Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures

 ,
,  ,
,  and
and
Abstract
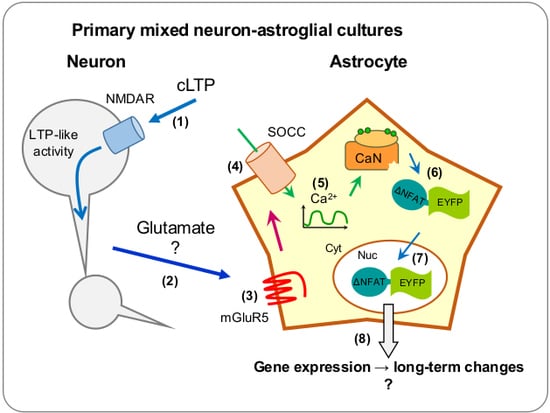
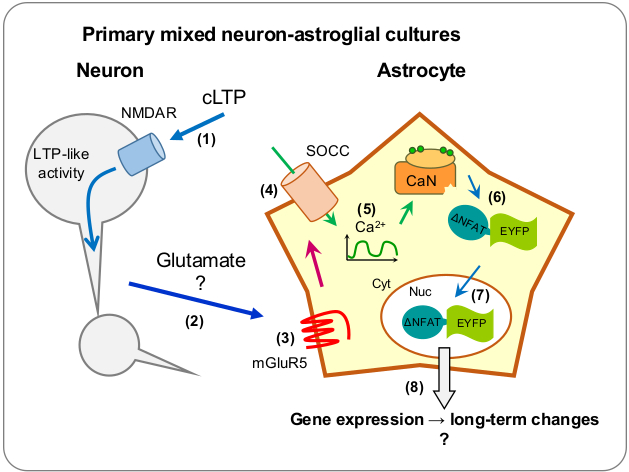
1. Introduction
2. Results
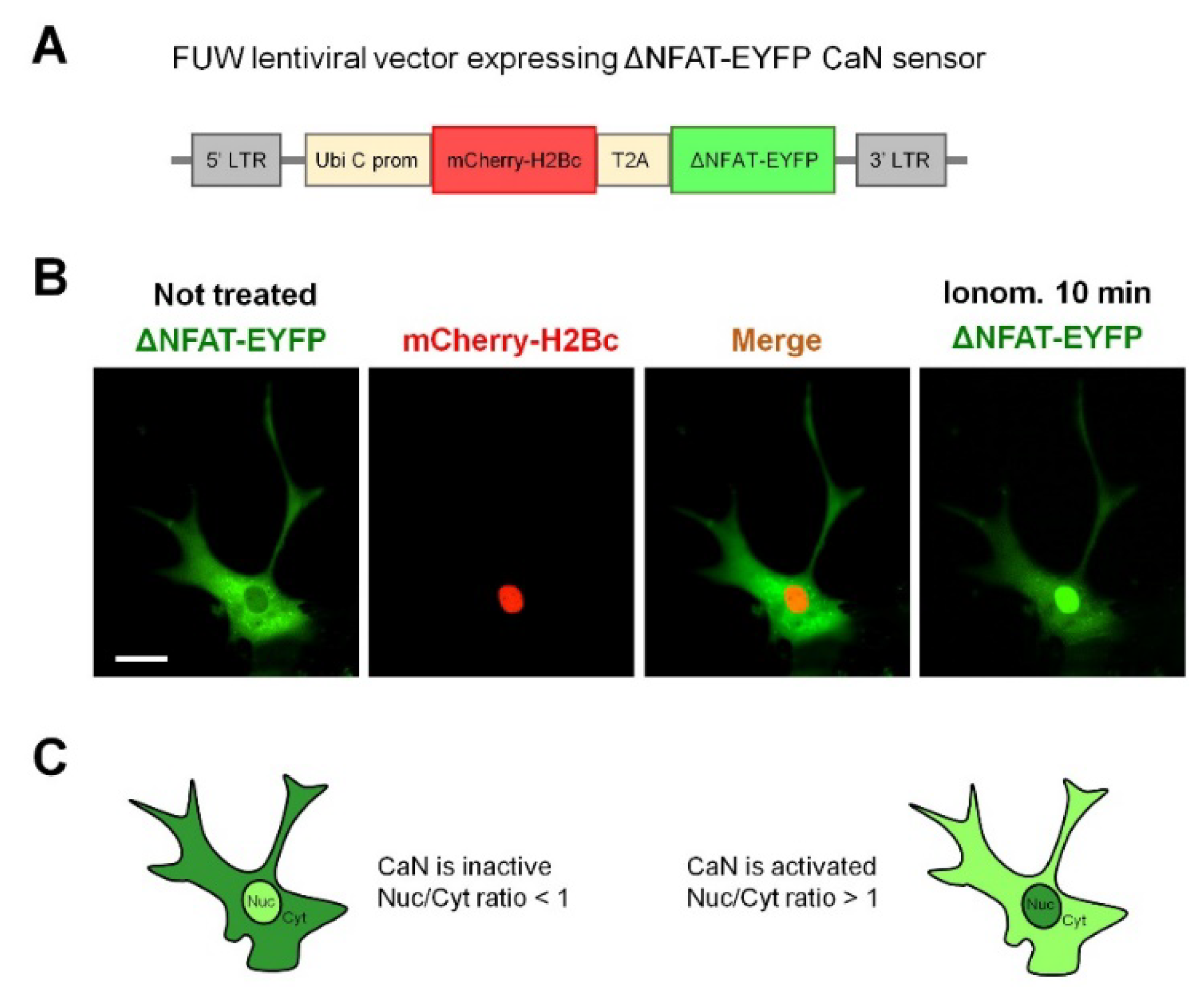
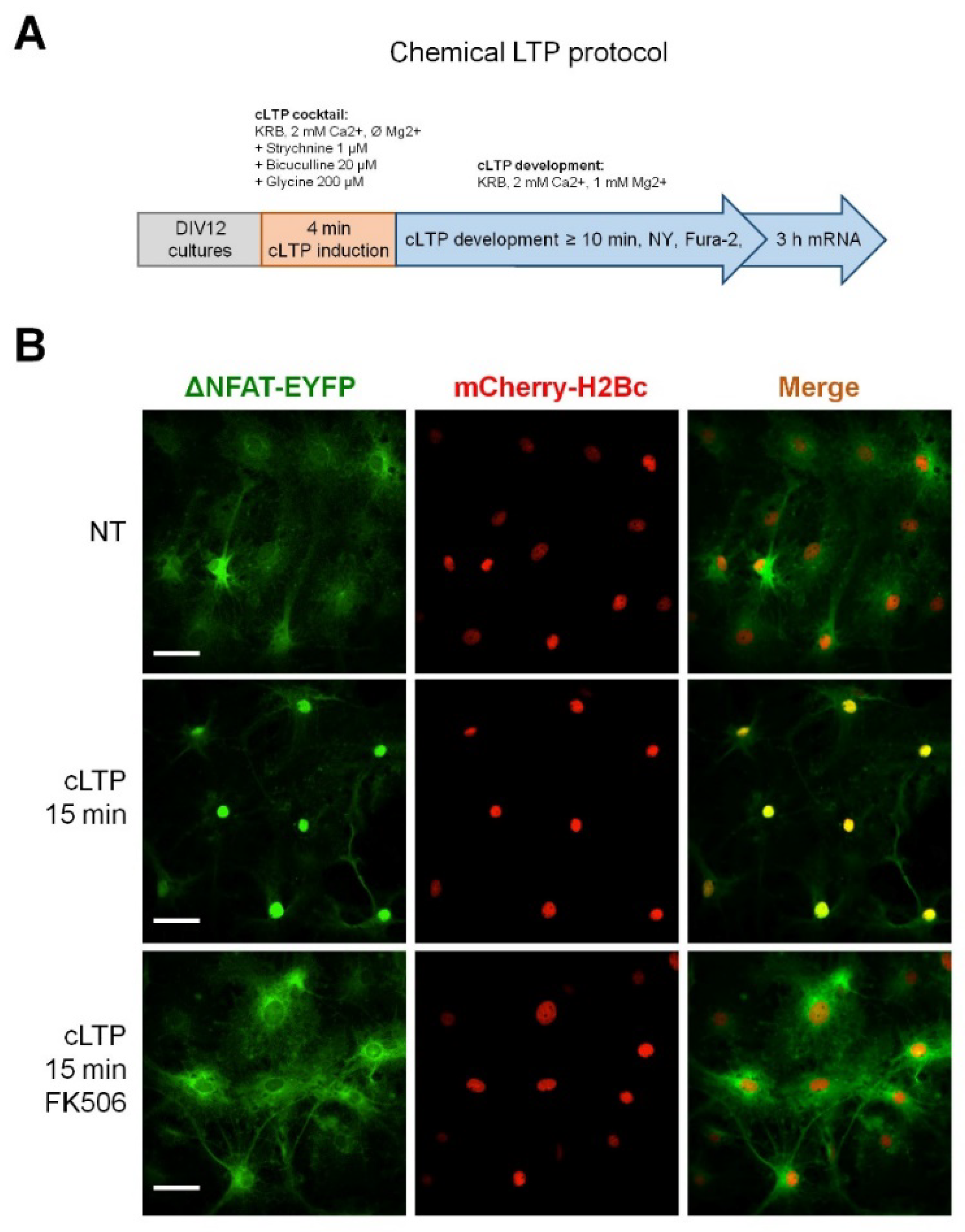
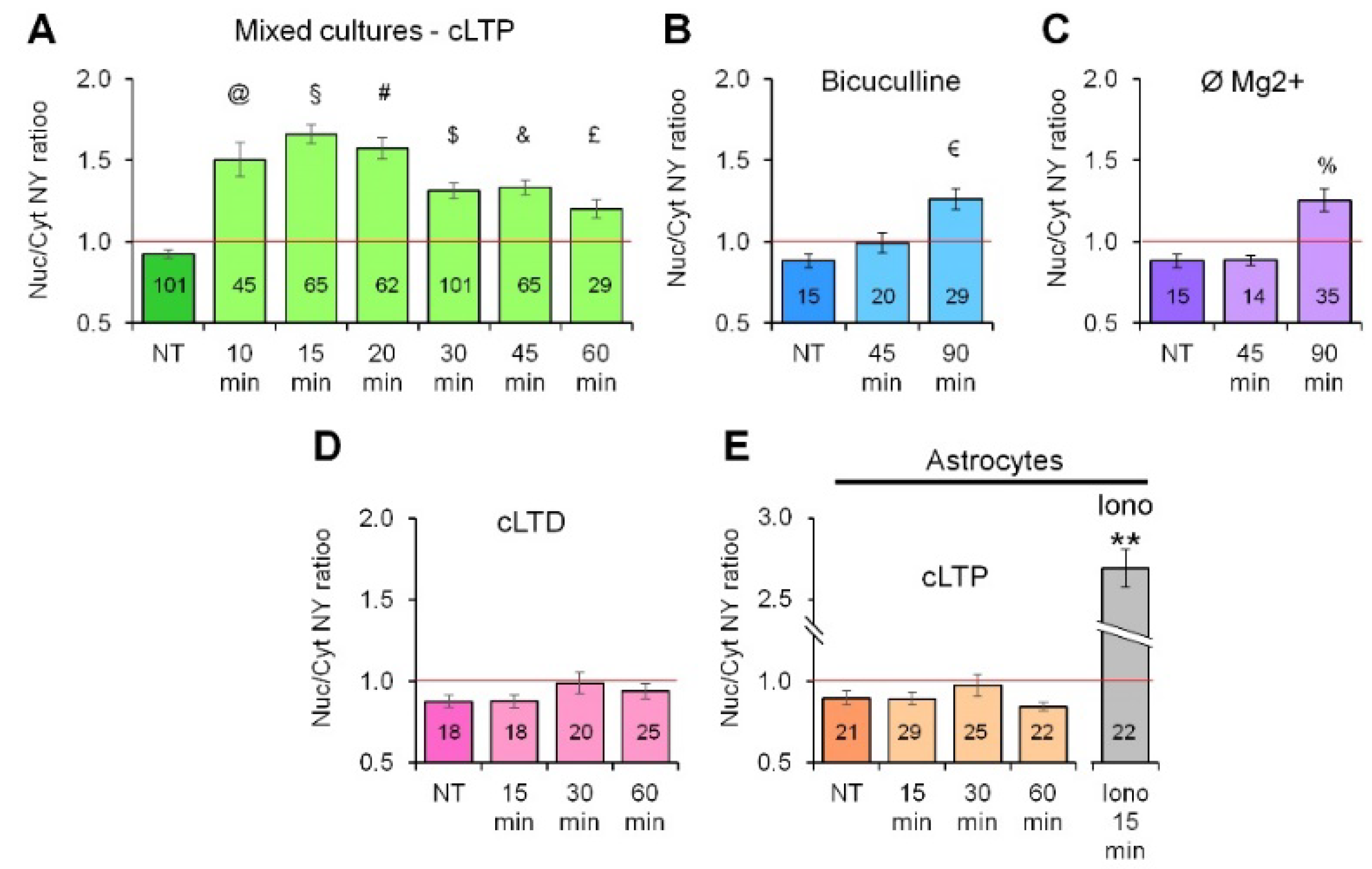
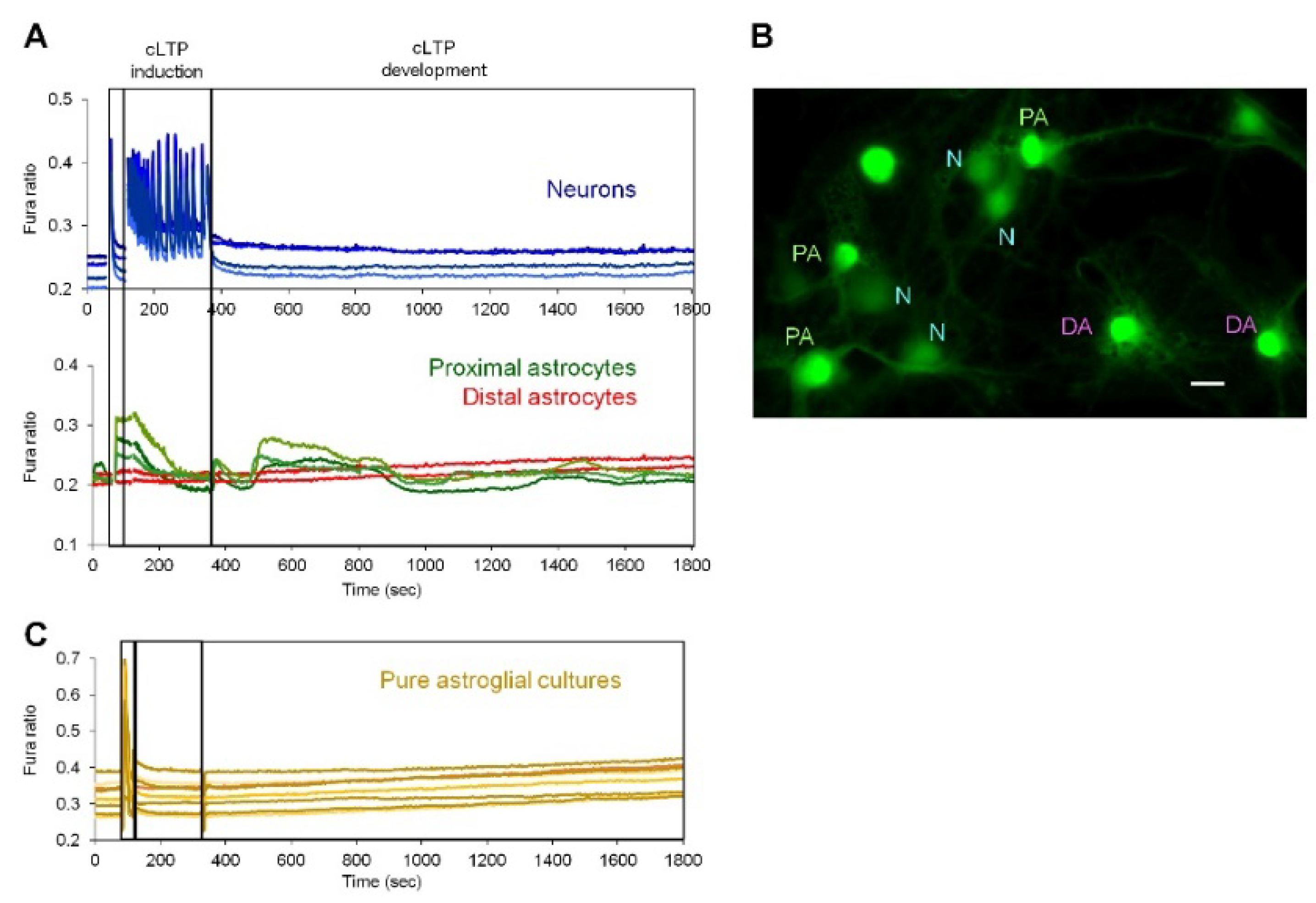
2.1. Neuronal Activity Leads to Activation of CaN in Astrocytes
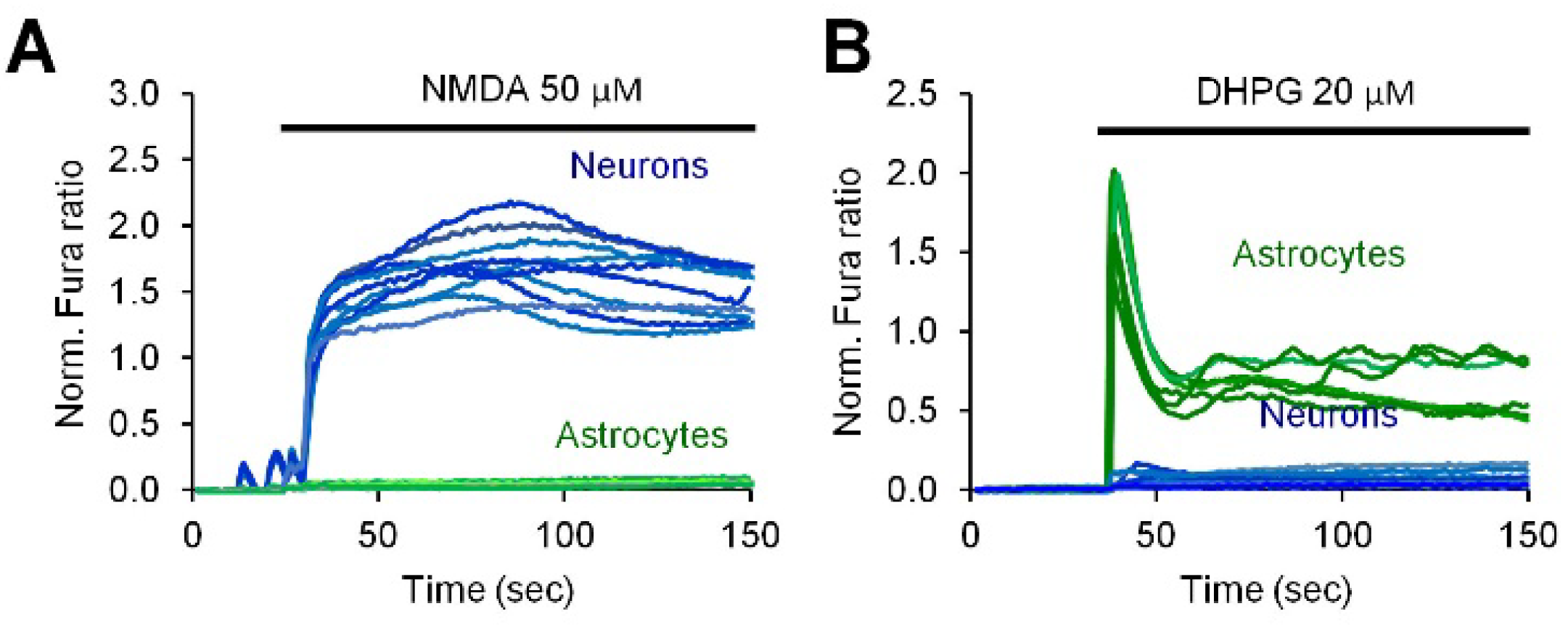
2.2. Neuronal Activity Induces Elevation of Cytosolic Calcium in Astrocytes
2.3. Activity-Induced Ca2+ Transients and CaN Activation in Astrocytes Depend on NMDA, mGluR5 and Store-Operated Ca2+ Entry
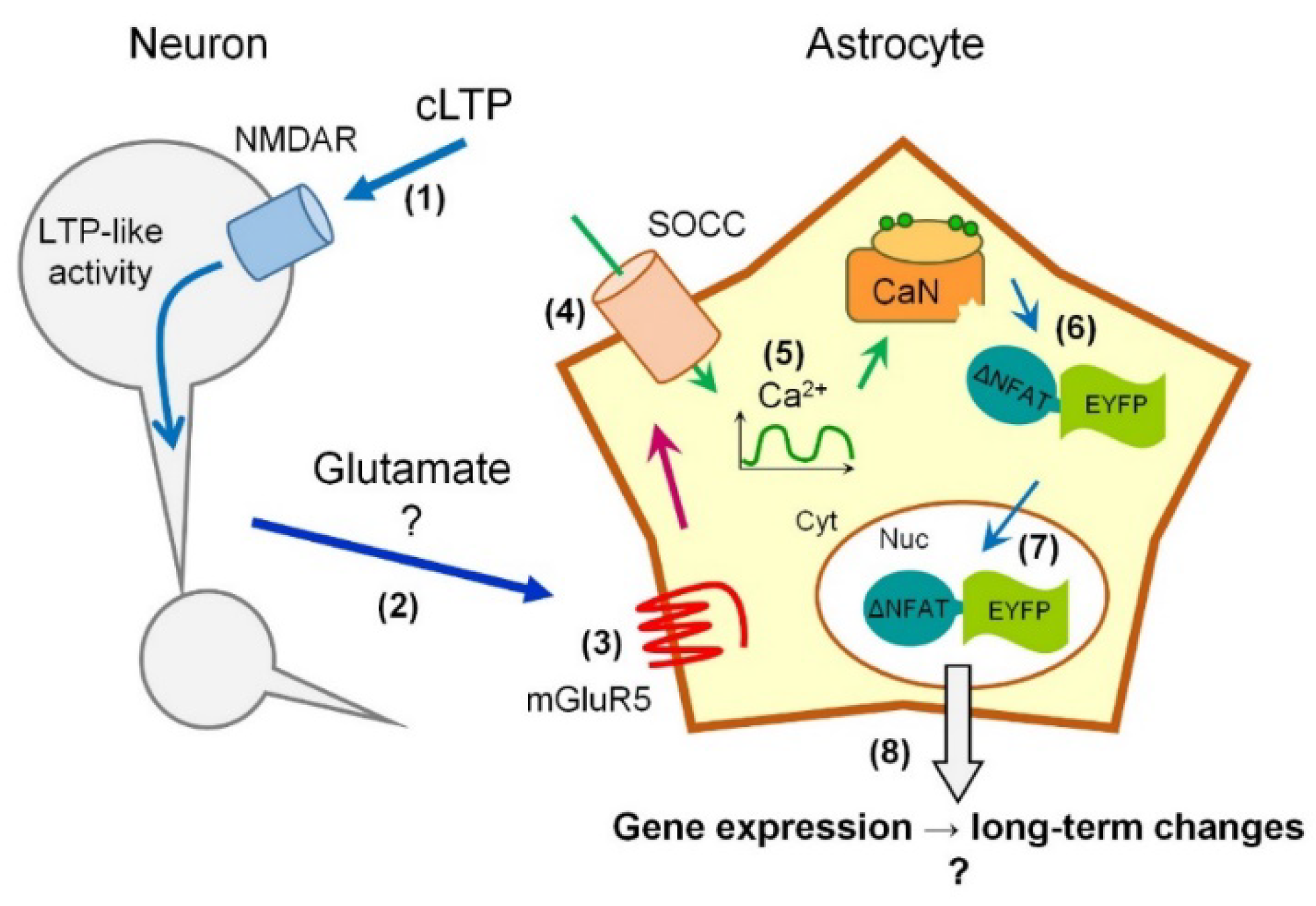
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Hippocampal Mixed and Astroglial Cultures
4.3. Lentivirus Production and Transduction
4.4. Induction of cLTP and NY Translocation Quantification
4.5. Induction of cLTP and NY Translocation Quantification
4.6. Pharmacological Reagents
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CaN | Calcineurin |
| cLTP | Chemical Long-Term Potentiation |
| cLTD | Chemical Long-Term Depression |
| KRB | Modified krebs-Ringer Buffer |
| MK801 | (5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine Maleate |
| MTEP | 3-((2-Methyl-1,3-thiazol-4-yl) ethynyl) Pyridine Hydrochloride |
| DHPG | (RS)-3,5-Dihydroxyphenylglycine |
| U73122 | 1-[6-[[(17β)-3-Methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione |
| NMDA | N-Methyl-d-aspartic Acid |
| TRPC3 | Transient Receptor Potential C3 |
| 2APB | 2-Aminoethoxydiphenylborane |
| Pyr3 | 1-[4-[(2,3,3-Trichloro-1-oxo-2-propen-1-yl)amino]phenyl]-5-(trifluoromethyl)-1H-pyrazole-4-carboxylic Acid |
| Pyr6 | N-[4-[3,5-Bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-3-fluoro-4-pyridinecarboxamide |
| Pyr10 | N-[4-[3,5-Bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-4-methyl-benzenesulfonamide |
| DMSO | Dimethyl Sulfoxide |
| SOCE | Store-Operated Calcium Entry |
References
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Di Castro, M.A.; Chuquet, J.; Liaudet, N.; Bhaukaurally, K.; Santello, M.; Bouvier, D.; Tiret, P.; Volterra, A. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 2011, 14, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, T.A.; McCarthy, K.D. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Grubišić, V.; Verkhratsky, A. Ca(2+) sources for the exocytotic release of glutamate from astrocytes. Biochim. Biophys. Acta 2011, 1813, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Petravicz, J.; McMullen, A.B.; Sweger, E.J.; Minton, S.K.; Taves, S.R.; Casper, K.B.; Fiacco, T.A.; McCarthy, K.D. What is the role of astrocyte calcium in neurophysiology? Neuron 2008, 59, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, T.A.; Agulhon, C.; McCarthy, K.D. Sorting out astrocyte physiology from pharmacology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Fiacco, T.A.; McCarthy, K.D. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science 2010, 327, 1250–1254. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Han, X.; Deane, R.; Zlokovic, B.; Nedergaard, M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2007, 1097, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Volterra, A. Astrocytic dysfunction: Insights on the role in neurodegeneration. Brain Res. Bull. 2009, 80, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.J.; Gasperini, R.; Foa, L.; Small, D.H. Astrocytes in Alzheimer’s disease: Emerging roles in calcium dysregulation and synaptic plasticity. J. Alzheimers Dis. 2010, 22, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Ronco, V.; Grolla, A.A.; Verkhratsky, A.; Genazzani, A.A. Glial calcium signalling in Alzheimer’s disease. Rev. Physiol. Biochem. Pharmacol. 2014, 167, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Zeidán-Chuliá, F.; Genazzani, A.A.; Verkhratsky, A. Calcium signalling toolkits in astrocytes and spatio-temporal progression of Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Verkhratsky, A.; Zorec, R. Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration. Int. J. Mol. Sci. 2017, 18, 358. [Google Scholar] [CrossRef] [PubMed]
- Klee, C.B.; Crouch, T.H.; Krinks, M.H. Calcineurin: A calcium- and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. USA 1979, 76, 6270–6273. [Google Scholar] [CrossRef] [PubMed]
- Baumgärtel, K.; Mansuy, I.M. Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 2012, 19, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chattarji, S.; Barbarosie, M.; Rondi-Reig, L.; Philpot, B.D.; Miyakawa, T.; Bear, M.F.; Tonegawa, S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 2001, 107, 617–629. [Google Scholar] [CrossRef]
- Baumgärtel, K.; Genoux, D.; Welzl, H.; Tweedie-Cullen, R.Y.; Koshibu, K.; Livingstone-Zatchej, M.; Mamie, C.; Mansuy, I.M. Control of the establishment of aversive memory by calcineurin and Zif268. Nat. Neurosci. 2008, 11, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Kimaro, W.H.; Seke Etet, P.F. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021. [Google Scholar] [CrossRef] [PubMed]
- Furman, J.L.; Norris, C.M. Calcineurin and glial signaling: Neuroinflammation and beyond. J. Neuroinflamm. 2014, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Rocchio, F.; Lisa, M.; Fcancesco, M. From Pathology to Physiology of Calcineurin Signalling in Astrocytes | Opera Medica et Physiologica. Available online: http://operamedphys.org/OMP_2016_02_0029 (accessed on 24 August 2018).
- Lodygin, D.; Odoardi, F.; Schläger, C.; Körner, H.; Kitz, A.; Nosov, M.; van den Brandt, J.; Reichardt, H.M.; Haberl, M.; Flügel, A. A combination of fluorescent NFAT and H2B sensors uncovers dynamics of T cell activation in real time during CNS autoimmunity. Nat. Med. 2013, 19, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Palkowitsch, L.; Marienfeld, U.; Brunner, C.; Eitelhuber, A.; Krappmann, D.; Marienfeld, R.B. The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J. Biol. Chem. 2011, 286, 7522–7534. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Pavlath, G.K. A calcineurin- and NFAT-dependent pathway regulates Myf5 gene expression in skeletal muscle reserve cells. J. Cell Sci. 2001, 114, 303–310. [Google Scholar] [PubMed]
- Hojayev, B.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. FHL2 binds calcineurin and represses pathological cardiac growth. Mol. Cell. Biol. 2012, 32, 4025–4034. [Google Scholar] [CrossRef] [PubMed]
- Halpain, S.; Hipolito, A.; Saffer, L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 9835–9844. [Google Scholar] [CrossRef]
- Dinamarca, M.C.; Guzzetti, F.; Karpova, A.; Lim, D.; Mitro, N.; Musardo, S.; Mellone, M.; Marcello, E.; Stanic, J.; Samaddar, T.; et al. Ring finger protein 10 is a novel synaptonuclear messenger encoding activation of NMDA receptors in hippocampus. eLife 2016, 5, e12430. [Google Scholar] [CrossRef] [PubMed]
- Grolla, A.A.; Fakhfouri, G.; Balzaretti, G.; Marcello, E.; Gardoni, F.; Canonico, P.L.; DiLuca, M.; Genazzani, A.A.; Lim, D. Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging 2013, 34, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca(2+) entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Bonev, A.D.; Nelson, M.T. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ. Res. 2004, 95, e73–e81. [Google Scholar] [CrossRef] [PubMed]
- Negulescu, P.A.; Shastri, N.; Cahalan, M.D. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 2873–2877. [Google Scholar] [CrossRef] [PubMed]
- Dolmetsch, R.E.; Lewis, R.S.; Goodnow, C.C.; Healy, J.I. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997, 386, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Fam, S.R.; Gallagher, C.J.; Kalia, L.V.; Salter, M.W. Differential frequency dependence of P2Y1- and P2Y2-mediated Ca2+ signaling in astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 4437–4444. [Google Scholar] [CrossRef]
- Hirase, H.; Qian, L.; Barthó, P.; Buzsáki, G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Winship, I.R.; Plaa, N.; Murphy, T.H. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 6268–6272. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Fellin, T.; Zhu, Y.; Lee, S.-Y.; Auberson, Y.P.; Meaney, D.F.; Coulter, D.A.; Carmignoto, G.; Haydon, P.G. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 10674–10684. [Google Scholar] [CrossRef] [PubMed]
- Atkin, S.D.; Patel, S.; Kocharyan, A.; Holtzclaw, L.A.; Weerth, S.H.; Schram, V.; Pickel, J.; Russell, J.T. Transgenic mice expressing a cameleon fluorescent Ca2+ indicator in astrocytes and Schwann cells allow study of glial cell Ca2+ signals in situ and in vivo. J. Neurosci. Methods 2009, 181, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Wyss, M.T.; Weber, B. Somatotopic astrocytic activity in the somatosensory cortex. Glia 2013, 61, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Sekiya, H.; Xu, M.; Satoh, K.; Kitajima, N.; Yoshida, K.; Okubo, Y.; Sasaki, T.; Moritoh, S.; Hasuwa, H.; et al. In vivo visualization of subtle, transient, and local activity of astrocytes using an ultrasensitive Ca(2+) indicator. Cell Rep. 2014, 8, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Otsu, Y.; Couchman, K.; Lyons, D.G.; Collot, M.; Agarwal, A.; Mallet, J.-M.; Pfrieger, F.W.; Bergles, D.E.; Charpak, S. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat. Neurosci. 2015, 18, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Ferrari, K.D.; Barrett, M.J.P.; Glück, C.; Stobart, M.J.; Zuend, M.; Weber, B. Cortical Circuit Activity Evokes Rapid Astrocyte Calcium Signals on a Similar Timescale to Neurons. Neuron 2018, 98, 726–735.e4. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; O’Donnell, J.; Thrane, A.S.; Zeppenfeld, D.; Kang, H.; Xie, L.; Wang, F.; Nedergaard, M. α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 2013, 54, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Ferrari, K.D.; Barrett, M.J.P.; Stobart, M.J.; Looser, Z.J.; Saab, A.S.; Weber, B. Long-term In Vivo Calcium Imaging of Astrocytes Reveals Distinct Cellular Compartment Responses to Sensory Stimulation. Cereb. Cortex 2018, 28, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.V.; Bonev, A.D.; Wilkerson, M.K.; Nelson, M.T. Dynamic inositol trisphosphate-mediated calcium signals within astrocytic endfeet underlie vasodilation of cerebral arterioles. J. Gen. Physiol. 2006, 128, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Piet, R.; Jahr, C.E. Glutamatergic and purinergic receptor-mediated calcium transients in Bergmann glial cells. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 4027–4035. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Shih, P.-Y.; Gomi, H.; Yoshida, T.; Nakai, J.; Ando, R.; Furuichi, T.; Mikoshiba, K.; Semyanov, A.; Itohara, S. Astrocytic Ca2+ signals are required for the functional integrity of tripartite synapses. Mol. Brain 2013, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Haustein, M.D.; Kracun, S.; Lu, X.-H.; Shih, T.; Jackson-Weaver, O.; Tong, X.; Xu, J.; Yang, X.W.; O’Dell, T.J.; Marvin, J.S.; et al. Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 2014, 82, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.; Sikdar, S.K. Astrocytes in 17beta-estradiol treated mixed hippocampal cultures show attenuated calcium response to neuronal activity. Glia 2006, 53, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Suadicani, S.O.; Cherkas, P.S.; Zuckerman, J.; Smith, D.N.; Spray, D.C.; Hanani, M. Bidirectional calcium signaling between satellite glial cells and neurons in cultured mouse trigeminal ganglia. Neuron Glia Biol. 2010, 6, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Sydekum, E.; Krueppel, R.; Engelbrecht, C.J.; Schlegel, F.; Schröter, A.; Rudin, M.; Helmchen, F. Simultaneous BOLD fMRI and fiber-optic calcium recording in rat neocortex. Nat. Methods 2012, 9, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca2+ signalling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, M.; Lewis, A.; Butt, A.M. Store-operated calcium entry is essential for glial calcium signalling in CNS white matter. Brain Struct. Funct. 2017, 222, 2993–3005. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Parpura, V. Store-operated calcium entry in neuroglia. Neurosci. Bull. 2014, 30, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xia, J.; Munoz, F.M.; Manners, M.T.; Pan, R.; Meucci, O.; Dai, Y.; Hu, H. STIMs and Orai1 regulate cytokine production in spinal astrocytes. J. Neuroinflamm. 2016, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.S.; Fox, R.; Schousboe, A.; Waagepetersen, H.S.; Bak, L.K. Astrocyte glycogenolysis is triggered by store-operated calcium entry and provides metabolic energy for cellular calcium homeostasis. Glia 2014, 62, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Bernier, L.-P.; Dissing-Olesen, L.; Groten, C.J.; LeDue, J.M.; Ko, R.; Drissler, S.; MacVicar, B.A. Ca2+ transients in astrocyte fine processes occur via Ca2+ influx in the adult mouse hippocampus. Glia 2016, 64, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.V.; Ormerod, K.G.; Littleton, J.T. Astrocyte Ca2+ Influx Negatively Regulates Neuronal Activity. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, C.; Ponzoni, L.; Lim, D.; Schmeisser, M.J.; Reim, D.; Morello, N.; Orellana, D.; Tozzi, A.; Durante, V.; Scalmani, P.; et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol. Psychiatry 2017, 22, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Oh-hora, M.; Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 2008, 20, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Eder, P. Cardiac Remodeling and Disease: SOCE and TRPC Signaling in Cardiac Pathology. Adv. Exp. Med. Biol. 2017, 993, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Finch, E.A.; Graham, V.; Zhang, Z.-S.; Ding, J.-D.; Burch, J.; Oh-hora, M.; Rosenberg, P. STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol. Cell. Biol. 2012, 32, 3009–3017. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Shum, A.K.; McBride, H.J.; Kessler, J.A.; Feske, S.; Miller, R.J.; Prakriya, M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 9107–9123. [Google Scholar] [CrossRef] [PubMed]
- Bernardinelli, Y.; Randall, J.; Janett, E.; Nikonenko, I.; König, S.; Jones, E.V.; Flores, C.E.; Murai, K.K.; Bochet, C.G.; Holtmaat, A.; et al. Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr. Biol. 2014, 24, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.; Sibille, J.; Zapata, J.; Rouach, N. Activity-Dependent Plasticity of Astroglial Potassium and Glutamate Clearance. Neural Plast. 2015, 2015, 109106. [Google Scholar] [CrossRef] [PubMed]
- Pirttimaki, T.M.; Parri, H.R. Astrocyte plasticity: Implications for synaptic and neuronal activity. Neurosci. Rev. J. Bring. Neurobiol. Neurol. Psychiatry 2013, 19, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Otmakhov, N.; Khibnik, L.; Otmakhova, N.; Carpenter, S.; Riahi, S.; Asrican, B.; Lisman, J. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J. Neurophysiol. 2004, 91, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.C.; Derkach, V.A.; Guire, E.S.; Soderling, T.R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 2006, 281, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jen, P.H. GABAergic and glycinergic neural inhibition in excitatory frequency tuning of bat inferior collicular neurons. Exp. Brain Res. 2001, 141, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Man, H.; Ju, W.; Trimble, W.S.; MacDonald, J.F.; Wang, Y.T. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 2001, 29, 243–254. [Google Scholar] [CrossRef]
- Behnisch, T.; Yuanxiang, P.; Bethge, P.; Parvez, S.; Chen, Y.; Yu, J.; Karpova, A.; Frey, J.U.; Mikhaylova, M.; Kreutz, M.R. Nuclear translocation of jacob in hippocampal neurons after stimuli inducing long-term potentiation but not long-term depression. PLoS ONE 2011, 6, e17276. [Google Scholar] [CrossRef]
- Mizui, T.; Sekino, Y.; Yamazaki, H.; Ishizuka, Y.; Takahashi, H.; Kojima, N.; Kojima, M.; Shirao, T. Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS ONE 2014, 9, e85367. [Google Scholar] [CrossRef] [PubMed]
- Sokal, D.M.; Mason, R.; Parker, T.L. Multi-neuronal recordings reveal a differential effect of thapsigargin on bicuculline- or gabazine-induced epileptiform excitability in rat hippocampal neuronal networks. Neuropharmacology 2000, 39, 2408–2417. [Google Scholar] [CrossRef]
- Oltedal, L.; Haglerød, C.; Furmanek, T.; Davanger, S. Vesicular release of glutamate from hippocampal neurons in culture: An immunocytochemical assay. Exp. Brain Res. 2008, 184, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.T.; McCarthy, K.D. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 5073–5081. [Google Scholar] [CrossRef]
- Sun, W.; McConnell, E.; Pare, J.-F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Ruffinatti, F.; Tapella, L.; Gregnanin, I.; Stevano, A.; Chiorino, G.; Canonico, P.L.; Distasi, C.; Genazzani, A.A.; Lim, D. Transcriptional remodeling in primary hippocampal astrocytes from an Alzheimer’s disease mouse model. Curr. Alzheimer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Bertoli, A.; Sorgato, M.C.; Moccia, F. Generation and usage of aequorin lentiviral vectors for Ca(2+) measurement in sub-cellular compartments of hard-to-transfect cells. Cell Calcium 2016, 59, 228–239. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Condition | Astroglial Ca2+ Elevation | NY Nuclear Translocation |
|---|---|---|
| cLTP | Yes Ca2+ signals | Translocation after 10 min |
| LTD | NA | No translocation |
| Bicuculline alone 40 μM | NA | Translocation after 1 h |
| Ø Mg2+ | NA | Translocation after 2 h |
| cLTP on pure astrocytes | No Ca2+ signals | No translocation |
| Pharmacological Treatment | Astroglial Ca2+ Elevation | NY Nuclear Translocation |
|---|---|---|
| cLTP → KRB + Mg + Ca | Yes | Yes |
| cLTP → Ø Ca2+ | No | No |
| cLTP → KRB + Mg + Ca + FK506 | Yes | No |
| cLTP → KRB + Mg + Ca + cyclosporine A | Yes | No |
| cLTP (MK801) → KRB + Mg + Ca | Yes | Yes |
| cLTP → KRB + Mg + Ca + MK801 | Yes | Yes |
| cLTP (MK801) → KRB + Mg + Ca + MK801 | No | No |
| cLTP → KRB + Mg + Ca + MTEP | No | No |
| cLTP → KRB + Mg + Ca + 2APB | No | No |
| cLTP → KRB + Mg + Ca + Pyr3 | No | No |
| cLTP → KRB + Mg + Ca + Pyr6 | No | No |
| cLTP → KRB + Mg + Ca + Pyr10 | Yes | Yes |
| cLTP → KRB + Mg + Ca + Xestospongin | Yes | Yes |
| cLTP → KRB + Mg + Ca + U73122 | Yes | Yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, D.; Mapelli, L.; Canonico, P.L.; Moccia, F.; Genazzani, A.A. Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. Int. J. Mol. Sci. 2018, 19, 2997. https://doi.org/10.3390/ijms19102997
Lim D, Mapelli L, Canonico PL, Moccia F, Genazzani AA. Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. International Journal of Molecular Sciences. 2018; 19(10):2997. https://doi.org/10.3390/ijms19102997
Chicago/Turabian StyleLim, Dmitry, Lisa Mapelli, Pier Luigi Canonico, Francesco Moccia, and Armando A. Genazzani. 2018. "Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures" International Journal of Molecular Sciences 19, no. 10: 2997. https://doi.org/10.3390/ijms19102997
APA StyleLim, D., Mapelli, L., Canonico, P. L., Moccia, F., & Genazzani, A. A. (2018). Neuronal Activity-Dependent Activation of Astroglial Calcineurin in Mouse Primary Hippocampal Cultures. International Journal of Molecular Sciences, 19(10), 2997. https://doi.org/10.3390/ijms19102997