6. Association of RA and Conventional Chemotherapies
4.7. Mechanisms of Resistance to ATRA
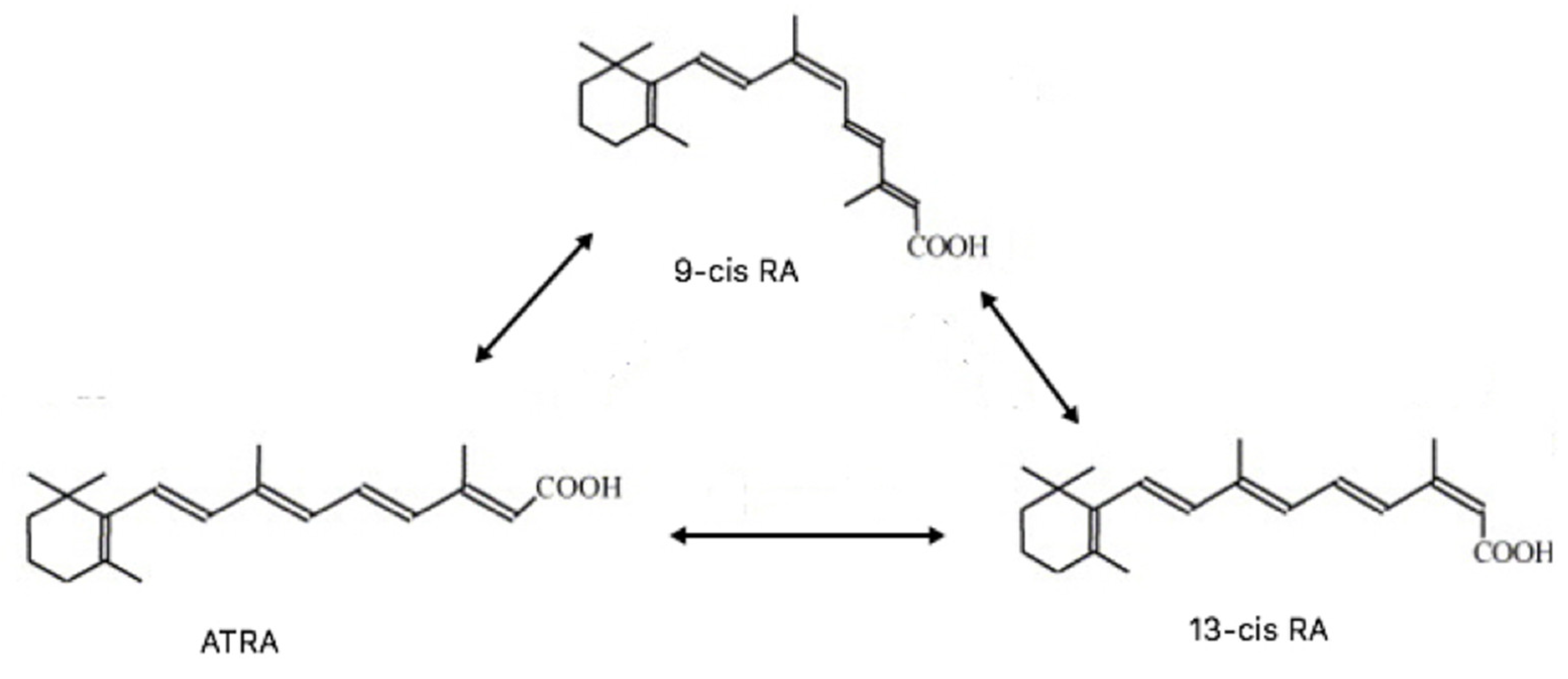
4.8. Other Retinoids
4.8.1. 13cisRA
4.8.2. 9cisRA
4.8.3. Fenretinide (RII)
4.8.4. ATPR
4.8.5. ATRA-Podophyllotoxin Conjugate
5. Implications and Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| 13cisRA | 13cis retinoic acid |
| 9cisRA | 9cis retinoic acid |
| ADH | Alcool dehydrogenase |
| ALDH | Aldehyde dehydrogenase |
| APL | Acute promyelotic leukemia |
| ATRA | All-trans retinoic acid |
| ATPR | 4-amino-2-trifluoromethyl-phenyl retinate |
| CD44 | Cluster of differentiation 44 |
| CRABP2 | Cellular retinoic acid binding protein 2 |
| CRBP | Cellular retinol binding protein |
| CSC | Cancer stem cell |
| CYP26 | Cytochrome P450 26A1 |
| CYP450 | Cytochrome P450 |
| EMT | Epithelial to mesenchymal transition |
| GC | Gastric cancers |
| GCSC | Gastric cancer stem cell |
| MALT | Mucosa associated lymphoid tissue |
| N-Cor | Nuclear corepressor |
| PDX | Patient derived xenograft |
| PML | Promyelotic leukemia |
| PLZF | Promyelotic leukemia zinc finger |
| RA | Retinoic acid |
| R-ALDH | Retinaldehyde dehydrogenase |
| RAR | Retinoic acid receptor |
| RARE | Retinoic acid response element |
| RII | N-4-(hydroxycarbophenyl) retinamide |
| RXR | Retinoid X receptor |
| SMRT | Silencing mediator of RA and thyroid hormone receptor |
References
- Fléjou, J.-F. WHO Classification of digestive tumors: The fourth edition. Ann. Pathol. 2011, 31, S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; Tavilla, A.; Brenner, H.; Luttmann, S.; Navarro, C.; Gavin, A.T.; Holleczek, B.; Johnston, B.T.; Cook, M.B.; Bannon, F.; et al. EUROCARE-5 Working Group Survival for oesophageal, stomach and small intestine cancers in Europe 1999–2007: Results from EUROCARE-5. Eur. J. Cancer Oxf. Engl. 1990 2015, 51, 2144–2157. [Google Scholar] [CrossRef]
- Chapelle, N.; Bouvier, A.-M.; Manfredi, S.; Drouillard, A.; Lepage, C.; Faivre, J.; Jooste, V. Early Gastric Cancer: Trends in Incidence, Management, and Survival in a Well-Defined French Population. Ann. Surg. Oncol. 2016, 23, 3677–3683. [Google Scholar] [CrossRef] [PubMed]
- Talley, N.J. Is it time to screen and treat H. pylori to prevent gastric cancer? Lancet Lond. Engl. 2008, 372, 350–352. [Google Scholar] [CrossRef]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Thompson, J.N.; Van de Velde, C.J.H.; Nicolson, M.; Scarffe, J.H.; Lofts, F.J.; Falk, S.J.; Iveson, T.J.; et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N. Engl. J. Med. 2006, 355, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ychou, M.; Boige, V.; Pignon, J.-P.; Conroy, T.; Bouché, O.; Lebreton, G.; Ducourtieux, M.; Bedenne, L.; Fabre, J.-M.; Saint-Aubert, B.; Genève, J.; Lasser, P.; et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: An FNCLCC and FFCD multicenter phase III trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.W.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.K.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells Dayt. Ohio 2009, 27, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Saikawa, Y.; Ohashi, M.; Kumagai, K.; Kitajima, M.; Okano, H.; Matsuzaki, Y.; Kitagawa, Y. Tumor initiating potential of side population cells in human gastric cancer. Int. J. Oncol. 2009, 34, 1201–1207. [Google Scholar] [PubMed]
- Nguyen, P.H.; Giraud, J.; Chambonnier, L.; Dubus, P.; Wittkop, L.; Belleannée, G.; Collet, D.; Soubeyran, I.; Evrard, S.; Rousseau, B.; Senant-Dugot, N.; Mégraud, F.; et al. Characterization of Biomarkers of Tumorigenic and Chemoresistant Cancer Stem Cells in Human Gastric Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Varon, C.; Dubus, P.; Mazurier, F.; Asencio, C.; Chambonnier, L.; Ferrand, J.; Giese, A.; Senant-Dugot, N.; Carlotti, M.; Mégraud, F. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology 2012, 142, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannée, G.; Mégraud, F.; Varon, C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 2014, 33, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Lanvers, C.; Hempel, G.; Blaschke, G.; Boos, J. Chemically induced isomerization and differential uptake modulate retinoic acid disposition in HL-60 cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1998, 12, 1627–1633. [Google Scholar] [CrossRef]
- Altucci, L.; Gronemeyer, H. The promise of retinoids to fight against cancer. Nat. Rev. Cancer 2001, 1, 181–193. [Google Scholar] [CrossRef] [PubMed]
- De Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Ammatuna, E. The biology of acute promyelocytic leukemia and its impact on diagnosis and treatment. Hematol. Am. Soc. Hematol. Educ. Program 2006, 2006, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Abaza, Y.; Kantarjian, H.; Garcia-Manero, G.; Estey, E.; Borthakur, G.; Jabbour, E.; Faderl, S.; O’Brien, S.; Wierda, W.; Pierce, S.; et al. Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood 2017, 129, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Bœuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; Soubeyran, I.; Belleannée, G.; et al. All-trans retinoic acid targets gastric cancer stem cells and inhibits patient-derived gastric carcinoma tumor growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Teelmann, K.; Tsukaguchi, T.; Klaus, M.; Eliason, J.F. Comparison of the therapeutic effects of a new arotinoid, Ro 40-8757, and all-trans- and 13-cis-retinoic acids on rat breast cancer. Cancer Res. 1993, 53, 2319–2325. [Google Scholar] [PubMed]
- Dillehay, D.L.; Shealy, Y.F.; Lamon, E.W. Inhibition of Moloney murine lymphoma and sarcoma growth in vivo by dietary retinoids. Cancer Res. 1989, 49, 44–50. [Google Scholar] [PubMed]
- Aebi, S.; Kröning, R.; Cenni, B.; Sharma, A.; Fink, D.; Los, G.; Weisman, R.; Howell, S.B.; Christen, R.D. All-trans retinoic acid enhances cisplatin-induced apoptosis in human ovarian adenocarcinoma and in squamous head and neck cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 2033–2038. [Google Scholar]
- Felix, E.L.; Loyd, B.; Cohen, M.H. Inhibition of the growth and development of a transplantable murine melanoma by vitamin A. Science 1975, 189, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric cancer originating from bone marrow-derived cells. Science 2004, 306, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Dubus, P.; Mégraud, F.; Varon, C. Helicobacter pylori infection and stem cells at the origin of gastric cancer. Oncogene 2015, 34, 2547–2555. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Dylla, S.J.; Beviglia, L.; Park, I.-K.; Chartier, C.; Raval, J.; Ngan, L.; Pickell, K.; Aguilar, J.; Lazetic, S.; Smith-Berdan, S.; et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 2008, 3, e2428. [Google Scholar] [CrossRef]
- Planque, C.; Rajabi, F.; Grillet, F.; Finetti, P.; Bertucci, F.; Gironella, M.; Lozano, J.J.; Beucher, B.; Giraud, J.; Garambois, V.; et al. Pregnane X-receptor promotes stem cell-mediated colon cancer relapse. Oncotarget 2016, 7, 56558–56573. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, L.; Sladek, N.E. Cellular levels of class 1 and class 3 aldehyde dehydrogenases and certain other drug-metabolizing enzymes in human breast malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 1901–1914. [Google Scholar]
- Sládek, N.E.; Kollander, R.; Sreerama, L.; Kiang, D.T. Cellular levels of aldehyde dehydrogenases (ALDH1A1 and ALDH3A1) as predictors of therapeutic responses to cyclophosphamide-based chemotherapy of breast cancer: A retrospective study. Rational individualization of oxazaphosphorine-based cancer chemotherapeutic regimens. Cancer Chemother. Pharmacol. 2002, 49, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.N.; Chow, E.K.-H. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Mégraud, F.; Bessède, E.; Varon, C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2015, 21, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Molina, S.; Acuña-Amador, L.; Dubus, P.; Staedel, C.; Chambonnier, L.; Buissonnière, A.; Sifré, E.; Giese, A.; Bénéjat, L.; et al. Deletion of IQGAP1 promotes Helicobacter pylori-induced gastric dysplasia in mice and acquisition of cancer stem cell properties in vitro. Oncotarget 2016, 7, 80688–80699. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Avigdor, A.; Goichberg, P.; Shivtiel, S.; Dar, A.; Peled, A.; Samira, S.; Kollet, O.; Hershkoviz, R.; Alon, R.; Hardan, I.; et al. CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood 2004, 103, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Ghatak, S.; Toole, B.P. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 2005, 280, 20310–20315. [Google Scholar] [CrossRef] [PubMed]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef] [PubMed]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W.K. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle Georget. Tex. 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Bollag, W.; Matter, A. From vitamin A to retinoids in experimental and clinical oncology: Achievements, failures, and outlook. Ann. N. Y. Acad. Sci. 1981, 359, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [PubMed]
- Runde, V.; Aul, C.; Südhoff, T.; Heyll, A.; Schneider, W. Retinoic acid in the treatment of acute promyelocytic leukemia: Inefficacy of the 13-cis isomer and induction of complete remission by the all-trans isomer complicated by thromboembolic events. Ann. Hematol. 1992, 64, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Pemrick, S.M.; Lucas, D.A.; Grippo, J.F. The retinoid receptors. Leukemia 1994, 8 (Suppl. 3), S1–S10. [Google Scholar] [PubMed]
- Pubchem Retinoic Acid Receptor Alpha (Human). Available online: https://pubchem.ncbi.nlm.nih.gov/target/gene/RARA/human (accessed on 29 September 2018).
- Delva, L.; Bastie, J.N.; Rochette-Egly, C.; Kraïba, R.; Balitrand, N.; Despouy, G.; Chambon, P.; Chomienne, C. Physical and functional interactions between cellular retinoic acid binding protein II and the retinoic acid-dependent nuclear complex. Mol. Cell. Biol. 1999, 19, 7158–7167. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Glass, C.K.; Rosenfeld, M.G. Coactivator and corepressor complexes in nuclear receptor function. Curr. Opin. Genet. Dev. 1999, 9, 140–147. [Google Scholar] [CrossRef]
- Apfel, C.; Bauer, F.; Crettaz, M.; Forni, L.; Kamber, M.; Kaufmann, F.; LeMotte, P.; Pirson, W.; Klaus, M. A retinoic acid receptor alpha antagonist selectively counteracts retinoic acid effects. Proc. Natl. Acad. Sci. USA 1992, 89, 7129–7133. [Google Scholar] [CrossRef] [PubMed]
- Marshall, G.M.; Cheung, B.; Stacey, K.P.; Norris, M.D.; Haber, M. Regulation of retinoic acid receptor alpha expression in human neuroblastoma cell lines and tumor tissue. Anticancer Res. 1994, 14, 437–441. [Google Scholar] [PubMed]
- Muindi, J.R.; Frankel, S.R.; Huselton, C.; DeGrazia, F.; Garland, W.A.; Young, C.W.; Warrell, R.P. Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res. 1992, 52, 2138–2142. [Google Scholar] [PubMed]
- Meyskens, F.L.; Surwit, E.; Moon, T.E.; Childers, J.M.; Davis, J.R.; Dorr, R.T.; Johnson, C.S.; Alberts, D.S. Enhancement of regression of cervical intraepithelial neoplasia II (moderate dysplasia) with topically applied all-trans-retinoic acid: A randomized trial. J. Natl. Cancer Inst. 1994, 86, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Boisnic, S.; Branchet, M.C.; Pascal, F.; Ben Slama, L.; Rostin, M.; Szpirglas, H. Topical tretinoin in the treatment of lichen planus and leukoplakia of the mouth mucosa. A clinical evaluation. Ann. Dermatol. Venereol. 1994, 121, 459–463. [Google Scholar] [PubMed]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.-H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Kirikoshi, H.; Katoh, M. Expression and regulation of WNT10B in human cancer: Up-regulation of WNT10B in MCF-7 cells by beta-estradiol and down-regulation of WNT10B in NT2 cells by retinoic acid. Int. J. Mol. Med. 2002, 10, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Regulation of WNT3 and WNT3A mRNAs in human cancer cell lines NT2, MCF-7, and MKN45. Int. J. Oncol. 2002, 20, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Liu, Y.; Liu, L.; Wu, K.; Wei, W.; Zhu, Y.; Wu, J. Retinoic acid activates human inducible nitric oxide synthase gene through binding of RARalpha/RXRalpha heterodimer to a novel retinoic acid response element in the promoter. Biochem. Biophys. Res. Commun. 2007, 355, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dawson, M.I.; Agadir, A.; Lee, M.O.; Jong, L.; Hobbs, P.D.; Zhang, X.K. Regulation of RAR beta expression by RAR- and RXR-selective retinoids in human lung cancer cell lines: Effect on growth inhibition and apoptosis induction. Int. J. Cancer 1998, 75, 88–95. [Google Scholar] [CrossRef]
- Liu, Y.; Lee, M.O.; Wang, H.G.; Li, Y.; Hashimoto, Y.; Klaus, M.; Reed, J.C.; Zhang, X. Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol. Cell. Biol. 1996, 16, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wu, M.; Levi, G.; Ferrari, N. Inhibition of cancer cell growth by all-trans retinoic acid and its analog N-(4-hydroxyphenyl) retinamide: A possible mechanism of action via regulation of retinoid receptors expression. Int. J. Cancer 1998, 78, 248–254. [Google Scholar] [CrossRef]
- Wuarin, L.; Chang, B.; Wada, R.; Sidell, N. Retinoic acid up-regulates nuclear retinoic acid receptor-alpha expression in human neuroblastoma cells. Int. J. Cancer 1994, 56, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Ong, E.S.; Dyck, J.A.; Evans, R.M. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 1990, 345, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.A.; Sturzenbecker, L.J.; Kazmer, S.; Bosakowski, T.; Huselton, C.; Allenby, G.; Speck, J.; Kratzeisen, C.; Rosenberger, M.; Lovey, A. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR alpha. Nature 1992, 355, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J. Vitamin A receptors. Nutr. Rev. 1994, 52, S32–S44. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, P.P. In vivo analysis of the molecular genetics of acute promyelocytic leukemia. Oncogene 2001, 20, 5726–5735. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Sternsdorf, T.; Tini, M.; Evans, R.M. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001, 20, 7204–7215. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Borgmeyer, U.; Heyman, R.A.; Zhou, J.Y.; Ong, E.S.; Oro, A.E.; Kakizuka, A.; Evans, R.M. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992, 6, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.P. Differentiating agents in pediatric malignancies: retinoids in neuroblastoma. Curr. Oncol. Rep. 2000, 2, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Idres, N.; Marill, J.; Flexor, M.A.; Chabot, G.G. Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J. Biol. Chem. 2002, 277, 31491–31498. [Google Scholar] [CrossRef] [PubMed]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Cheung, B.; Hocker, J.E.; Smith, S.A.; Norris, M.D.; Haber, M.; Marshall, G.M. Favorable prognostic significance of high-level retinoic acid receptor beta expression in neuroblastoma mediated by effects on cell cycle regulation. Oncogene 1998, 17, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Clagett-Dame, M.; Verhalen, T.J.; Biedler, J.L.; Repa, J.J. Identification and characterization of all-trans-retinoic acid receptor transcripts and receptor protein in human neuroblastoma cells. Arch. Biochem. Biophys. 1993, 300, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, K.; Wan, Y.-J.Y. Retinoids activate RXR/CAR-mediated pathway and induce CYP3A. Biochem. Pharmacol. 2010, 79, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, N.; Tsurumi, H.; Okuno, M.; Matsushima-Nishiwaki, R.; Shimizu, M.; Moriwaki, H. Retinoid X receptor alpha is highly phosphorylated in retinoic acid-resistant HL-60R cells and the combination of 9-cis retinoic acid plus MEK inhibitor induces apoptosis in the cells. Leuk. Res. 2008, 32, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liao, Q.; Ren, C.-F.; Wei, J. Identification of ligand binding site on RXRγ using molecular docking and dynamics methods. J. Mol. Model. 2011, 17, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.T.; Fisher, G.J.; Zhang, Q.Y.; Eisen, D.; Krust, A.; Kastner, P.; Chambon, P.; Voorhees, J.J. Retinoic acid receptor gene expression in human skin. J. Investig. Dermatol. 1991, 96, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.-W.; Chen, F.-H.; Ge, J.-F.; Cao, L.-Y.; Li, H. Retinoid receptors in gastric cancer: Expression and influence on prognosis. Asian Pac. J. Cancer Prev. 2012, 13, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Schmutzler, C.; Brtko, J.; Winzer, R.; Jakobs, T.C.; Meissner-Weigl, J.; Simon, D.; Goretzki, P.E.; Köhrle, J. Functional retinoid and thyroid hormone receptors in human thyroid-carcinoma cell lines and tissues. Int. J. Cancer 1998, 76, 368–376. [Google Scholar] [CrossRef]
- Lotan, R.; Xu, X.C.; Lippman, S.M.; Ro, J.Y.; Lee, J.S.; Lee, J.J.; Hong, W.K. Suppression of retinoic acid receptor-beta in premalignant oral lesions and its up-regulation by isotretinoin. N. Engl. J. Med. 1995, 332, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Lotan, Y.; Xu, X.C.; Shalev, M.; Lotan, R.; Williams, R.; Wheeler, T.M.; Thompson, T.C.; Kadmon, D. Differential expression of nuclear retinoid receptors in normal and malignant prostates. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Zhang, W.; El-Naggar, A.K.; Lippman, S.M.; Lin, P.; Lotan, R.; Xu, X.C. Loss of retinoic acid receptor-beta expression is an early event during esophageal carcinogenesis. Am. J. Pathol. 1999, 155, 1519–1523. [Google Scholar] [CrossRef]
- Houle, B.; Rochette-Egly, C.; Bradley, W.E. Tumor-suppressive effect of the retinoic acid receptor beta in human epidermoid lung cancer cells. Proc. Natl. Acad. Sci. USA 1993, 90, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, N.; Mathur, M.; Bahadur, S.; Kumar Shukla, N.; Ralhan, R. Retinoic acid receptor-alpha as a prognostic indicator in oral squamous cell carcinoma. Int. J. Cancer 2003, 103, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Van der Leede, B.M.; Geertzema, J.; Vroom, T.M.; Décimo, D.; Lutz, Y.; van der Saag, P.T.; van der Burg, B. Immunohistochemical analysis of retinoic acid receptor-alpha in human breast tumors: Retinoic acid receptor-alpha expression correlates with proliferative activity. Am. J. Pathol. 1996, 148, 1905–1914. [Google Scholar] [PubMed]
- Fujimaki, Y. Formation of Gastric Carcinoma in Albino Rats Fed on Deficient Diets. J. Cancer Res. 1926, 10, 469–477. [Google Scholar] [CrossRef]
- Haenszel, W.; Correa, P.; López, A.; Cuello, C.; Zarama, G.; Zavala, D.; Fontham, E. Serum micronutrient levels in relation to gastric pathology. Int. J. Cancer 1985, 36, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Y.; Meng, Y.-P.; Bo, L.-S.; Ke, W.-B. miR-542-3p Appended Sorafenib/All-trans Retinoic Acid (ATRA)-Loaded Lipid Nanoparticles to Enhance the Anticancer Efficacy in Gastric Cancers. Pharm. Res. 2017, 34, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Yokozaki, H.; Domen, T.; Hayashi, K.; Kuniyasu, H.; Yasui, W.; Lotan, R.; Tahara, E. Growth inhibition of cultured human gastric cancer cells by 9-cis-retinoic acid with induction of cdk inhibitor Waf1/Cip1/Sdi1/p21 protein. Differ. Res. Biol. Divers. 1997, 61, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Birrer, M.J.; Watts, R.G.; Matrisian, L.M.; Colburn, N.H. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc. Natl. Acad. Sci. USA 1994, 91, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Curran, T.; Franza, B.R. Fos and Jun: The AP-1 connection. Cell 1988, 55, 395–397. [Google Scholar] [CrossRef]
- Huang, S.-L.; Shyu, R.-Y.; Yeh, M.-Y.; Jiang, S.-Y. The retinoid-inducible gene I: Effect on apoptosis and mitogen-activated kinase signal pathways. Anticancer Res. 2002, 22, 799–804. [Google Scholar] [PubMed]
- Ye, X.; Wu, Q.; Liu, S.; Lin, X.; Zhang, B.; Wu, J.; Cai, J.; Zhang, M.; Su, W. Distinct role and functional mode of TR3 and RARalpha in mediating ATRA-induced signalling pathway in breast and gastric cancer cells. Int. J. Biochem. Cell Biol. 2004, 36, 98–113. [Google Scholar] [CrossRef]
- Lin, X.-F.; Zhao, B.-X.; Chen, H.-Z.; Ye, X.-F.; Yang, C.-Y.; Zhou, H.-Y.; Zhang, M.-Q.; Lin, S.-C.; Wu, Q. RXRalpha acts as a carrier for TR3 nuclear export in a 9-cis retinoic acid-dependent manner in gastric cancer cells. J. Cell Sci. 2004, 117, 5609–5621. [Google Scholar] [CrossRef] [PubMed]
- Patrad, E.; Niapour, A.; Farassati, F.; Amani, M. Combination treatment of all-trans retinoic acid (ATRA) and γ-secretase inhibitor (DAPT) cause growth inhibition and apoptosis induction in the human gastric cancer cell line. Cytotechnology 2018, 70, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Y.; Shyu, R.Y.; Chen, H.Y.; Lee, M.M.; Wu, K.L.; Yeh, M.Y. In vitro and in vivo growth inhibition of SC-M1 gastric cancer cells by retinoic acid. Oncology 1996, 53, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, Y.-Q.; Chen, Z.-M.; Chen, F.; Su, W.-J. Effects of retinoic acid on metastasis and its related proteins in gastric cancer cells in vivo and in vitro. Acta Pharmacol. Sin. 2002, 23, 835–841. [Google Scholar] [PubMed]
- Karam, S.M.; Hassan, W.M.; John, R. Expression of retinoid receptors in multiple cell lineages in the gastric mucosae of mice and humans. J. Gastroenterol. Hepatol. 2005, 20, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, X.; Xing, L.; Chang, Y.; Wu, L.; Jin, Z.; Su, X.; Bai, Y.; Zheng, Y.; Jiang, Y.; et al. Addition of all-trans-retinoic acid to omeprazole and sucralfate therapy improves the prognosis of gastric dysplasia. J. Int. Med. Res. 2015, 43, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Quach, M.A.; Ake, C.D.; Chen, M.; Wang, J. Gastrointestinal lymphomas: Morphology, immunophenotype and molecular features. J. Gastrointest. Oncol. 2012, 3, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Wotherspoon, A.C. Helicobacter pylori infection and gastric lymphoma. Br. Med. Bull. 1998, 54, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Salcedo, L.M.; Sokol, L.; Chavez, J.C.; Dalia, S. Primary Gastric Lymphoma, Epidemiology, Clinical Diagnosis, and Treatment. Cancer Control J. Moffitt Cancer Cent. 2018, 25, 1073274818778256. [Google Scholar] [CrossRef]
- Sacks, P.G.; Harris, D.; Chou, T.C. Modulation of growth and proliferation in squamous cell carcinoma by retinoic acid: A rationale for combination therapy with chemotherapeutic agents. Int. J. Cancer 1995, 61, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Shalinsky, D.R.; Bischoff, E.D.; Gregory, M.L.; Lamph, W.W.; Heyman, R.A.; Hayes, J.S.; Thomazy, V.; Davies, P.J. Enhanced antitumor efficacy of cisplatin in combination with ALRT1057 (9-cis retinoic acid) in human oral squamous carcinoma xenografts in nude mice. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 511–520. [Google Scholar]
- Goncalves, A.; Camerlo, J.; Bun, H.; Gravis, G.; Genre, D.; Bertucci, F.; Resbeut, M.; Pech-Gourg, F.; Durand, A.; Maraninchi, D.; et al. Phase II study of a combination of cisplatin, all-trans-retinoic acid and interferon-alpha in squamous cell carcinoma: Clinical results and pharmacokinetics. Anticancer Res. 2001, 21, 1431–1437. [Google Scholar] [PubMed]
- Scribner, D.R.; Benbrook, D.M. Retinoids enhance cisplatin-based chemoradiation in cervical cancer cells in vitro. Gynecol. Oncol. 2002, 85, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chan, S.Y.; Ho, P.C.-L. Comparison of the in vitro and in vivo effects of retinoids either alone or in combination with cisplatin and 5-fluorouracil on tumor development and metastasis of melanoma. Cancer Chemother. Pharmacol. 2008, 63, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; González-De la Rosa, C.H.; Aréchaga-Ocampo, E.; Villanueva-Rodríguez, G.; Cerón-Lizárraga, T.L.; Martínez-Barrera, L.; Vázquez-Manríquez, M.E.; Ríos-Trejo, M.A.; Alvarez-Avitia, M.A.; Hernández-Pedro, N.; et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.; Bertolini, G.; Pastorino, U.; Roz, L.; Sozzi, G. Combination Treatment with All-Trans Retinoic Acid Prevents Cisplatin-Induced Enrichment of CD133+ Tumor-Initiating Cells and Reveals Heterogeneity of Cancer Stem Cell Compartment in Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Najafzadeh, N.; Mazani, M.; Abbasi, A.; Farassati, F.; Amani, M. Low-dose all-trans retinoic acid enhances cytotoxicity of cisplatin and 5-fluorouracil on CD44(+) cancer stem cells. Biomed. Pharmacother. Biomed. Pharmacother. 2015, 74, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Li, Y.P.; Nobile, L.M.; Grills, G.; Carrera, I.; Paietta, E.; Tallman, M.S.; Wiernik, P.H.; Gallagher, R.E. Leukemic cellular retinoic acid resistance and missense mutations in the PML-RARalpha fusion gene after relapse of acute promyelocytic leukemia from treatment with all-trans retinoic acid and intensive chemotherapy. Blood 1998, 92, 1172–1183. [Google Scholar] [PubMed]
- Gallagher, R.E. Retinoic acid resistance in acute promyelocytic leukemia. Leukemia 2002, 16, 1940–1958. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Redfern, C.P.F.; Veal, G.J. 13-cis retinoic acid and isomerisation in paediatric oncology—Is changing shape the key to success? Biochem. Pharmacol. 2005, 69, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Njar, V.C.O. Cytochrome p450 retinoic acid 4-hydroxylase inhibitors: Potential agents for cancer therapy. Mini Rev. Med. Chem. 2002, 2, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Van Heusden, J.; Van Ginckel, R.; Bruwiere, H.; Moelans, P.; Janssen, B.; Floren, W.; van der Leede, B.J.; van Dun, J.; Sanz, G.; Venet, M.; et al. Inhibition of all-TRANS-retinoic acid metabolism by R116010 induces antitumour activity. Br. J. Cancer 2002, 86, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Takatsuka, J.; Takahashi, N.; de Luca, L.M. Retinoic acid metabolism and inhibition of cell proliferation: An unexpected liaison. Cancer Res. 1996, 56, 675–678. [Google Scholar] [PubMed]
- Idres, N.; Benoît, G.; Flexor, M.A.; Lanotte, M.; Chabot, G.G. Granulocytic differentiation of human NB4 promyelocytic leukemia cells induced by all-trans retinoic acid metabolites. Cancer Res. 2001, 61, 700–705. [Google Scholar] [PubMed]
- Reynolds, N.J.; Fisher, G.J.; Griffiths, C.E.; Tavakkol, A.; Talwar, H.S.; Rowse, P.E.; Hamilton, T.A.; Voorhees, J.J. Retinoic acid metabolites exhibit biological activity in human keratinocytes, mouse melanoma cells and hairless mouse skin in vivo. J. Pharmacol. Exp. Ther. 1993, 266, 1636–1642. [Google Scholar] [PubMed]
- Kim, S.Y.; Yoo, S.J.; Kwon, H.J.; Kim, S.H.; Byun, Y.; Lee, K.-S. Retinoic acid 4-hydroxylase-mediated catabolism of all-trans retinoic acid and the cell proliferation in head and neck squamous cell carcinoma. Metabolism 2002, 51, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Kizaki, M.; Ueno, H.; Yamazoe, Y.; Shimada, M.; Takayama, N.; Muto, A.; Matsushita, H.; Nakajima, H.; Morikawa, M.; Koeffler, H.P.; et al. Mechanisms of retinoid resistance in leukemic cells: Possible role of cytochrome P450 and P-glycoprotein. Blood 1996, 87, 725–733. [Google Scholar] [PubMed]
- Cornic, M.; Delva, L.; Castaigne, S.; Lefebvre, P.; Balitrand, N.; Degos, L.; Chomienne, C. In vitro all-trans retinoic acid (ATRA) sensitivity and cellular retinoic acid binding protein (CRABP) levels in relapse leukemic cells after remission induction by ATRA in acute promyelocytic leukemia. Leukemia 1994, 8, 914–917. [Google Scholar] [PubMed]
- Imaizumi, M.; Suzuki, H.; Yoshinari, M.; Sato, A.; Saito, T.; Sugawara, A.; Tsuchiya, S.; Hatae, Y.; Fujimoto, T.; Kakizuka, A.; et al. Mutations in the E-domain of RAR portion of the PML/RAR chimeric gene may confer clinical resistance to all-trans retinoic acid in acute promyelocytic leukemia. Blood 1998, 92, 374–382. [Google Scholar] [PubMed]
- Levin, A.A. Receptors as tools for understanding the toxicity of retinoids. Toxicol. Lett. 1995, 82–83, 91–97. [Google Scholar] [CrossRef]
- Thiele, C.J.; Reynolds, C.P.; Israel, M.A. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature 1985, 313, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Villablanca, J.G.; Khan, A.A.; Avramis, V.I.; Seeger, R.C.; Matthay, K.K.; Ramsay, N.K.; Reynolds, C.P. Phase I trial of 13-cis-retinoic acid in children with neuroblastoma following bone marrow transplantation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1995, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; Gerbing, R.B. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Kraemer, K.H.; DiGiovanna, J.J.; Moshell, A.N.; Tarone, R.E.; Peck, G.L. Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N. Engl. J. Med. 1988, 318, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Somos, S.; Farkas, B. Immunomodulatory treatment with low-dose interferon-alpha and oral retinoic acid in lymphangioma-like Kaposi’s sarcoma. Anticancer Res. 2000, 20, 541–545. [Google Scholar] [PubMed]
- Lippman, S.M.; Parkinson, D.R.; Itri, L.M.; Weber, R.S.; Schantz, S.P.; Ota, D.M.; Schusterman, M.A.; Krakoff, I.H.; Gutterman, J.U.; Hong, W.K. 13-cis-retinoic acid and interferon alpha-2a: Effective combination therapy for advanced squamous cell carcinoma of the skin. J. Natl. Cancer Inst. 1992, 84, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Halter, S.A.; Fraker, L.D.; Adcock, D.; Vick, S. Effect of retinoids on xenotransplanted human mammary carcinoma cells in athymic mice. Cancer Res. 1988, 48, 3733–3736. [Google Scholar] [PubMed]
- Napoli, J.L. Retinoic acid biosynthesis and metabolism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 993–1001. [Google Scholar] [CrossRef]
- Fowler, J.F.; Graff, O.; Hamedani, A.G. A phase 3, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of alitretinoin (BAL4079) in the treatment of severe chronic hand eczema refractory to potent topical corticosteroid therapy. J. Drugs Dermatol. 2014, 13, 1198–1204. [Google Scholar] [PubMed]
- Papi, A.; Rocchi, P.; Ferreri, A.M.; Guerra, F.; Orlandi, M. Enhanced effects of PPARgamma ligands and RXR selective retinoids in combination to inhibit migration and invasiveness in cancer cells. Oncol. Rep. 2009, 21, 1083–1089. [Google Scholar] [PubMed]
- Haugen, B.R.; Larson, L.L.; Pugazhenthi, U.; Hays, W.R.; Klopper, J.P.; Kramer, C.A.; Sharma, V. Retinoic acid and retinoid X receptors are differentially expressed in thyroid cancer and thyroid carcinoma cell lines and predict response to treatment with retinoids. J. Clin. Endocrinol. Metab. 2004, 89, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Ponthan, F.; Borgström, P.; Hassan, M.; Wassberg, E.; Redfern, C.P.; Kogner, P. The vitamin A analogues: 13-cis retinoic acid, 9-cis retinoic acid, and Ro 13-6307 inhibit neuroblastoma tumour growth in vivo. Med. Pediatr. Oncol. 2001, 36, 127–131. [Google Scholar] [CrossRef]
- Maurer, B.J.; Metelitsa, L.S.; Seeger, R.C.; Cabot, M.C.; Reynolds, C.P. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)- retinamide in neuroblastoma cell lines. J. Natl. Cancer Inst. 1999, 91, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.P.; Kurie, J.M.; Lotan, D.; Zou, C.C.; Hong, W.K.; Lotan, R. Higher potency of N-(4-hydroxyphenyl)retinamide than all-trans-retinoic acid in induction of apoptosis in non-small cell lung cancer cell lines. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 1345–1355. [Google Scholar]
- Delia, D.; Aiello, A.; Lombardi, L.; Pelicci, P.G.; Grignani, F.; Grignani, F.; Formelli, F.; Menard, S.; Costa, A.; Veronesi, U. N-(4-hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res. 1993, 53, 6036–6041. [Google Scholar] [PubMed]
- Sabichi, A.L.; Xu, H.; Fischer, S.; Zou, C.; Yang, X.; Steele, V.E.; Kelloff, G.J.; Lotan, R.; Clifford, J.L. Retinoid receptor-dependent and independent biological activities of novel fenretinide analogues and metabolites. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 4606–4613. [Google Scholar]
- Formelli, F.; Cleris, L. Synthetic retinoid fenretinide is effective against a human ovarian carcinoma xenograft and potentiates cisplatin activity. Cancer Res. 1993, 53, 5374–5376. [Google Scholar] [PubMed]
- Decensi, A.; Formelli, F.; Torrisi, R.; Costa, A. Breast cancer chemoprevention: Studies with 4-HPR alone and in combination with tamoxifen using circulating growth factors as potential surrogate endpoints. J. Cell. Biochem. Suppl. 1993, 17G, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.J.; Kang, M.H.; Villablanca, J.G.; Janeba, J.; Groshen, S.; Matthay, K.K.; Sondel, P.M.; Maris, J.M.; Jackson, H.A.; Goodarzian, F.; et al. Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: A report from the New Approaches to Neuroblastoma Therapy (NANT) consortium. Pediatr. Blood Cancer 2013, 60, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Camerini, T.; Mariani, L.; De Palo, G.; Marubini, E.; Di Mauro, M.G.; Decensi, A.; Costa, A.; Veronesi, U. Safety of the synthetic retinoid fenretinide: Long-term results from a controlled clinical trial for the prevention of contralateral breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.D.; Cobleigh, M.A.; Gray, R.; Graham, M.L.; Norton, L.; Martino, S.; Budd, G.T.; Ingle, J.N.; Wood, W.C. Phase III double-blind, placebo-controlled, prospective randomized trial of adjuvant tamoxifen vs. tamoxifen and fenretinide in postmenopausal women with positive receptors (EB193): An intergroup trial coordinated by the Eastern Cooperative Oncology Group. Med. Oncol. Northwood Lond. Engl. 2011, 28 (Suppl. 1), S39–S47. [Google Scholar] [CrossRef]
- Ju, J.; Wang, N.; Wang, X.; Chen, F. A novel all-trans retinoic acid derivative inhibits proliferation and induces differentiation of human gastric carcinoma xenografts via up-regulating retinoic acid receptor β. Am. J. Transl. Res. 2015, 7, 856–865. [Google Scholar] [PubMed]
- Xia, Q.; Zhao, Y.; Wang, J.; Qiao, W.; Zhang, D.; Yin, H.; Xu, D.; Chen, F. Proteomic analysis of cell cycle arrest and differentiation induction caused by ATPR, a derivative of all-trans retinoic acid, in human gastric cancer SGC-7901 cells. Proteomics Clin. Appl. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yan, Y.; Zhou, J.; Zhou, Q.; Gui, S.; Wang, Y. A novel all-trans retinoid acid derivatives inhibits the migration of breast cancer cell lines MDA-MB-231 via myosin light chain kinase involving p38-MAPK pathway. Biomed. Pharmacother. Biomed. Pharmacother. 2013, 67, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gui, S.-Y.; Chen, F.-H.; Zhou, Q.; Wang, Y. New insights into 4-amino-2-tri-fluoromethyl-phenyl ester inhibition of cell growth and migration in the A549 lung adenocarcinoma cell line. Asian Pac. J. Cancer Prev. 2013, 14, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Yang, Y.; Zhang, S.; Zhou, Q.; Wei, W.; Wang, Y. 4-Amino-2-trifluoromethyl-phenyl retinate inhibits the migration of BGC-823 human gastric cancer cells by downregulating the phosphorylation level of MLC II. Oncol. Rep. 2014, 32, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, F.; Zhang, L.; Zhou, Q.; Gui, S.; Wang, Y. A novel all-trans retinoic acid derivative 4-amino-2-trifluoromethyl-phenyl retinate inhibits the proliferation of human hepatocellular carcinoma HepG2 cells by inducing G0/G1 cell cycle arrest and apoptosis via upregulation of p53 and ASPP1 and downregulation of iASPP. Oncol. Rep. 2016, 36, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Liu, L.; Zheng, C.; Wang, Y. Synthesis and Antiproliferative Activity of Novel All-Trans-Retinoic Acid-Podophyllotoxin Conjugate towards Human Gastric Cancer Cells. Mol. Basel Switz. 2017, 22, 628. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ATRA | 13cisRA | 9cisRA | |
|---|---|---|---|
| RARα | [50], [65,66,67,68] | [64],[69],[70] | [71] |
| RARβ | [51], [62], [61], [72], [65], [66], [73] | [64],[69],[70] | [71] |
| RARγ | [65], [66] | [64],[69],[70] | [71] |
| RXRα | [63],[74],[65] | [74] | [64], [74], [65], [68], [71], [75] |
| RXRβ | [63], [65] | ||
| RXRγ | [63],[74],[65] | [74] | [74], [65], [68], [76] |
| Fonction | Model of Study | Comments |
|---|---|---|
| Cell cycle blocking and differentiation initiation | GC cell lines Tumorspheres PDX | Inhibition of the cell cycle progression by p21WAF1/CIP1 induction [89]. Inhibition of cell proliferation by the inhibition of AP1 transcription factor [90,91]. Downregulation of ERK/MAPK pathway [92]. Induction of the expression of GCSCs differentiation markers [19]. Inhibition of GC cells proliferation in combination with Sorafenib and miRNA in encapsulated nanoparticles [88]. |
| Apoptosis initiation | GC cell lines Tumorspheres PDX | Induction of PDCD4 and cleaved caspase 3 apoptosis markers in GC cell lines, tumorspheres and in PDX models [19]. Induction of apoptosis in GC cell lines by the translocation of TR3 orphan receptor in the mitochondria [93]. Initiation of apoptosis in combination with Sorafenib and miRNA in encapsulated nanoparticles [88]. |
| CSC properties inhibition | GC cell lines Tumorspheres PDX | Inhibition of tumorspheres formation and survival [19]. Reduction of tumor growth in mice subcutaneous xenografts models [19,88,96]. Diminution of liver metastasis after intrasplenic xenograft of GC cells [97]. Reduction of tumor growth in mice subcutaneous xenograft models in combination with Sorafenib and miRNA in encapsulated nanoparticles [88]. Downregulation of the expression of the CSC markers and stemness genes [19]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouriez, D.; Giraud, J.; Gronnier, C.; Varon, C. Efficiency of All-Trans Retinoic Acid on Gastric Cancer: A Narrative Literature Review. Int. J. Mol. Sci. 2018, 19, 3388. https://doi.org/10.3390/ijms19113388
Bouriez D, Giraud J, Gronnier C, Varon C. Efficiency of All-Trans Retinoic Acid on Gastric Cancer: A Narrative Literature Review. International Journal of Molecular Sciences. 2018; 19(11):3388. https://doi.org/10.3390/ijms19113388
Chicago/Turabian StyleBouriez, Damien, Julie Giraud, Caroline Gronnier, and Christine Varon. 2018. "Efficiency of All-Trans Retinoic Acid on Gastric Cancer: A Narrative Literature Review" International Journal of Molecular Sciences 19, no. 11: 3388. https://doi.org/10.3390/ijms19113388
APA StyleBouriez, D., Giraud, J., Gronnier, C., & Varon, C. (2018). Efficiency of All-Trans Retinoic Acid on Gastric Cancer: A Narrative Literature Review. International Journal of Molecular Sciences, 19(11), 3388. https://doi.org/10.3390/ijms19113388