Roles of Gut-Derived Secretory Factors in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Their Possible Clinical Applications

 , ,
, ,
Abstract
1. Introduction
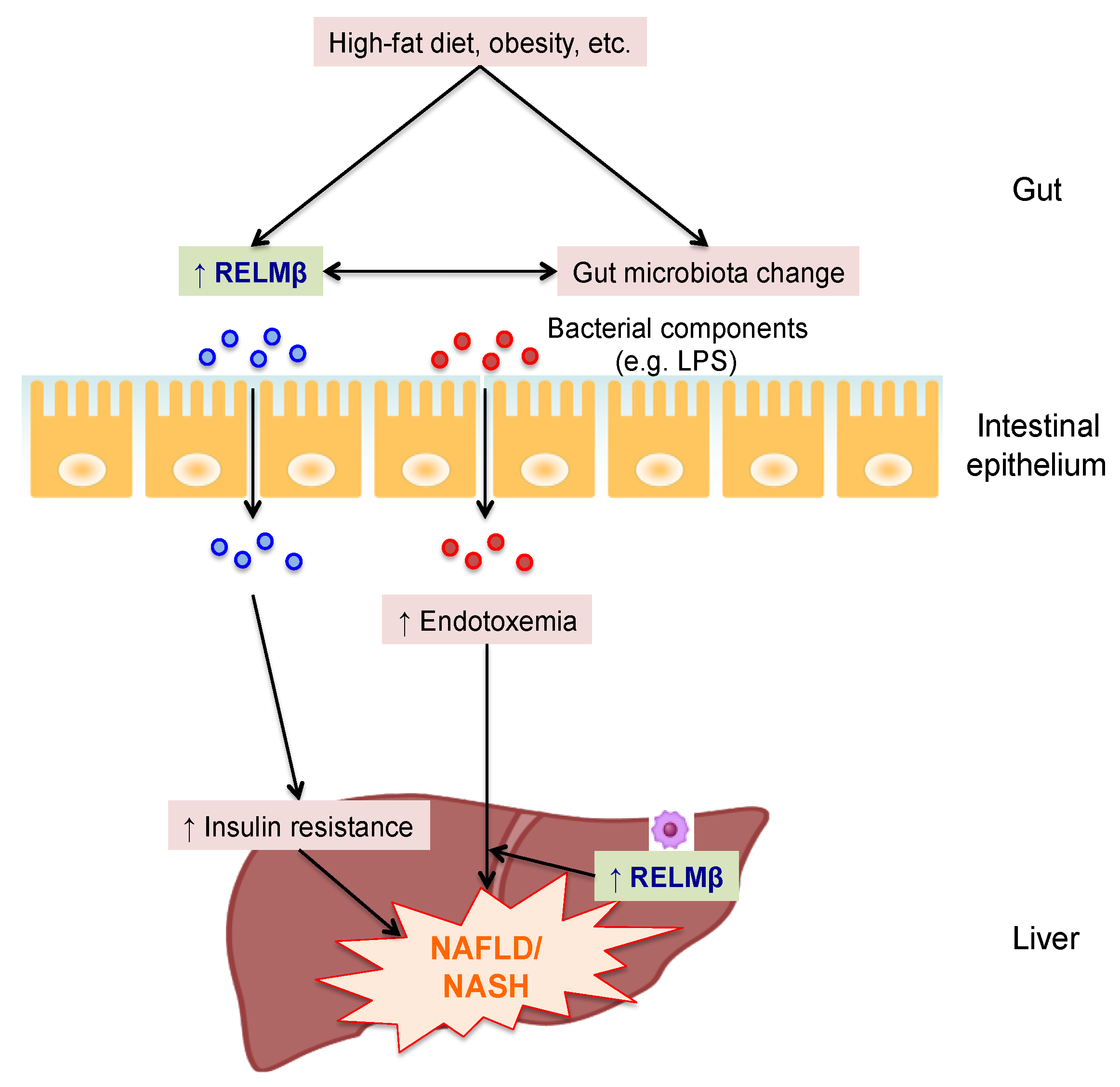
2. RELMβ
3. Glucagon-Like Peptide-1
4. Glucagon-Like Peptide-2
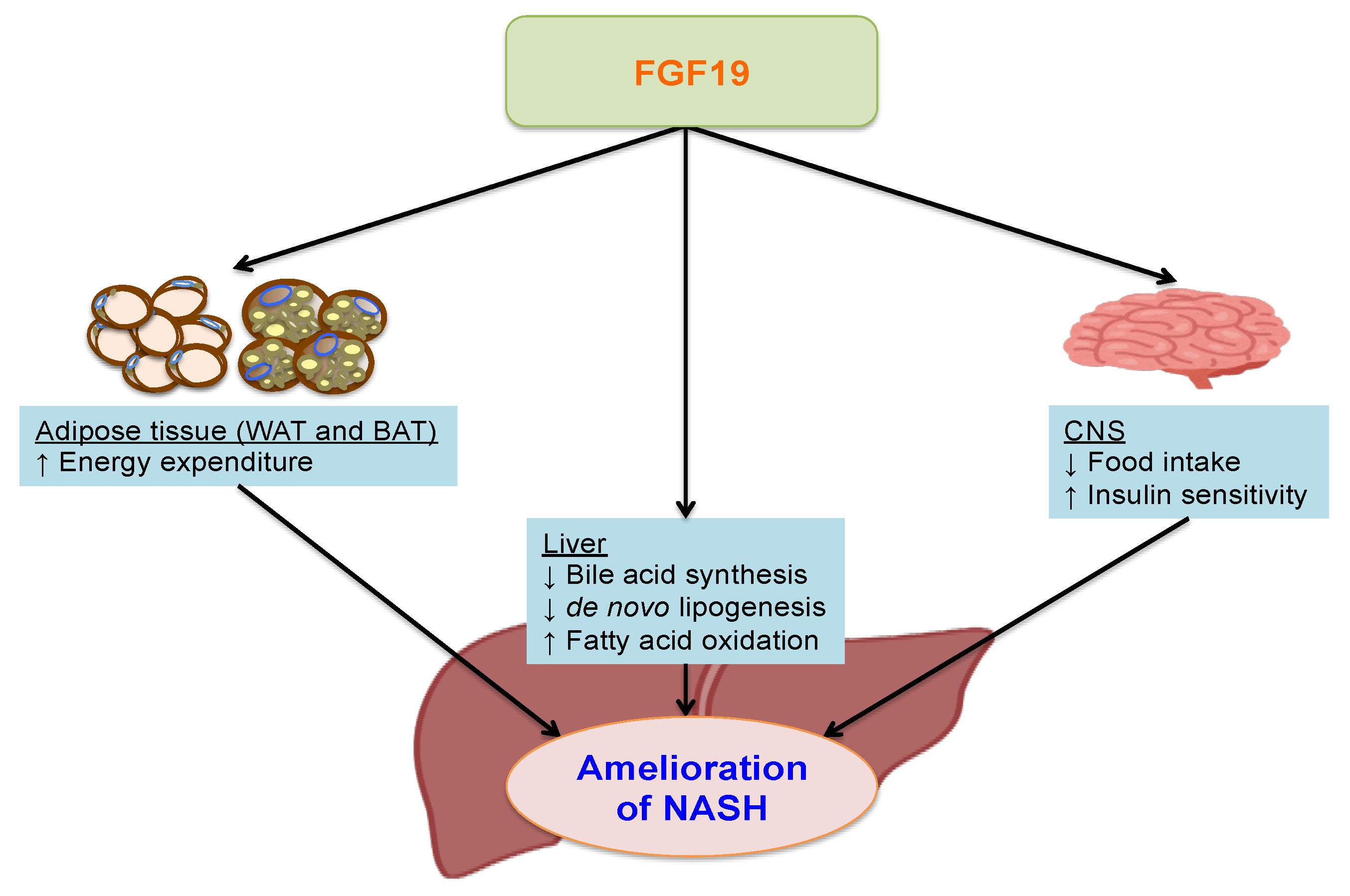
5. Fibroblast Growth Factor 19
6. Neurotensin
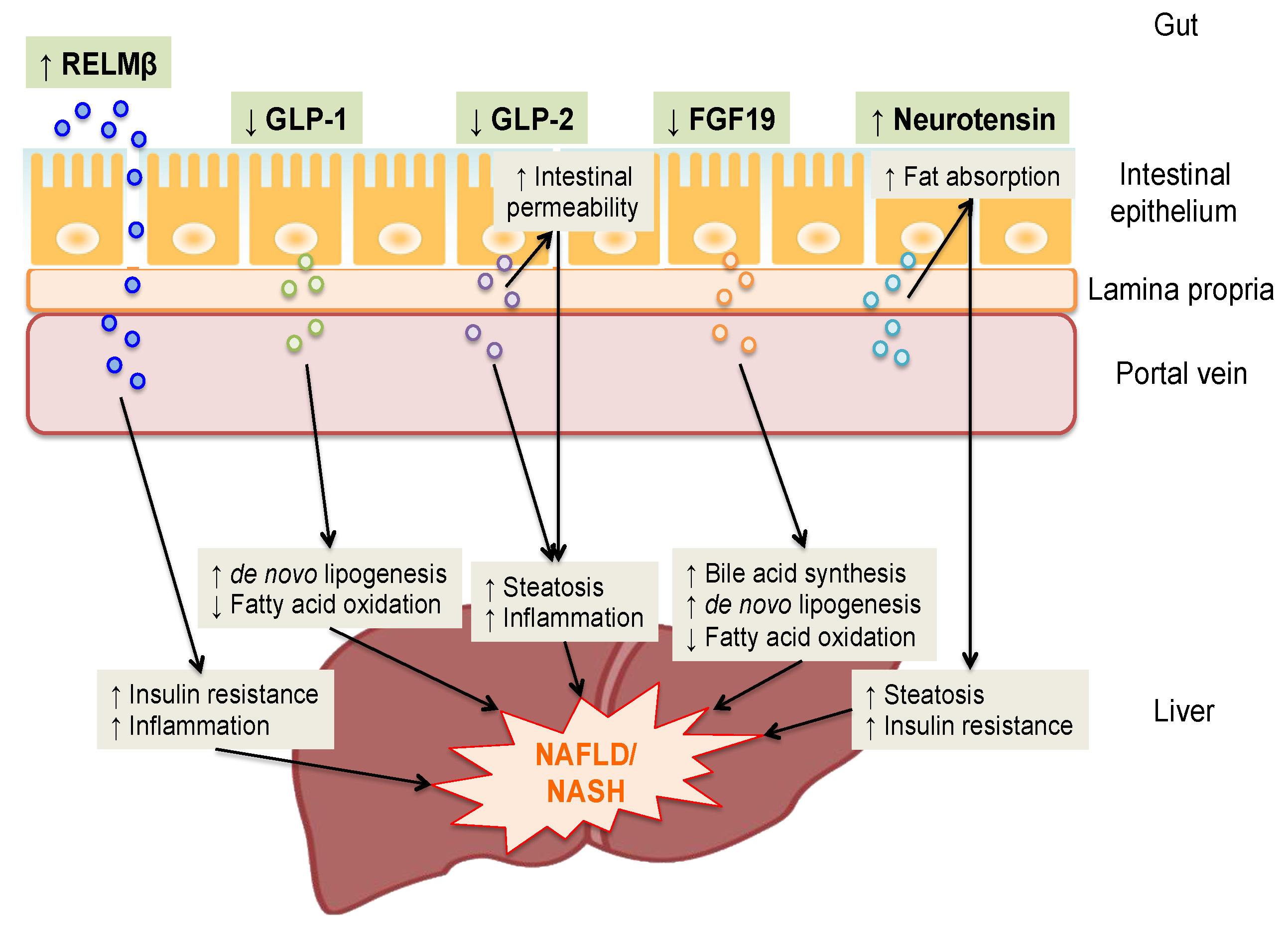
7. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| BMI | Body mass index |
| CNS | Central nervous system |
| DPP-4 | Dipeptidyl peptidase-4 |
| FXR | Farnesoid X receptor |
| FGF | Fibroblast growth factor |
| GLP-1 | Glucagon-like peptide-1 |
| GLP-2 | Glucagon-like peptide 2 |
| GLA | Gut-liver axis |
| HNF4α | Hepatocyte nuclear factor 4α |
| HFD | High fat diet |
| KO | Knock-out |
| LPS | Lipopolysaccharide |
| MCD | Methionine choline-deficient |
| NT | Neurotensin |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| RELMβ | Resistin like molecule β |
| SBS | Short bowel syndrome |
| T2DM | Type 2 diabetes mellitus |
References
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, E.; Taniai, M.; Tokushige, K. Characteristics and diagnosis of NAFLD/NASH. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 4), 64–70. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Brodosi, L.; Marchignoli, F.; Petroni, M.L.; Marchesini, G. NASH: A glance at the landscape of pharmacological treatment. Ann. Hepatol. 2016, 15, 673–681. [Google Scholar] [PubMed]
- Zorn, A.M.; Wells, J.M. Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 221–251. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Mandato, C.; Pierri, L.; Poeta, M.; Di Stasi, M.; Vajro, P. Gut-liver axis and probiotics: Their role in non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 15518–15531. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Kushiyama, A.; Sakoda, H.; Nakatsu, Y.; Iizuka, M.; Taki, N.; Fujishiro, M.; Fukushima, T.; Kamata, H.; Nagamachi, A.; et al. Involvement of resistin-like molecule beta in the development of methionine-choline deficient diet-induced non-alcoholic steatohepatitis in mice. Sci. Rep. 2016, 6, 20157. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, M.L.; Jiang, H.Q.; Steppan, C.M.; Shin, M.E.; Thurnheer, M.C.; Cebra, J.J.; Lazar, M.A.; Wu, G.D. Bacterial colonization leads to the colonic secretion of RELMbeta/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 2003, 125, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, A.; Sakoda, H.; Oue, N.; Okubo, M.; Nakatsu, Y.; Ono, H.; Fukushima, T.; Kamata, H.; Nishimura, F.; Kikuchi, T.; et al. Resistin-like molecule beta is abundantly expressed in foam cells and is involved in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Shojima, N.; Ogihara, T.; Inukai, K.; Fujishiro, M.; Sakoda, H.; Kushiyama, A.; Katagiri, H.; Anai, M.; Ono, H.; Fukushima, Y.; et al. Serum concentrations of resistin-like molecules beta and gamma are elevated in high-fat-fed and obese db/db mice, with increased production in the intestinal tract and bone marrow. Diabetologia 2005, 48, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Renigunta, A.; Hild, C.; Rose, F.; Klepetko, W.; Grimminger, F.; Seeger, W.; Hanze, J. Human RELMbeta is a mitogenic factor in lung cells and induced in hypoxia. FEBS Lett. 2006, 580, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Brown, E.J.; Wright, C.M.; Bhat, S.; Banerjee, R.R.; Dai, C.Y.; Enders, G.H.; Silberg, D.G.; Wen, X.; Wu, G.D.; et al. A family of tissue-specific resistin-like molecules. Proc. Natl. Acad. Sci. USA 2001, 98, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Propheter, D.C.; Chara, A.L.; Harris, T.A.; Ruhn, K.A.; Hooper, L.V. Resistin-like molecule beta is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc. Natl. Acad. Sci. USA 2017, 114, 11027–11033. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 13596–13600. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Wang, M.L.; Keilbaugh, S.A.; He, W.; Brenes, M.; Swain, G.P.; Knight, P.A.; Donaldson, D.D.; Lazar, M.A.; Miller, H.R.; et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2004, 101, 13596–13600. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.R.; Yang, J.Q.; Hogan, S.P.; Groschwitz, K.; Khodoun, M.; Munitz, A.; Orekov, T.; Perkins, C.; Wang, Q.; Brombacher, F.; et al. Intestinal epithelial cell secretion of RELM-beta protects against gastrointestinal worm infection. J. Exp. Med. 2009, 206, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.L.; Vidrich, A.; Wang, M.L.; Wu, G.D.; Cominelli, F.; Rivera-Nieves, J.; Bamias, G.; Cohn, S.M. Resistin-like molecule beta (RELMbeta/FIZZ2) is highly expressed in the ileum of SAMP1/YitFc mice and is associated with initiation of ileitis. J. Immunol. 2007, 179, 7012–7020. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.S.; Morampudi, V.; Chan, J.M.; Bhinder, G.; Lau, J.; Yang, H.; Ma, C.; Huang, T.; Ryz, N.; Sham, H.P.; et al. Goblet Cell Derived RELM-beta Recruits CD4+ T Cells during Infectious Colitis to Promote Protective Intestinal Epithelial Cell Proliferation. PLoS Pathog. 2015, 11, e1005108. [Google Scholar] [CrossRef] [PubMed]
- McVay, L.D.; Keilbaugh, S.A.; Wong, T.M.; Kierstein, S.; Shin, M.E.; Lehrke, M.; Lefterova, M.I.; Shifflett, D.E.; Barnes, S.L.; Cominelli, F.; et al. Absence of bacterially induced RELMbeta reduces injury in the dextran sodium sulfate model of colitis. J. Clin. Investig. 2006, 116, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.G.; Guild, K.J.; Du, Y.; Zaph, C.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.; Stevens, S.; Karow, M.; Artis, D. Goblet cell-derived resistin-like molecule beta augments CD4+ T cell production of IFN-gamma and infection-induced intestinal inflammation. J. Immunol. 2008, 181, 4709–4715. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, K.; Deol, P.; Evans, J.R.; Vuong, L.M.; Chen, G.; Briancon, N.; Bolotin, E.; Lytle, C.; Nair, M.G.; Sladek, F.M. Opposing roles of nuclear receptor HNF4alpha isoforms in colitis and colitis-associated colon cancer. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Morampudi, V.; Dalwadi, U.; Bhinder, G.; Sham, H.P.; Gill, S.K.; Chan, J.; Bergstrom, K.S.; Huang, T.; Ma, C.; Jacobson, K.; et al. The goblet cell-derived mediator RELM-beta drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal Immunol. 2016, 9, 1218–1233. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Obici, S.; Scherer, P.E.; Rossetti, L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J. Clin. Investig. 2003, 111, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, A.; Shojima, N.; Ogihara, T.; Inukai, K.; Sakoda, H.; Fujishiro, M.; Fukushima, Y.; Anai, M.; Ono, H.; Horike, N.; et al. Resistin-like molecule beta activates MAPKs, suppresses insulin signaling in hepatocytes, and induces diabetes, hyperlipidemia, and fatty liver in transgenic mice on a high fat diet. J. Biol. Chem. 2005, 280, 42016–42025. [Google Scholar] [CrossRef] [PubMed]
- Montalto, M.; Maggiano, N.; Ricci, R.; Curigliano, V.; Santoro, L.; Di Nicuolo, F.; Vecchio, F.M.; Gasbarrini, A.; Gasbarrini, G. Lactobacillus acidophilus protects tight junctions from aspirin damage in HT-29 cells. Digestion 2004, 69, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Ahrne, S.; Hagslatt, M.L. Effect of lactobacilli on paracellular permeability in the gut. Nutrients 2011, 3, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Sakoda, H.; Kushiyama, A.; Fujishiro, M.; Nakatsu, Y.; Fukushima, T.; Matsunaga, Y.; Kamata, H.; Asahara, T.; Yoshida, Y.; et al. Lactobacillus casei strain Shirota protects against nonalcoholic steatohepatitis development in a rodent model. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G911–G918. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.P.; Seidu, L.; Blanchard, C.; Groschwitz, K.; Mishra, A.; Karow, M.L.; Ahrens, R.; Artis, D.; Murphy, A.J.; Valenzuela, D.M.; et al. Resistin-like molecule beta regulates innate colonic function: Barrier integrity and inflammation susceptibility. J. Allergy Clin. Immunol. 2006, 118, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Neilson, A.P.; Djuric, Z.; Land, S.; Kato, I. Plasma levels of resistin-like molecule beta in humans. Cancer Epidemiol. 2011, 35, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Greig, N.H. The glucagon-like peptides: A double-edged therapeutic sword? Trends Pharmacol. Sci. 2003, 24, 377–383. [Google Scholar] [CrossRef]
- Koliaki, C.; Doupis, J. Incretin-based therapy: A powerful and promising weapon in the treatment of type 2 diabetes mellitus. Diabetes Ther. 2011, 2, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamdah, R.; Rabiee, A.; Meneilly, G.S.; Shannon, R.P.; Andersen, D.K.; Elahi, D. Clinical review: The extrapancreatic effects of glucagon-like peptide-1 and related peptides. J. Clin. Endocrinol. Metab. 2009, 94, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.D.; Salter, B.M.; Oliveria, J.P.; El-Gammal, A.; Tworek, D.; Smith, S.G.; Sehmi, R.; Gauvreau, G.M.; Butler, M.; O’Byrne, P.M. Glucagon-like peptide-1 receptor expression on human eosinophils and its regulation of eosinophil activation. Clin. Exp. Allergy 2017, 47, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Orskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.J. Long-Acting Glucagon-Like Peptide 1 Receptor Agonists A review of their efficacy and tolerability. Diabetes Care 2011, 34, S279–S284. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M. Incretin therapies: Highlighting common features and differences in the modes of action of glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes Obes. Metab. 2016, 18, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Bernsmeier, C.; Meyer-Gerspach, A.C.; Blaser, L.S.; Jeker, L.; Steinert, R.E.; Heim, M.H.; Beglinger, C. Glucose-induced glucagon-like Peptide 1 secretion is deficient in patients with non-alcoholic fatty liver disease. PLoS ONE 2014, 9, e87488. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment Pharmacol. Ther. 2013, 37, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.J.; Irwin, A.; Gardner, C.J.; Daousi, C.; Purewal, T.; Furlong, N.; Goenka, N.; Thomas, E.L.; Adams, V.L.; Pushpakom, S.P.; et al. Improved glycaemia correlates with liver fat reduction in obese, type 2 diabetes, patients given glucagon-like peptide-1 (GLP-1) receptor agonists. PLoS ONE 2012, 7, e50117. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Eguchi, Y.; Kitajima, Y.; Hyogo, H.; Takahashi, H.; Kojima, M.; Ono, M.; Araki, N.; Tanaka, K.; Yamaguchi, M.; Matsuda, Y.; et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2015, 45, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Sorli, C.; Harashima, S.; Tsoukas, G.M.; Unger, J.; Karsbol, J.D.; Hansen, T.; Bain, S.C. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): A double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017, 5, 251–260. [Google Scholar] [CrossRef]
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of Oral Semaglutide Compared with Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients with Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Philo, L.; Nguyen, P.; Hofflich, H.; Hernandez, C.; Bettencourt, R.; Richards, L.; Salotti, J.; Bhatt, A.; Hooker, J.; et al. Sitagliptin vs placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2016, 65, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.R.; McKenzie, C.A.; Tirona, R.G.; Summers, K.; Seney, S.; Chakrabarti, S.; Malhotra, N.; Beaton, M.D. Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J. Gastroenterol. 2017, 23, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Grasset, E.; Puel, A.; Charpentier, J.; Collet, X.; Christensen, J.E.; Terce, F.; Burcelin, R. A Specific Gut Microbiota Dysbiosis of Type 2 Diabetic Mice Induces GLP-1 Resistance through an Enteric NO-Dependent and Gut-Brain Axis Mechanism. Cell Metab. 2017, 25, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Nakatsu, Y.; Kushiyama, A.; Yamamotoya, T.; Matsunaga, Y.; Inoue, M.K.; Fujishiro, M.; Sakoda, H.; Ohno, H.; Yoneda, M.; et al. Gut Microbiota as a Therapeutic Target for Metabolic Disorders. Curr. Med. Chem. 2018, 25, 984–1001. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Yusta, B. Physiology and Pharmacology of the Enteroendocrine Hormone Glucagon-Like Peptide-2. Ann. Rev. Physiol. 2014, 76, 561–583. [Google Scholar] [CrossRef] [PubMed]
- Ugleholdt, R.; Zhu, X.; Deacon, C.F.; Orskov, C.; Steiner, D.F.; Holst, J.J. Impaired intestinal proglucagon processing in mice lacking prohormone convertase 1. Endocrinology 2004, 145, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.A.; Boushey, R.P.; Drucker, D.J.; Brubaker, P.L. Secretion of the intestinotropic hormone glucagon-like peptide 2 is differentially regulated by nutrients in humans. Gastroenterology 1999, 117, 99–105. [Google Scholar] [CrossRef]
- Hartmann, B.; Harr, M.B.; Jeppesen, P.B.; Wojdemann, M.; Deacon, C.F.; Mortensen, P.B.; Holst, J.J. In vivo and in vitro degradation of glucagon-like peptide-2 in humans. J. Clin. Endocr. Metab. 2000, 85, 2884–2888. [Google Scholar] [CrossRef] [PubMed]
- Wallis, K.; Walters, J.R.F.; Gabe, S. Short bowel syndrome: The role of GLP-2 on improving outcome. Curr. Opin. Clin. Nutr. 2009, 12, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Munroe, D.G.; Gupta, A.K.; Kooshesh, F.; Vyas, T.B.; Rizkalla, G.; Wang, H.; Demchyshyn, L.; Yang, Z.J.; Kamboj, R.K.; Chen, H.Y.; et al. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 1999, 96, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Huang, L.; Munroe, D.; Wolff, G.; Fantaske, R.; Sharma, S.; Demchyshyn, L.; Asa, S.L.; Drucker, D.J. Enteroendocrine localization of GLP-2 receptor expression in humans and rodents. Gastroenterology 2000, 119, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.E.; Brubaker, P.L. Frontiers in glucagon-like peptide-2: Multiple actions, multiple mediators. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E460–E465. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.W.; Sharp, J.W.; Brownfield, M.S.; Raybould, H.E.; Ney, D.M. Localization and activation of glucagon-like peptide-2 receptors on vagal afferents in the rat. Endocrinology 2007, 148, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Angelone, T.; Filice, E.; Quintieri, A.M.; Imbrogno, S.; Amodio, N.; Pasqua, T.; Pellegrino, D.; Mule, F.; Cerra, M.C. Receptor identification and physiological characterisation of glucagon-like peptide-2 in the rat heart. Nutr. Metab. Cardiovasc. Dis. NMCD 2012, 22, 486–494. [Google Scholar] [CrossRef] [PubMed]
- El-Jamal, N.; Erdual, E.; Neunlist, M.; Koriche, D.; Dubuquoy, C.; Maggiotto, F.; Chevalier, J.; Berrebi, D.; Dubuquoy, L.; Boulanger, E.; et al. Glugacon-like peptide-2: Broad receptor expression, limited therapeutic effect on intestinal inflammation and novel role in liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G274–G285. [Google Scholar] [CrossRef] [PubMed]
- Tang-Christensen, M.; Larsen, P.J.; Thulesen, J.; Romer, J.; Vrang, N. The proglucagon-derived peptide, glucagon-like peptide-2, is a neurotransmitter involved in the regulation of food intake. Nat. Med. 2000, 6, 802. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.B.; Flint, A.; Raben, A.; Hartmann, B.; Holst, J.J.; Astrup, A. No effect of physiological concentrations of glucagon-like peptide-2 on appetite and energy intake in normal weight subjects. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Ehrlich, P.; Asa, S.L.; Brubaker, P.L. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 1996, 93, 7911–7916. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Hill, M.; Asa, S.L.; Brubaker, P.L.; Drucker, D.J. Intestinal growth-promoting properties of glucagon-like peptide-2 in mice. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E77–E84. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R.; Wallace, L.E.; Sigalet, D.L. Glucagon-like peptide-2 induces intestinal adaptation in parenterally fed rats with short bowel syndrome. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G964–G972. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, P.B.; Gilroy, R.; Pertkiewicz, M.; Allard, J.P.; Messing, B.; O'Keefe, S.J. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut 2011, 60, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, P.B.; Pertkiewicz, M.; Messing, B.; Iyer, K.; Seidner, D.L.; O’Keefe, S.J.; Forbes, A.; Heinze, H.; Joelsson, B. Teduglutide reduces need for parenteral support among patients with short bowel syndrome with intestinal failure. Gastroenterology 2012, 143, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Au, A.; Gupta, A.; Schembri, P.; Cheeseman, C.I. Rapid insertion of GLUT2 into the rat jejunal brush-border membrane promoted by glucagon-like peptide 2. Biochem. J. 2002, 367, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Ramsanahie, A.P.; Berger, U.V.; Zinner, M.J.; Whang, E.E.; Rhoads, D.B.; Ashley, S.W. Effect of glucagon-like peptide-2 (GLP-2) on diurnal SGLT1 expression. Dig. Dis. Sci. 2004, 49, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Petersen, Y.M. Glucagon-Like Peptide 2 Enhances Maltase-Glucoamylase and Sucrase-Isomaltase Gene Expression and Activity in Parenterally Fed Premature Neonatal Piglets. Pediatr. Res. 2002, 52, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Nauck, M.A.; Pott, A.; Heinze, K.; Goetze, O.; Bulut, K.; Schmidt, W.E.; Gallwitz, B.; Holst, J.J. Glucagon-like peptide 2 stimulates glucagon secretion, enhances lipid absorption, and inhibits gastric acid secretion in humans. Gastroenterology 2006, 130, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Xiao, C.; Morgantini, C.; Connelly, P.W.; Patterson, B.W.; Lewis, G.F. Glucagon-like peptide-2 regulates release of chylomicrons from the intestine. Gastroenterology 2014, 147, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, M.A.; McKay, D.M.; Yang, P.C.; Cameron, H.; Perdue, M.H. Glucagon-like peptide-2 enhances intestinal epithelial barrier function of both transcellular and paracellular pathways in the mouse. Gut 2000, 47, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Moran, G.W.; O'Neill, C.; McLaughlin, J.T. GLP-2 enhances barrier formation and attenuates TNFalpha-induced changes in a Caco-2 cell model of the intestinal barrier. Regul. Pept. 2012, 178, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Verdam, F.J.; Rensen, S.S.; Driessen, A.; Greve, J.W.; Buurman, W.A. Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2011, 45, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.A.; Peffer, N.; Pirone, A.; Bassiri, A.; Sague, S.; Palmer, J.M.; Johnson, D.L.; Nesspor, T.; Kliwinski, C.; Hornby, P.J. GLP-2 receptor agonism ameliorates inflammation and gastrointestinal stasis in murine postoperative ileus. J. Pharmacol. Exp. Ther. 2010, 333, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Baldassano, S.; Liu, S.; Qu, M.H.; Mule, F.; Wood, J.D. Glucagon-like peptide-2 modulates neurally evoked mucosal chloride secretion in guinea pig small intestine in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G800–G805. [Google Scholar] [CrossRef] [PubMed]
- Baldassano, S.; Amato, A.; Rappa, F.; Cappello, F.; Mule, F. Influence of endogenous glucagon-like peptide-2 on lipid disorders in mice fed a high-fat diet. Endocr. Res. 2016, 41, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Valderas, J.P.; Padilla, O.; Solari, S.; Escalona, M.; Gonzalez, G. Feeding and bone turnover in gastric bypass. J. Clin. Endocrinol. Metab. 2014, 99, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.S.; Schwartz, R.P.; Ferraz-de-Souza, B.; da Silva, M.E.; Correa, P.H.; Nery, M. The role of enteric hormone GLP-2 in the response of bone markers to a mixed meal in postmenopausal women with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2015, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Wallis, K.; Walters, J.R.; Forbes, A. Review article: Glucagon-like peptide 2--current applications and future directions. Aliment Pharmacol. Ther. 2007, 25, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Holmstrom, S.R.; Fon Tacer, K.; Bookout, A.L.; Kliewer, S.A.; Mangelsdorf, D.J. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J. Biol. Chem. 2010, 285, 14486–14494. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.S.; Anderson, K.A.; Dewey, A.M.; Kavesh, M.H.; Green, R.M. A chronic high-cholesterol diet paradoxically suppresses hepatic CYP7A1 expression in FVB/NJ mice. J. Lipid Res. 2011, 52, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.D.; Wang, F.; Kan, M.; Jin, C.L.; Jones, R.B.; Weinstein, M.; Deng, C.X.; McKeehan, W.L. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J. Biol. Chem. 2000, 275, 15482–15489. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Fujimori, T.; Furuya, A.; Satoh, J.; Nabeshima, Y.; Nabeshima, Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J. Clin. Investig. 2005, 115, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, K.; Maeda, R.; Urakawa, I.; Yamazaki, Y.; Tanaka, T.; Ito, S.; Nabeshima, Y.; Tomita, T.; Odori, S.; Hosoda, K.; et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.L.; Coulter, S.; Liddle, C.; Wong, A.; Eastham-Anderson, J.; French, D.M.; Peterson, A.S.; Sonoda, J. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4-dependent and independent pathways. PLoS ONE 2011, 6, e17868. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.J.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S., Jr. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metab. 2014, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Straka, M.S.; Dahl, R.; Devereaux, M.W.; Yerushalmi, B.; Gumpricht, E.; Elkins, N.; Everson, G. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr. Res. 2001, 49, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kaminaga, T.; Yasuda, H.; Kamiya, T.; Hara, H. The involvement of endoplasmic reticulum stress in bile acid-induced hepatocellular injury. J. Clin. Biochem. Nutr. 2014, 54, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Aranha, M.M.; Cortez-Pinto, H.; Costa, A.; da Silva, I.B.; Camilo, M.E.; de Moura, M.C.; Rodrigues, C.M. Bile acid levels are increased in the liver of patients with steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2008, 20, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Janus, D.; Dolezal-Oltarzewska, K.; Kalicka-Kasperczyk, A.; Poplawska, K.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. J. Pediatr. Endocrinol. Metab. 2012, 25, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Ceccarelli, S.; Panera, N.; Prono, F.; Petrini, S.; De Stefanis, C.; Pezzullo, M.; Tozzi, A.; Villani, A.; Bedogni, G.; et al. Association between Serum Atypical Fibroblast Growth Factors 21 and 19 and Pediatric Nonalcoholic Fatty Liver Disease. PLoS ONE 2013, 8, e67160. [Google Scholar] [CrossRef] [PubMed]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.-P.; et al. A Mouse Model of Hepatocellular Carcinoma. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Pai, R.; Dunlap, D.; Qing, J.; Mohtashemi, I.; Hotzel, K.; French, D.M. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating beta-catenin signaling. Cancer Res. 2008, 68, 5086–5095. [Google Scholar] [CrossRef] [PubMed]
- French, D.M.; Lin, B.C.; Wang, M.; Adams, C.; Shek, T.; Hotzel, K.; Bolon, B.; Ferrando, R.; Blackmore, C.; Schroeder, K.; et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE 2012, 7, e36713. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, X.; Phung, V.; Lindhout, D.A.; Mondal, K.; Hsu, J.Y.; Yang, H.; Humphrey, M.; Ding, X.; Arora, T.; et al. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19. Cancer Res. 2014, 74, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ko, B.; Elliott, M.; Zhou, M.; Lindhout, D.A.; Phung, V.; To, C.; Learned, R.M.; Tian, H.; DePaoli, A.M.; et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci. Transl. Med. 2014, 6, 247ra100. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Polak, J.M.; Sullivan, S.N.; Bloom, S.R.; Buchan, A.M.; Facer, P.; Brown, M.R.; Pearse, A.G. Specific localisation of neurotensin to the N cell in human intestine by radioimmunoassay and immunocytochemistry. Nature 1977, 270, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.P.; Mazella, J.; Kitabgi, P. Neurotensin and neurotensin receptors. Trends Pharmacol. Sci. 1999, 20, 302–309. [Google Scholar] [CrossRef]
- Kalafatakis, K.; Triantafyllou, K. Contribution of neurotensin in the immune and neuroendocrine modulation of normal and abnormal enteric function. Regul. Pept. 2011, 170, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.F.; Hammer, R.A.; Leeman, S.E. Elevation of plasma neurotensin during lipid perfusion of rat small intestine. Peptides 1981, 2 (Suppl. 2), 263–266. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Parker, M.C.; Ferris, C.F.; Leeman, S.E. Neurotensin stimulates [3H]oleic acid translocation across rat small intestine. Am. J. Physiol. 1986, 251, G823–G829. [Google Scholar] [CrossRef] [PubMed]
- Hammer, R.A.; Matsumoto, B.K.; Blei, A.T.; Pearl, G.; Ingram, H. Local Effect of Neurotensin on Canine Ileal Blood-Flow, and Its Release by Luminal Lipid. Scand. J. Gastroenterol. 1988, 23, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Hoang, H.D.; Bussjaeger, L.J.; Solomon, T.E. Effect of Neurotensin on Pancreatic and Gastric-Secretion and Growth in Rats. Pancreas 1988, 3, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bugescu, R.; Mayer, T.A.; Gata-Garcia, A.; Kurt, G.; Woodworth, H.L.; Leinninger, G.M. Loss of Action via Neurotensin-Leptin Receptor Neurons Disrupts Leptin and Ghrelin-Mediated Control of Energy Balance. Endocrinology 2017, 158, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Melander, O.; Maisel, A.S.; Almgren, P.; Manjer, J.; Belting, M.; Hedblad, B.; Engstrom, G.; Kilger, U.; Nilsson, P.; Bergmann, A.; et al. Plasma proneurotensin and incidence of diabetes, cardiovascular disease, breast cancer, and mortality. JAMA 2012, 308, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, J.; Zaytseva, Y.Y.; Liu, Y.; Rychahou, P.; Jiang, K.; Starr, M.E.; Kim, J.T.; Harris, J.W.; Yiannikouris, F.B.; et al. An obligatory role for neurotensin in high-fat-diet-induced obesity. Nature 2016, 533, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Leonetti, F.; Capoccia, D.; Di Cristofano, C.; Silecchia, G.; Orho-Melander, M.; Melander, O.; Cavallo, M.G. Increased Plasma Proneurotensin Levels Identify NAFLD in Adults with and Without Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 2253–2260. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Capoccia, D.; Bertoccini, L.; Ceccarelli, V.; Chiappetta, C.; Leonetti, F.; Di Cristofano, C.; Silecchia, G.; Orho-Melander, M.; et al. Neurotensin Is a Lipid-Induced Gastrointestinal Peptide Associated with Visceral Adipose Tissue Inflammation in Obesity. Nutrients 2018, 10, 526. [Google Scholar] [CrossRef] [PubMed]
- Auguet, T.; Aragones, G.; Berlanga, A.; Martinez, S.; Sabench, F.; Aguilar, C.; Villar, B.; Sirvent, J.J.; Del Castillo, D.; Richart, C. Low Circulating Levels of Neurotensin in Women with Nonalcoholic Fatty Liver Disease Associated with Severe Obesity. Obesity 2018, 26, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Hellmich, S.; Bergmann, A. Proneurotensin 1-117, a stable neurotensin precursor fragment identified in human circulation. Peptides 2006, 27, 1787–1793. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| References | Study Design | Study Subjects | Therapy and Follow-Up Duration | Outcomes |
|---|---|---|---|---|
| Cuthbertson et al. 2012 [46] | SA | 25 (T2DM); NAFLD | Exenatide 20 μg (n = 19) or Liraglutide 1.2 mg (n = 6); 6 months | ↓ALT; ↓Liver fat (1H MRS) |
| Armstrong et al. 2016 [47] | DB, RAND, PLAC | 23; NASH (biopsy proven) | Liraglutide 1.8 mg vs. placebo; 12 months | Histology (disappearance of ballooning without worsening of fibrosis) improved |
| Eguchi et al. 2015 [48] | SA | 19 (T2DM); NASH (biopsy proven) | Liraglutide 0.9 mg; 6 months | ↓AST, ALT; ↓Liver fat (CT) |
| Cui et al. 2016 [52] | DB, RAND, PLAC | 24 (prediabetes or early diabetes); NAFLD | Sitagliptin 100 mg; 6 months | Liver fat (MRI) not improved |
| Joy et al. 2017 [53] | DB, RAND, PLAC | 6 (T2DM); NASH (biopsy proven) | Sitagliptin 100 mg; 6 months | Histology (Fibrosis and NAS) not improved |
| References | Groups | Findings |
|---|---|---|
| Barchetta et al. 2018 [130] | 28 Obesity without NAFLD; 32 Obesity with NAFLD | Obesity with NAFLD vs. Obesity without NAFLD, ↑Plasma pro-NT; Plasma pro-NT correlated positively with NAFLD, presence and severity |
| Barchetta et al. 2018 [131] | 40 MO | Plasma pro-NT correlated positively with NAFLD presence and severity, and VAT inflammation. |
| Auguet et al. 2018 [132] | 20 Normal weight; 18 MO without NAFLD; 33 MO with NAFLD | MO with NAFLD vs. Normal weight, ↓Plasma NT; MO with NAFLD vs. MO without NAFLD, ↓Plasma NT; No difference in plasma NT between SS and NASH. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okubo, H.; Kushiyama, A.; Nakatsu, Y.; Yamamotoya, T.; Matsunaga, Y.; Fujishiro, M.; Sakoda, H.; Ohno, H.; Yoneda, M.; Asano, T. Roles of Gut-Derived Secretory Factors in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Their Possible Clinical Applications. Int. J. Mol. Sci. 2018, 19, 3064. https://doi.org/10.3390/ijms19103064
Okubo H, Kushiyama A, Nakatsu Y, Yamamotoya T, Matsunaga Y, Fujishiro M, Sakoda H, Ohno H, Yoneda M, Asano T. Roles of Gut-Derived Secretory Factors in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Their Possible Clinical Applications. International Journal of Molecular Sciences. 2018; 19(10):3064. https://doi.org/10.3390/ijms19103064
Chicago/Turabian StyleOkubo, Hirofumi, Akifumi Kushiyama, Yusuke Nakatsu, Takeshi Yamamotoya, Yasuka Matsunaga, Midori Fujishiro, Hideyuki Sakoda, Haruya Ohno, Masayasu Yoneda, and Tomoichiro Asano. 2018. "Roles of Gut-Derived Secretory Factors in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Their Possible Clinical Applications" International Journal of Molecular Sciences 19, no. 10: 3064. https://doi.org/10.3390/ijms19103064
APA StyleOkubo, H., Kushiyama, A., Nakatsu, Y., Yamamotoya, T., Matsunaga, Y., Fujishiro, M., Sakoda, H., Ohno, H., Yoneda, M., & Asano, T. (2018). Roles of Gut-Derived Secretory Factors in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Their Possible Clinical Applications. International Journal of Molecular Sciences, 19(10), 3064. https://doi.org/10.3390/ijms19103064