Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver


Abstract

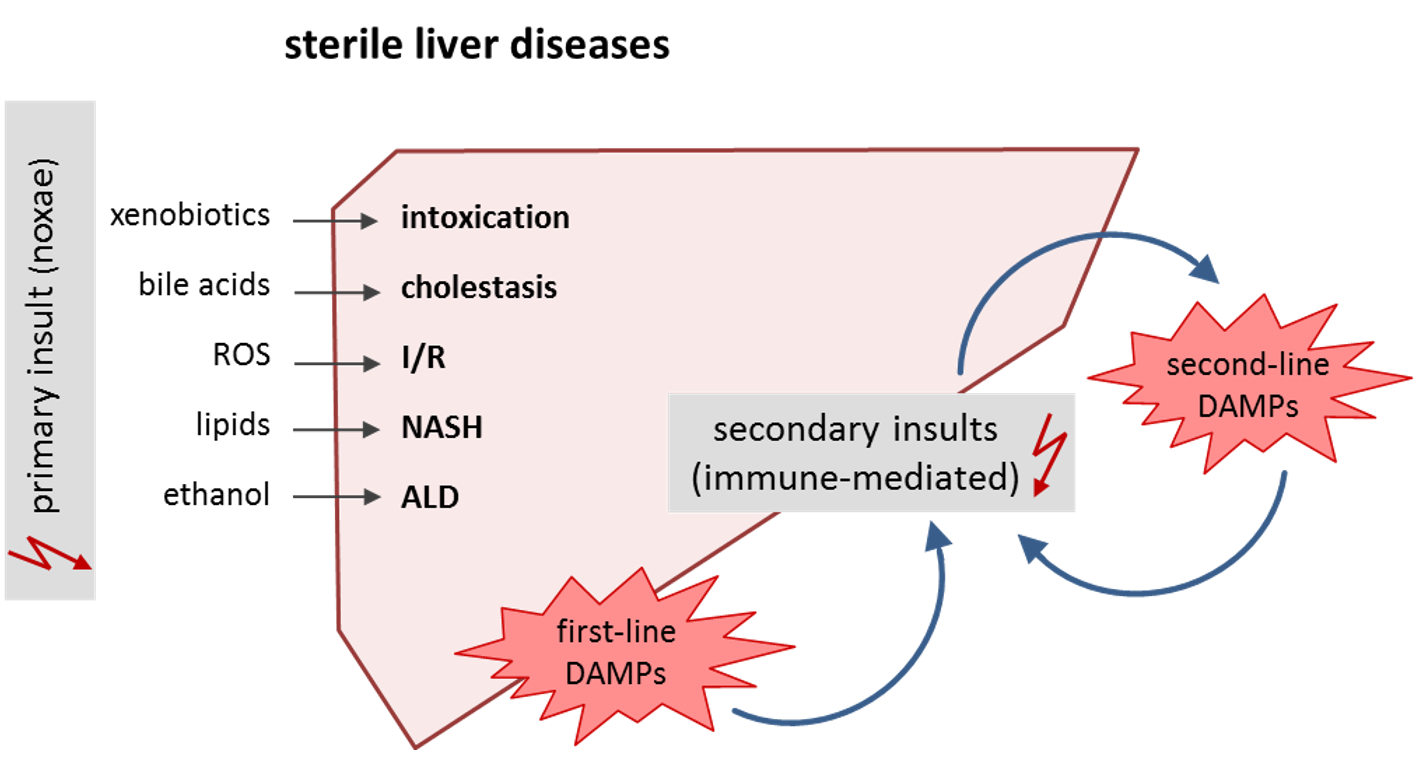
1. Introduction: Danger Self-Patterns in Sterile Liver Disease
2. High Mobility Group Box-1 (HMGB1)
2.1. HMGB1 in APAP Intoxication: A Passive Release from Dying Hepatocytes Followed by an Active Release from KCs
2.2. HMGB1 in Cholestatic Liver Injury
2.3. HMGB1 in I/R Injury: Early Active Secretion by Hepatocytes
3. Keratin 18 (K18)
3.1. NASH: K18 Fragments as an Indicator for Apoptotic Cell Death
3.2. ALD: Roles for Cleaved and Native K18 as Diagnostic and Prognostic Biomarkers
4. Adenosine Triphosphate (ATP)
4.1. NASH: Confirmatory Evidence for Apoptotic Hepatocellular Death along with Necrotic Cell Death
4.2. ALD: DAMPs and PAMPs
5. Mitochondrial DAMPs
5.1. MtDAMPs in APAP Intoxication: A Further Piece of Evidence for a Primarily Necrotic Cell Death and a Role for IFNs
5.2. MtDAMPs in NASH: Evidence for Necrotic Cell Death Too
5.3. MtDAMPs in I/R Injury: Contribution of IFNs
6. Conclusions
Funding
Acknowledgments
Abbreviations
| acHMGB1 | acetylated high mobility group box-1 |
| ALT | Alanine aminotransferase |
| ALD | Alcoholic liver disease |
| APAP | para-Acetaminophenol |
| ATP | Adenosine triphosphate |
| cGAS | cyclic GMP-AMP synthase |
| CXCL | CXC motif chemokine |
| DAMP | Danger-associated molecular pattern |
| DC | Dendritic cell |
| FFA | Free fatty acid |
| HMGB1 | High mobility group box-1 |
| K18 | Keratin-18 |
| IFN | Interferon |
| IFNAR | IFN-α/β receptor |
| IFP35 | IFN-induced protein 35 |
| IL | interleukin |
| I/R | Ischemia/reperfusion |
| IRF | Interferon regulatory factor |
| ISG | IFN-stimulated gene |
| KC | Kupffer cell |
| LPS | Lipopolysaccharide |
| mtDNA | mitochondrial DNA |
| NASH | Non-alcoholic liver disease |
| NFP | N-formyl peptides |
| NLR | NOD (nucleotide-binding oligomerization domain)-like receptor |
| NLRP3 | pyrin domain containing-3 protein |
| NMI | N-myc and STAT interactor |
| NOD | Nucleotide-binding oligomerization domain |
| P1R | Purinergic receptor, nucleoside binding |
| P2R | Purinergic receptor, nucleotide binding |
| PAMP | Pathogen-associated molecular pattern |
| PRR | Pattern recognition receptor |
| RLR | RIG-I (retinoic acid-inducible gene-I)-like receptor |
| ROS | Reactive oxygen species |
| RAGE | Receptor for advanced glycation endproduct |
| RIG-1 | Retinoic acid-inducible gene-1 |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of interferon genes |
| TLR | Toll-like receptor |
References
- Eguchi, A.; Wree, A.; Feldstein, A.E. Biomarkers of liver cell death. J. Hepatol. 2014, 60, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Dolasia, K.; Bisht, M.K.; Pradhan, G.; Udgata, A.; Mukhopadhyay, S. TLRs/NLRs: Shaping the landscape of host immunity. Int. Rev. Immunol. 2018, 37, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Almeda-Valdes, P.; Aguilar Olivos, N.E.; Barranco-Fragoso, B.; Uribe, M.; Mendez-Sanchez, N. The Role of Dendritic Cells in Fibrosis Progression in Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2015, 2015, 768071. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Sterile inflammation in acute liver injury: Myth or mystery? Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Nam, C.M.; Jee, S.H.; Han, K.H.; Oh, D.K.; Suh, I. Normal serum aminotransferase concentration and risk of mortality from liver diseases: Prospective cohort study. BMJ 2004, 328, 983. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, C.E.; Everhart, J.E. Elevated serum alanine aminotransferase and gamma-glutamyltransferase and mortality in the United States population. Gastroenterology 2009, 136, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Kim, W.R.; Benson, J.T.; Therneau, T.M.; Melton, L.J., 3rd. Serum aminotransferase activity and mortality risk in a United States community. Hepatology 2008, 47, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Apekey, T.A.; Seddoh, D.; Walley, J. Liver enzymes and risk of all-cause mortality in general populations: A systematic review and meta-analysis. Int. J. Epidemiol. 2014, 43, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundback, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. HMGB1 loves company. J. Leukoc. Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Chavan, S.S.; Andersson, U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol. Med. 2015, 21, S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Tirone, M.; Tran, N.L.; Ceriotti, C.; Gorzanelli, A.; Canepari, M.; Bottinelli, R.; Raucci, A.; Di Maggio, S.; Santiago, C.; Mellado, M.; et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J. Exp. Med. 2018, 215, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ju, Z.; Ragab, A.A.; Lundback, P.; Long, W.; Valdes-Ferrer, S.I.; He, M.; Pribis, J.P.; Li, J.; et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 2015, 212, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Billiar, T.R.; Lotze, M.T. A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol. Med. 2012, 18, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
- Ostapowicz, G.; Fontana, R.J.; Schiodt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar] [CrossRef]
- Antoine, D.J.; Jenkins, R.E.; Dear, J.W.; Williams, D.P.; McGill, M.R.; Sharpe, M.R.; Craig, D.G.; Simpson, K.J.; Jaeschke, H.; Park, B.K. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol. 2012, 56, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Antoine, D.J.; Dear, J.W.; Lewis, P.S.; Platt, V.; Coyle, J.; Masson, M.; Thanacoody, R.H.; Gray, A.J.; Webb, D.J.; Moggs, J.G.; et al. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology 2013, 58, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Dear, J.W.; Clarke, J.I.; Francis, B.; Allen, L.; Wraight, J.; Shen, J.; Dargan, P.I.; Wood, D.; Cooper, J.; Thomas, S.H.L.; et al. Risk stratification after paracetamol overdose using mechanistic biomarkers: Results from two prospective cohort studies. Lancet Gastroenterol. Hepatol. 2018, 3, 104–113. [Google Scholar] [CrossRef]
- Huebener, P.; Hernandez, C.; Schwabe, R.F. HMGB1 and injury amplification. Oncotarget 2015, 6, 23048–23049. [Google Scholar] [CrossRef] [PubMed]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Molteni, R.; Suda, T.; Samaniego, S.; Raucci, A.; Habiel, D.M.; Miller, F.; Jiang, H.P.; Li, J.; Pardi, R.; et al. Inhibitor of NF-kappa B kinases alpha and beta are both essential for high mobility group box 1-mediated chemotaxis [corrected]. J. Immunol. 2010, 184, 4497–4509. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Shen, Z.; Song, F. Autophagy and acetaminophen-induced hepatotoxicity. Arch. Toxicol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Therapeutic targets for cholestatic liver injury. Expert Opin. Ther. Targets 2016, 20, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.L.; Ghallab, A.; Vartak, N.; Reif, R.; Schaap, F.G.; Hampe, J.; Hengstler, J.G. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017, 65, 722–738. [Google Scholar] [CrossRef] [PubMed]
- Ghallab, A.; Hofmann, U.; Sezgin, S.; Vartak, N.; Hassan, R.; Zaza, A.; Godoy, P.; Schneider, K.M.; Guenther, G.; Ahmed, Y.A.; et al. Bile micro-infarcts in cholestasis are initiated by rupture of the apical hepatocyte membrane and cause shunting of bile to sinusoidal blood. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Dorko, K.; Antoine, D.J.; Clarke, J.I.; Gholami, P.; Li, F.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Fan, F.; et al. Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol. Appl. Pharmacol. 2015, 283, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Antoine, D.J.; Jenkins, R.E.; Bajt, M.L.; Park, B.K.; Jaeschke, H. Plasma biomarkers of liver injury and inflammation demonstrate a lack of apoptosis during obstructive cholestasis in mice. Toxicol. Appl. Pharmacol. 2013, 273, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cai, S.Y.; Boyer, J.L. Mechanisms of bile acid mediated inflammation in the liver. Mol. Asp. Med. 2017, 56, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Calmus, Y.; Poupon, R. Shaping macrophages function and innate immunity by bile acids: Mechanisms and implication in cholestatic liver diseases. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Haselow, K.; Bode, J.G.; Wammers, M.; Ehlting, C.; Keitel, V.; Kleinebrecht, L.; Schupp, A.K.; Haussinger, D.; Graf, D. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J. Leukoc. Biol. 2013, 94, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Hall, C.; Glaser, S.; Francis, H.; Meng, F.; Alpini, G. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am. J. Pathol. 2016, 186, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Evankovich, J.; Cho, S.W.; Zhang, R.; Cardinal, J.; Dhupar, R.; Zhang, L.; Klune, J.R.; Zlotnicki, J.; Billiar, T.; Tsung, A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J. Biol. Chem. 2010, 285, 39888–39897. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Nace, G.W.; McDonald, K.A.; Tai, S.; Klune, J.R.; Rosborough, B.R.; Ding, Q.; Loughran, P.; Zhu, X.; Beer-Stolz, D.; et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: A role for intracellular high-mobility group box 1 in cellular protection. Hepatology 2014, 59, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Ku, N.O.; Strnad, P.; Hanada, S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Investig. 2009, 119, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Merrison, W.; Dinsdale, D.; Cohen, G.M. Active caspases and cleaved cytokeratins are sequestered into cytoplasmic inclusions in TRAIL-induced apoptosis. J. Cell Biol. 2000, 148, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Weerasinghe, S.V.; Ku, N.O.; Altshuler, P.J.; Kwan, R.; Omary, M.B. Mutation of caspase-digestion sites in keratin 18 interferes with filament reorganization, and predisposes to hepatocyte necrosis and loss of membrane integrity. J. Cell Sci. 2014, 127, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.O.; Strnad, P.; Bantel, H.; Omary, M.B. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 2016, 64, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Mofrad, P.; Contos, M.J.; Haque, M.; Sargeant, C.; Fisher, R.A.; Luketic, V.A.; Sterling, R.K.; Shiffman, M.L.; Stravitz, R.T.; Sanyal, A.J. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003, 37, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Is Keratin-18 only a Marker of Cell Death in Acute-On-Chronic Liver Failure? J. Lab. Precis. Med. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.; Andreola, F.; Bachtiger, P.; Amoros, A.; Pavesi, M.; Mookerjee, R.; Zheng, Y.B.; Gronbaek, H.; Gerbes, A.L.; Sola, E.; et al. Cell death markers in patients with cirrhosis and acute decompensation. Hepatology 2018, 67, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Bridges, B.W.; Dunn, W.; Olson, J.C.; Weinman, S.A.; Jaeschke, H. Cell Death and Prognosis of Mortality in Alcoholic Hepatitis Patients Using Plasma Keratin-18. Gene Expr. 2017, 17, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, J.; Altamirano, J.; Devue, C.; Roux, O.; Payance, A.; Lebrec, D.; Bedossa, P.; Valla, D.; Durand, F.; Ait-Oufella, H.; et al. A prospective study of the utility of plasma biomarkers to diagnose alcoholic hepatitis. Hepatology 2017, 66, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Katsnelson, M.A.; Dubyak, G.R.; Pearlman, E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat. Commun. 2016, 7, 10555. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Loos, F.; Liu, P.; Kroemer, G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol. Rev. 2017, 280, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Waldrop, S.L.; Khimji, A.K.; Kilic, G. Pannexin1 contributes to pathophysiological ATP release in lipoapoptosis induced by saturated free fatty acids in liver cells. Am. J. Physiol. Cell Physiol. 2012, 303, C1034–C1044. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Waldrop, S.L.; Bronk, S.F.; Gores, G.J.; Davis, L.S.; Kilic, G. Lipoapoptosis induced by saturated free fatty acids stimulates monocyte migration: A novel role for Pannexin1 in liver cells. Purinergic Signal 2015, 11, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernandez, A.; De Lacy, A.M.; Fernandez-Checa, J.C.; Caballeria, J.; Garcia-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, L.; Hu, L.; Liu, Y.; Sun, H.Y.; Tang, J.; Hou, Y.J.; Chang, Y.X.; Tu, Q.Q.; Feng, G.S.; et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011, 54, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Parlesak, A.; Schafer, C.; Schutz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Panaro, M.A.; Acquafredda, A.; Sisto, M.; Lisi, S.; Maffione, A.B.; Mitolo, V. Biological role of the N-formyl peptide receptors. Immunopharmacol. Immunotoxicol. 2006, 28, 103–127. [Google Scholar] [CrossRef] [PubMed]
- Gabl, M.; Sundqvist, M.; Holdfeldt, A.; Lind, S.; Martensson, J.; Christenson, K.; Marutani, T.; Dahlgren, C.; Mukai, H.; Forsman, H. Mitocryptides from Human Mitochondrial DNA-Encoded Proteins Activate Neutrophil Formyl Peptide Receptors: Receptor Preference and Signaling Properties. J. Immunol. 2018, 200, 3269–3282. [Google Scholar] [CrossRef] [PubMed]
- Heit, B.; Robbins, S.M.; Downey, C.M.; Guan, Z.; Colarusso, P.; Miller, B.J.; Jirik, F.R.; Kubes, P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat. Immunol. 2008, 9, 743–752. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Grunvogel, O.; Esser-Nobis, K.; Windisch, M.P.; Frese, M.; Trippler, M.; Bartenschlager, R.; Lohmann, V.; Binder, M. Type I and type II interferon responses in two human liver cell lines (Huh-7 and HuH6). Genom. Data 2016, 7, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, B.; Yu, Y.; Iyer, S.S.; Sun, M.; Cheng, G. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. 2015, 16, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Xiahou, Z.; Wang, X.; Shen, J.; Zhu, X.; Xu, F.; Hu, R.; Guo, D.; Li, H.; Tian, Y.; Liu, Y.; et al. NMI and IFP35 serve as proinflammatory DAMPs during cellular infection and injury. Nat. Commun. 2017, 8, 950. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Qiao, B.; Gao, F.; Shen, X.; Vardanian, A.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology 2008, 47, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Castellaneta, A.; Yoshida, O.; Kimura, S.; Yokota, S.; Geller, D.A.; Murase, N.; Thomson, A.W. Plasmacytoid dendritic cell-derived IFN-alpha promotes murine liver ischemia/reperfusion injury by induction of hepatocyte IRF-1. Hepatology 2014, 60, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.M.; Antunes, M.M.; Mattos, M.S.; Diniz, A.B.; Alvarenga, D.M.; Nakagaki, B.N.; Carvalho, E.; Lacerda, V.A.S.; Carvalho-Gontijo, R.; Goulart, J.; et al. Liver Immune Cells Release Type 1 Interferon Due to DNA Sensing and Amplify Liver Injury from Acetaminophen Overdose. Cells 2018, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Ueki, S.; Dhupar, R.; Cardinal, J.; Tsung, A.; Yoshida, J.; Ozaki, K.S.; Klune, J.R.; Murase, N.; Geller, D.A. Critical role of interferon regulatory factor-1 in murine liver transplant ischemia reperfusion injury. Hepatology 2010, 51, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yoshida, O.; Dou, L.; Spadaro, A.V.; Isse, K.; Ross, M.A.; Stolz, D.B.; Kimura, S.; Du, Q.; Demetris, A.J.; et al. IRF-1 promotes liver transplant ischemia/reperfusion injury via hepatocyte IL-15/IL-15Ralpha production. J. Immunol. 2015, 194, 6045–6056. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Q.; Du, Q.; Goswami, J.; Varley, P.R.; Chen, B.; Wang, R.H.; Morelli, A.E.; Stolz, D.B.; Billiar, T.R.; Li, J.; et al. Interferon regulatory factor 1-Rab27a regulated extracellular vesicles promote liver ischemia/reperfusion injury. Hepatology 2018, 67, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, S.; Wang, Z.; He, J.; Ding, Y.; Zhang, H.; Yu, W.; Shi, Y.; Cui, Z.; Wang, X.; et al. Interferon regulatory factor-1 activates autophagy to aggravate hepatic ischemia-reperfusion injury via the P38/P62 pathway in mice. Sci. Rep. 2017, 7, 43684. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Li, S.; Liu, Z.; Zhang, Y.; Zhang, H. Interferon Regulatory Factor 1 Activates Autophagy to Aggravate Hepatic Ischemia-Reperfusion Injury by Increasing High Mobility Group Box 1 Release. Cell. Physiol. Biochem. 2018, 48, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Klune, J.R.; Dhupar, R.; Kimura, S.; Ueki, S.; Cardinal, J.; Nakao, A.; Nace, G.; Evankovich, J.; Murase, N.; Tsung, A.; et al. Interferon regulatory factor-2 is protective against hepatic ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G666–G673. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.X.; Zhang, R.; Huang, L.; Zhu, L.H.; Jiang, D.S.; Chen, H.Z.; Zhang, Y.; Tian, S.; Zhang, X.F.; Zhang, X.D.; et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J. Hepatol. 2015, 62, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Avila, T.V.; Melgaco, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Deng, M.; Yi, Z.; Sun, Q.; Shapiro, R.A.; Xu, H.; Li, T.; Loughran, P.A.; Griepentrog, J.E.; Huang, H.; et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G655–G667. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R.; Staggs, V.S.; Sharpe, M.R.; Lee, W.M.; Jaeschke, H.; Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology 2014, 60, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Handa, P.; Vemulakonda, A.; Kowdley, K.V.; Uribe, M.; Mendez-Sanchez, N. Mitochondrial DNA from hepatocytes as a ligand for TLR9: Drivers of nonalcoholic steatohepatitis? World J. Gastroenterol. 2016, 22, 6965–6971. [Google Scholar] [CrossRef] [PubMed]
- Inzaugarat, M.E.; Wree, A.; Feldstein, A.E. Hepatocyte mitochondrial DNA released in microparticles and toll-like receptor 9 activation: A link between lipotoxicity and inflammation during nonalcoholic steatohepatitis. Hepatology 2016, 64, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Kano, A.; Haruyama, T.; Akaike, T.; Watanabe, Y. IRF-1 is an essential mediator in IFN-gamma-induced cell cycle arrest and apoptosis of primary cultured hepatocytes. Biochem. Biophys. Res. Commun. 1999, 257, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, D.; Tamura, T.; Yanai, H.; Taniguchi, T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 2010, 59, 489–510. [Google Scholar] [CrossRef] [PubMed]
- Dhupar, R.; Klune, J.R.; Evankovich, J.; Cardinal, J.; Zhang, M.; Ross, M.; Murase, N.; Geller, D.A.; Billiar, T.R.; Tsung, A. Interferon regulatory factor 1 mediates acetylation and release of high mobility group box 1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock 2011, 35, 293–301. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| General Role | ISG | Specific Role in Sterile Liver Disease | Ref | |
|---|---|---|---|---|
| IFN signaling | JAK2 STAT1/2 | IFNAR-deficiency protects from liver injury | I/R | [99] |
| IFN-α promotes liver injury | I/R | [100] | ||
| IFNAR-deficiency delays experimental intoxication injury | APAP | [101] | ||
| Transcription factors | IRF-1 | hepatocellular graft IRF-1 promotes experimental liver transplant injury | I/R | [102] |
| promotes experimental liver injury by enhancing hepatocyte apoptosis | I/R | [100] | ||
| promotes injury via IL-15 | I/R | [103] | ||
| promotes injury via activation of PMNs | I/R | [104] | ||
| activates autophagic cell death, aggravates hepatic injury | I/R | [105,106] | ||
| mediates HMGB1 release | I/R | [105,106] | ||
| IRF-2 | IRF-1 antagonist, protective against liver injury | I/R | [107] | |
| IRF-3 | linking alcohol-induced ER stress with hepatocyte apoptosis | ALD | [80] | |
| IRF-9 | mediator of liver injury | I/R | [108] | |
| Sensitizing for DAMPs | TLR4 | involved in ROS-mediated HMGB1 release in liver injury | I/R | [48] |
| engages disulfide-HMGB1 thereby promoting liver injury | I/R | [24] | ||
| mediates KC type I IFN activation in experimental liver injury | I/R | [99] | ||
| facilitates HMGB1-driven paracrine cytolytic effect on cholesterol-loaded hepatocytes | NASH | [78,79] | ||
| senses NMI and IFP35 in experimental liver intoxication | APAP | [98] | ||
| TLR9 | senses mtDNA in experimental intoxication | APAP | [109] | |
| promotes experimental liver injury | NASH | [110] | ||
| cGAS | promotes exp. liver injury by sensing extracellular DNA | APAP | [101] | |
| deletion of cGAS aggravates experimental liver injury independent of STING | I/R | [111] | ||
| STING | promotes exp. liver injury by sensing extracellular DNA | APAP | [101] | |
| promotes IRF-3-mediated parenchymal apoptotic cell death in response to alcohol | ALD | [80] | ||
| Acting as DAMPs | NMI | released by activated macrophages acting on TLR4 in experimental intoxication | APAP | [98] |
| NMI deficiency reduces liver injury and mortality | APAP | [98] | ||
| IFP35 | released by activated macrophages acting on TLR4 in experimental intoxication | APAP | [98] | |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. https://doi.org/10.3390/ijms19103104
Mihm S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. International Journal of Molecular Sciences. 2018; 19(10):3104. https://doi.org/10.3390/ijms19103104
Chicago/Turabian StyleMihm, Sabine. 2018. "Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver" International Journal of Molecular Sciences 19, no. 10: 3104. https://doi.org/10.3390/ijms19103104
APA StyleMihm, S. (2018). Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. International Journal of Molecular Sciences, 19(10), 3104. https://doi.org/10.3390/ijms19103104