Ageing, Cellular Senescence and Neurodegenerative Disease



Abstract
1. Ageing and Neurodegeneration
2. Cellular Senescence
3. The Senescence Phenotype
4. Markers of Cellular Senescence
- p16INK4a: This member of the INK4a family is a cyclin D-dependent kinase CDK4 and CDK6 inhibitor, which prevents the phosphorylation of the retinoblastoma protein (Rb), therefore leading to suspension of the cell cycle before the S-phase [78,79]. Increased levels of p16INK4a have been documented in aged and stressed tissues, compared to younger, healthy tissues, whereas the removal of p16INK4a-expressing senescent cells in mice prevented or delayed tissue dysfunction and age-related disorders [38,80]. This evidence has established p16INK4a as a widely-accepted marker of ageing and cellular senescence [81,82]. Limitations include poor detection of p16INK4a in mice by the currently available antibodies using immunohistochemistry [79], as well as some situations, where p16INK4a levels are increased, in the absence of other signs of cellular senescence [81,83,84].
- p21CIP1/WAF1/SDI1: p21 is a member of the second group of CDK inhibitors, namely the CIP/KIP (CDK interacting protein/kinase inhibitory protein) family and can inhibit a variety of CDKs [85]. Apart from its role in cellular senescence, it is a key mediator in several biological functions, including cell cycle arrest, cell death, DNA repair processes and even reprogramming of differentiated somatic cells into pluripotent stem cells [86]. In the context of senescence, stress-induced p53 activates p21 in order to trigger cell cycle arrest [39]. Although both p21 and p16INK4a upregulation lead to cell cycle arrest, they act through different pathways and have distinct roles in the induction and progression of cellular senescence [36].
- SA-β-gal: The activity of β-galactosidase detectable at pH 6.0, which is measured using in situ staining with the chromogenic substrate X-gal [senescence-associated β-galactosidase activity (SA-β-gal)] is today the most widely used biomarker for detecting senescent cells [79,87]. The lysosomal enzyme β-galactosidase encoded by the GLB1 (galactosidase beta 1) gene, is the source of SA-β-gal activity and it can, therefore, be elevated in any situation with increased lysosome number or activity [54,88]. It has also been reported that certain cell culture conditions can increase the level of SA-β-gal, giving a false positive result [88,89]. Another drawback of the SA-β-gal assay is that it can only be used on fresh or frozen tissues and not on formalin-fixed paraffin-embedded archival tissue samples, which significantly limits its spectrum of application [15,57].
- Lipofuscin: Intracellular lipofuscin aggregates consist of oxidized protein and lipid degradation residues and metal cations that cannot be degraded by lysosomal enzymes. Lipofuscin accumulates with age and its accumulation is a documented hallmark of senescent cells [90,91,92]. GL-13 (SenTraGorTM) is a biotinylated chemical compound derived from Sudan Black-B that specifically and strongly binds to lipofuscin [57,58]. Its ability to detect lipofuscin, not only on fresh tissues, but also on formalin-fixed paraffin-embedded archival samples and biological fluids gives a new perspective in the field of senescence markers [57,58,90,93].
5. Cellular Senescence and Its Putative Role in Neurodegeneration
- Promotion of chronic inflammation: Senescence-associated secretory phenotype (SASP) converts senescent cells into continuous sources of pro-inflammatory mediators, reactive oxygen species and metalloproteinases [43]. Senescent cells may sustain a proinflammatory milieu, which can be damaging for neighboring cells or “contaminating” in the sense of converting neighboring cells to senescent ones in a paracrine manner [44,94,95]. Interleukin-6, a typical SASP mediator, is upregulated in the aged brain and in AD [96,97,98] and its overexpression has been shown to drive neurodegeneration in vivo [99]. The ageing brain has higher background levels of low-grade inflammation primarily in the form of dystrophic microglia and increased levels pro-inflammatory cytokines and other mediators, a state known as inflammaging [100,101,102]. SASP-related mediators from increased numbers of senescent cells may be what underlies inflammaging [103,104]. There are many links between inflammaging and AD and PD pathologies [105,106], which suggest that SASP may contribute to the pathogenesis of neurodegeneration and may determine disease susceptibility or aggravate the course of the disease.
- Exhaustion of the regenerative capacities of the nervous system: There is evidence of neurogenesis from adult neural stem cells deriving from the subventricular zone (SVZ) and the subgranular zone (SGZ) of the hippocampal dentate gyrus, that can give rise to neurons, oligodendrocytes and astrocytes [107,108]. Ageing has been shown to significantly reduce adult hippocampal neurogenesis [109]. Cell cycle arrest of adult progenitor cells in the context of cellular senescence may reduce the regenerative capacities of the CNS. This notion is supported by recent in vivo evidence from the BUBR1 progeroid mouse model in which adult progenitor proliferation was impaired in the SGZ and SVZ in an age-dependent manner [110]. Although the role of adult neurogenesis in AD remains contentious, studies from animal models indicate that ablation of adult neurogenesis exacerbates memory deficits and upregulates hyperphosphorylated tau [111], whereas implantation of human neural stem cells alleviates memory deficits and AD pathology [112].Furthermore, oligodendrocyte progenitor cells (OPCs) are a population of adult stem cells responsible for mediating CNS myelin repair in demyelinating conditions such as MS [113,114]. However, despite remyelination being very efficient at the early stages of the disease, this process gradually fails over time [115,116]. Evidence from MS and its animal models suggests that remyelination can protect demyelinated axons and even correlates with greater age at death, whereas chronically demyelinated axons are prone to degeneration [117,118]. Inability to replenish adult progenitor cells due to cellular senescence could render the CNS susceptible to neurodegeneration.
- Loss of function: The functional state of senescent cells has not been fully elucidated. However, cell-cycle arrest, changes in gene expression and phenotypic changes that accompany cellular senescence constitute serious restrictions in the functionality of different cell types [119]. The number of senescent cells increases with age [30]. At the same time it must be noted that ageing is associated with loss of brain cells to an extent which may amount to up to 0.4% of brain volume, annually [120]. The processes that lead to loss of brain cells with normal ageing are unclear. Both apoptotic and senescent cells can be cleared by the immune system in a highly regulated manner [35]. Thus, brain volume loss may at least partly be due to immune-mediated clearance of senescent cells. It is also conceivable, that when the number of dysfunctional senescent cells exceeds a certain threshold in a brain with reduced reserves due to age-related cell loss, nervous tissue function is likely to become compromised. Senescent cell accumulation may occur preferentially in some brain regions e.g., substantia nigra, that are more susceptible to particular stressors, which could explain the ensuing functional deficits.
- Cerebral hypoperfusion and blood-brain barrier (BBB) dysfunction: Cerebral function depends on an adequate blood supply and an intact BBB, which is crucial for maintaining homeostasis of brain microenvironment and protecting the parenchyma from pathogens, circulating immune cells, ionic changes and toxic metabolites [121]. There is evidence of an age-related decline in cerebral microvascular structure [122] and vascular pathology has been shown to accompany age-related cognitive impairment and neurodegeneration [123]. BBB leakiness is seen both with normal ageing and AD [124,125]. In a model of AD in transgenic mice BBB permeability increase even preceded neuritic plaque formation [126] and in a neuropathological study, the ApoE4 allele, which is a major risk factor for developing AD, was associated with greater likelihood of BBB disruption [127]. Vascular cells and specifically endothelial cells and pericytes have been shown to undergo senescence in vitro and in vivo [128]. Accumulation of senescent endothelial cells is associated with impaired tight junction structure and compromised blood-brain barrier integrity [129] and it is linked to Sirt1 downregulation in senescent endothelial cells [130]. Although senescence has not been studied in the cellular components of the choroid plexus, it is known to undergo several age and disease-related structural and functional alterations [131,132]. The choroid plexus produces CSF and forms an interface between blood and CSF. It secretes trophic factors, such as epidermal growth factor (EGF) and fibroblast growth factor-2 (FGF-2) and may be a route for trafficking lymphocytes to and from the CNS with roles in immune surveillance and neuroinflammation [133,134]. In addition, a shift towards an interferon I-dependent expression profile is seen with ageing in human and mouse choroid plexus, which may adversely affect cognitive function and hippocampal neurogenesis [135]. It is conceivable that compromised cerebrovascular perfusion and altered function of the BBB and/or choroid plexus may adversely affect neuronal and glial survival.
6. Evidence of Cellular Senescence in CNS Cell Types
6.1. Astrocytes
6.2. Microglia
6.3. Oligodendrocytes
6.4. Oligodendrocyte Progenitor Cells
6.5. Neurons
6.6. Neural Stem Cells (NSCs)
7. Cellular Senescence in Alzheimer’s Disease, Parkinson’s Disease and Multiple Sclerosis
7.1. Alzheimer’s Disease
7.2. Parkinson’s Disease
7.3. Multiple Sclerosis
8. Cellular Senescence as a Therapeutic Target
9. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid-beta |
| AD | Alzheimer’s disease |
| Akt | protein kinase B |
| ALL | acute lymphocytic leukemia |
| APP | amyloid beta precursor protein |
| ATF6α | activating transcription factor 6 isoform α |
| ATM | ataxia-telangiectasia mutated |
| BBB | blood-brain barrier |
| Bcl-2 | B-cell lymphoma 2 |
| CDK | cyclin-dependent kinase |
| CDKI | cyclin-dependent kinase inhibitor protein |
| cGAS | cyclic GMP-AMP synthase |
| CIP/KIP | CDK interacting protein/kinase inhibitory protein |
| CML | chronic myelogenous leukemia |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DDR | DNA damage response |
| Ecrg4 | esophageal cancer-related gene 4 |
| EGF | epidermal growth factor |
| ER | endoplasmic reticulum |
| FGF-2 | fibroblast growth factor 2 |
| GLB1 | galactosidase beta 1 |
| HLA-DR | human leukocyte antigen-DR isotype |
| γH2AX | phosphorylated H2A histone family member X |
| IkB | inhibitor of kappa B |
| IL | Interleukin |
| MBP | myelin basic protein |
| MECP | methyl-CpG-binding protein |
| MMP | matrix metalloproteinase |
| MRI | magnetic resonance imaging |
| mTOR | mammalian target of rapamycin |
| MS | multiple sclerosis |
| NAWM | normal appearing white matter |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSCs | neural stem cells |
| OIS | oncogene-induced senescence |
| OPC | oligodendrocyte progenitor cell |
| PD | Parkinson’s disease |
| PS1 | presenilin-1 |
| p38MAPK | p38 mitogen-activated protein kinases |
| RB | Retinoblastoma |
| ROS | reactive oxygen species |
| RR-MS | relapsing-remitting multiple sclerosis |
| SA-β-Gal | senescence-associated β-galactosidase |
| SAHF | senescence-associated heterochromatin formation |
| SAMD | senescence-associated mitochondrial dysfunction |
| SASP | senescence-associated secretory phenotype |
| SIPS | stress-induced premature senescence |
| SIRT | Sirtuin |
| SP-MS | secondary progressive multiple sclerosis |
| SGZ | subgranular zone |
| STING | stimulator of IFN genes |
| SVZ | subventricular zone |
| S1P | sphingosine 1-phosphate receptor |
| TCDD | 2,3,7,8-Tetrachlorodibenzo-p-Dioxin |
| Terc | human telomerase gene |
| TGF-β | transforming growth factor beta |
| TNF-α | tumour necrosis factor alpha |
| UPR | unfolded protein response |
| 53BP1 | p53-binding protein 1 |
References
- Johnson, I.P. Age-related neurodegenerative disease research needs aging models. Front. Aging Neurosci. 2015, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, E.M.; Verhaaren, B.F.; Koudstaal, P.J.; Hofman, A.; Ikram, M.A.; Breteler, M.M. Is dementia incidence declining? Trends in dementia incidence since 1990 in the Rotterdam Study. Neurology 2012, 78, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Rocca, W.A.; Petersen, R.C.; Knopman, D.S.; Hebert, L.E.; Evans, D.A.; Hall, K.S.; Gao, S.; Unverzagt, F.W.; Langa, K.M.; Larson, E.B.; et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimers Dement. 2011, 7, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s Disease and Parkinson’s Disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Lockrow, J.P.; Fortress, A.M.; Granholm, A.C.E. Age-related neurodegeneration and memory loss in down syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Hosokawa, M.; Takeshita, S.; Irino, M.; Higuchi, K.; Matsushita, T.; Tomita, Y.; Yasuhira, K.; Hamamoto, H.; Shimizu, K.; et al. A new murine model of accelerated senescence. Mech. Ageing Dev. 1981, 17, 183–194. [Google Scholar] [CrossRef]
- Morley, J.E.; Kumar, V.B.; Bernardo, A.E.; Farr, S.A.; Uezu, K.; Tumosa, N.; Flood, J.F. Beta-amyloid precursor polypeptide in SAMP8 mice affects learning and memory. Peptides 2000, 21, 1761–1767. [Google Scholar] [CrossRef]
- Blin, P.; Dureau-Pournin, C.; Foubert-Samier, A.; Grolleau, A.; Corbillon, E.; Jové, J.; Lassalle, R.; Robinson, P.; Poutignat, N.; Droz-Perroteau, C.; Moore, N. Parkinson’s disease incidence and prevalence assessment in France using the national healthcare insurance database. Eur. J. Neurol. 2015, 22, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.W.; Khoo, T.K.; Coleman, S.Y.; Brayne, C.; Yarnall, A.J.; O’Brien, J.T.; Barker, R.A.; Burn, D.J. The incidence of Parkinson’s disease in the North-East of England. Age Ageing 2014, 43, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.-A. Multiple Sclerosis: An Immune or Neurodegenerative Disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Scalfari, A.; Neuhaus, A.; Daumer, M.; Ebers, G.C.; Muraro, P.A. Age and disability accumulation in multiple sclerosis. Neurology 2011, 77, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L.; Melov, S. On the Programmed/Non-Programmed Nature of Ageing within the Life History. Curr. Biol. 2011, 21, R701–R707. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L.; Feder, M.; Finch, C.E.; Franceschi, C.; Globerson, A.; Klingenberg, C.P.; LaMarco, K.; Omholt, S.; Westendorp, R.G. What accounts for the wide variation in life span of genetically identical organisms reared in a constant environment? Mech. Ageing Dev. 2005, 126, 439–443. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Barbouti, A.; Evangelou, K.; Pateras, I.S.; Papoudou-Bai, A.; Patereli, A.; Stefanaki, K.; Rontogianni, D.; Muñoz-Espín, D.; Kanavaros, P.; Gorgoulis, V.G. In situ evidence of cellular senescence in Thymic Epithelial Cells (TECs) during human thymic involution. Mech. Ageing Dev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef] [PubMed]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging (Albany NY) 2013, 5, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr. Opin. Cell Biol. 2010, 22, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence—Its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; Sedelnikova, O.A.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenet. Chromatin 2008, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.D.; Morales, C.P.; Herbert, B.S.; Rohde, J.M.; Passons, C.; Shay, J.W.; Wright, W.E. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001, 15, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Turenne, G.A.; Paul, P.; Laflair, L.; Price, B.D. Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53. Oncogene 2001, 20, 5100–5110. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells—Biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21CIP1, but not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Lawless, C.; Wang, C.; Jurk, D.; Merz, A.; Zglinicki, T. von; Passos, J.F. Quantitative assessment of markers for cell senescence. Exp. Gerontol. 2010, 45, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a -positive senescent cells delays ageing- associated disorders. Nature 2012, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; VandenBerg, J.; Medema, R.H. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Gilhotra, R.; Dhingra, D.; Gilhotra, N. Possible underlying influence of p38MAPK and NF-κB in the diminished anti-anxiety effect of diazepam in stressed mice. J. Pharmacol. Sci. 2011, 116, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-Associated Exosome Release from Human Prostate Cancer Cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef] [PubMed]
- Webley, K.; Bond, J.A.; Jones, C.J.; Blaydes, J.P.; Craig, A.; Hupp, T.; Wynford-Thomas, D. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell. Biol. 2000, 20, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Tavana, O.; Benjamin, C.L.; Puebla-Osorio, N.; Sang, M.; Ullrich, S.E.; Ananthaswamy, H.N.; Zhu, C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 2010, 9, 3328–3336. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-Dependent Acetylation of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez-Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ye, H.; Shao, C.; Zheng, X.; Li, Q.; Wang, L.; Zhao, M.; Lu, G.; Chen, B.; Zhang, J.; et al. Metabolomics–Proteomics Combined Approach Identifies Differential Metabolism-Associated Molecular Events between Senescence and Apoptosis. J. Proteome Res. 2017, 16, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeong, S.-Y.; Lim, W.-C.; Kim, S.; Park, Y.-Y.; Sun, X.; Youle, R.J.; Cho, H. Mitochondrial Fission and Fusion Mediators, hFis1 and OPA1, Modulate Cellular Senescence. J. Biol. Chem. 2007, 282, 22977–22983. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Karbowski, M.; Yamaguchi, H.; Kazi, A.; Wu, J.; Sebti, S.M.; Youle, R.J.; Wang, H.-G. Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 2005, 25, 9369–9382. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta-Bioenerg. 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; von Zglinicki, T. DNA damage in telomeres and mitochondria during cellular senescence: Is there a connection? Nucleic Acids Res. 2007, 35, 7505–7513. [Google Scholar] [CrossRef] [PubMed]
- Tasselli, L.; Xi, Y.; Zheng, W.; Tennen, R.I.; Odrowaz, Z.; Simeoni, F.; Li, W.; Chua, K.F. SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat. Struct. Mol. Biol. 2016, 23, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Komseli, E.-S.; Pateras, I.S.; Krejsgaard, T.; Stawiski, K.; Rizou, S.V.; Polyzos, A.; Roumelioti, F.-M.; Chiourea, M.; Mourkioti, I.; Paparouna, E.; et al. A prototypical non-malignant epithelial model to study genome dynamics and concurrently monitor micro-RNAs and proteins in situ during oncogene-induced senescence. BMC Genomics 2018, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Druelle, C.; Drullion, C.; Deslé, J.; Martin, N.; Saas, L.; Cormenier, J.; Malaquin, N.; Huot, L.; Slomianny, C.; Bouali, F.; et al. ATF6α regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 2016, 7, 67699–67715. [Google Scholar] [CrossRef] [PubMed]
- Cormenier, J.; Martin, N.; Deslé, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6α arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E 2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16(INK4a) tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16 INK4a-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.; Edwards, C.; Kobzik, L. Reciprocal Rb Inactivation and p16 INK4 Expression in Primary Lung Cancers and Cell Lines. Cancer Res. 1995, 55, 505–509. [Google Scholar] [PubMed]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16 ink4a expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [PubMed]
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is β-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 2000, 257, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Pefani, D.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA damage and protein stress responses during cancer development and treatment. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: Formation, distribution, and metabolic consequences. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Rizou, S.V.; Evangelou, K.; Myrianthopoulos, V.; Mourouzis, I.; Havaki, S.; Vasileiou, P.; Kotsinas, A.; Kastrinakis, N.; Sfikakis, P.; Townsend, P.; Mikros, E.; Pantos, C.; Gorgoulis, V.G. A novel quantitative method for the detection of lipofuscin, the main byproduct of cellular senescence, in fluids. Meth. Mol. Biol. 2018, in press. [Google Scholar]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37–38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Strauss, S.; Schreiter-Gasser, U.; Ganter, U.; Schlegel, P.; Witt, I.; Yolk, B.; Berger, M. Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 1991, 285, 111–114. [Google Scholar] [CrossRef]
- Huell, M.; Strauss, S.; Volk, B.; Berger, M.; Bauer, J. Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol. 1995, 89, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Kiecolt-Glaser, J.K.; Preacher, K.J.; MacCallum, R.C.; Atkinson, C.; Malarkey, W.B.; Glaser, R. Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc. Natl. Acad. Sci. USA 2003, 100, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic Microglia in the Aging Human Brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Rawji, K.S.; Mishra, M.K.; Michaels, N.J.; Rivest, S.; Stys, P.K.; Yong, V.W. Immunosenescence of microglia and macrophages: Impact on the ageing central nervous system. Brain 2016, 139, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Med. Inflamm. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Giunta, B.; Fernandez, F.; Nikolic, W.V.; Obregon, D.; Rrapo, E.; Town, T.; Tan, J. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflamm. 2008, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.; Song, H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The Glial Nature of Embryonic and Adult Neural Stem Cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Ferrer, I.; Aronica, E.; Kovacs, G.G.; Verney, C.; Nardelli, J.; Khung, S.; Delezoide, A.L.; Milenkovic, I.; Rasika, S.; et al. Hippocampal Radial Glial Subtypes and Their Neurogenic Potential in Human Fetuses and Healthy and Alzheimer’s Disease Adults. Cereb Cortex 2018, 28, 2458–2478. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jun, H.; Choi, C.-I.; Yoo, K.H.; Cho, C.H.; Hussaini, S.M.Q.; Simmons, A.J.; Kim, S.; van Deursen, J.M.; Baker, D.J.; et al. Age-related decline in BubR1 impairs adult hippocampal neurogenesis. Aging Cell 2017, 16, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Hollands, C.; Tobin, M.K.; Hsu, M.; Musaraca, K.; Yu, T.-S.; Mishra, R.; Kernie, S.G.; Lazarov, O. Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol. Neurodegener. 2017, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-S.; Jung, K.; Kim, I.-S.; Lee, H.; Kim, M.; Yun, S.; Hwang, K.; Shin, J.E.; Park, K.I. Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol. Neurodegener. 2015, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.; Dawson, M.; Papadopoulos, D.; Polito, A.; Di Bello, I.C.; Pham-Dinh, D.; Levine, J. The response of NG2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J. Neurocytol. 2002, 31, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating Oligodendrocytes in Chronic Lesions of Multiple Sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.-S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J.M. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple Sclerosis and Chronic Autoimmune Encephalomyelitis. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Purcell, M.; Kruger, A.; Tainsky, M.A. Gene expression profiling of replicative and induced senescence. Cell Cycle 2014, 13, 3927–3937. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, N.; Stromillo, M.L.; Giorgio, A.; Bartolozzi, M.L.; Battaglini, M.; Baldini, M.; Portaccio, E.; Amato, M.P.; Sormani, M.P. Establishing pathological cut-offs of brain atrophy rates in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Thore, C.R. Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 2011, 37, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003, 10, 463–470. [Google Scholar] [PubMed]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hulette, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.C.; van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 2016, 47, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Stamatanovic, S.; Martinez, G.; Hu, A.; Choi, J.; Keep, R.; Andjelkovic, A. Decline in Sirtuin-1 expression and activity plays a critical role in blood-brain barrier permeability in aging. Neurobiol Dis. 2018. [Google Scholar] [CrossRef]
- Rubenstein, E. Relationship of senescence of cerebrospinal fluid circulatory system to dementias of the aged. Lancet 1998, 351, 283–285. [Google Scholar] [CrossRef]
- Ott, B.R.; Jones, R.N.; Daiello, L.A.; de la Monte, S.M.; Stopa, E.G.; Johanson, C.E.; Denby, C.; Grammas, P. Blood-Cerebrospinal Fluid Barrier Gradients in Mild Cognitive Impairment and Alzheimer’s Disease: Relationship to Inflammatory Cytokines and Chemokines. Front Aging Neurosci. 2018, 10, 245. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+ T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, J.; Damkier, H.H. Transport across the choroid plexus epithelium. Am. J. Physiol. Cell Physiol. 2017, 312, C673–C686. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; García-Matas, S.; Rodríguez-Farré, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.; Chamberlain, C.G.; Weible, M.W., II; Hughes, S.; Chu, Y.; Chan-Ling, T. Aging-related changes in astrocytes in the rat retina: Imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell 2008, 7, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.J.; Wyllie, F.S.; Wynford-Thomas, D.; Kipling, D.; Jones, C.J. A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res. 2003, 63, 4854–4861. [Google Scholar] [PubMed]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Liang, L.; Xi, H.; Jiang, S.; Jiang, J.; Tang, C.; Liu, X.; Liu, S.; Wan, C.; Zhao, J.; et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces premature senescence of astrocytes via WNT/β-catenin signaling and ROS production. J. Appl. Toxicol. 2015, 35, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Görg, B.; Karababa, A.; Shafigullina, A.; Bidmon, H.J.; Häussinger, D. Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Glia 2015, 63, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; Mcquade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Crowe, E.P.; Tuzer, F.; Gregory, B.D.; Donahue, G.; Gosai, S.J.; Cohen, J.; Leung, Y.Y.; Yetkin, E.; Nativio, R.; Wang, L.-S.; et al. Changes in the Transcriptome of Human Astrocytes Accompanying Oxidative Stress-Induced Senescence. Front. Aging Neurosci. 2016, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte Senescence as a Component of Alzheimer’s Disease. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Cui, C.; Kim, S.; Sung, C.; Choi, C. Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem. Biol. Interact. 2018, 283, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Mombach, J.C.M.M.; Vendrusculo, B.; Bugs, C.A. A model for p38MAPK-induced astrocyte senescence. PLoS ONE 2015, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.K.; Wengenack, T.M.; Poduslo, J.F.; van Deursen, J.M. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol. Aging 2007, 28, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Invest. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Flanary, B.E.; Streit, W.J. Progressive Telomere Shortening Occurs in Cultured Rat Microglia, but Not Astrocytes. Glia 2004, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence That Aging and Amyloid Promote Microglial Cell Senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-M.; Zhao, Y.-M.; Luo, X.-G.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.-Y.; Luo, X.-M.; Chen, S.-D.; Wang, X.-Y. Repeated Lipopolysaccharide Stimulation Induces Cellular Senescence in BV2 Cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.R.; Streit, W.J. Microglia in the Aging Brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.-S.; Bechmann, I.; Qing-Shan, A.; Ae, X.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed]
- Giacci, M.K.; Bartlett, C.A.; Smith, N.M.; Iyer, K.S.; Toomey, L.M.; Jiang, H.; Guagliardo, P.; Kilburn, M.R.; Fitzgerald, M. Oligodendroglia Are Particularly Vulnerable to Oxidative Damage after Neurotrauma In Vivo. J. Neurosci. 2018, 38, 6491–6504. [Google Scholar] [CrossRef] [PubMed]
- Al-Mashhadi, S.; Simpson, J.E.; Heath, P.R.; Dickman, M.; Forster, G.; Matthews, F.E.; Brayne, C.; Ince, P.G.; Wharton, S.B. Oxidative glial cell damage associated with white matter lesions in the aging human brain. Brain Pathol. 2015, 25, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M. Contemplating Alzheimer’s Disease and the Contribution of White Matter Hyperintensities. Curr. Neurol. Neurosci. Rep. 2013, 13, 415. [Google Scholar] [CrossRef] [PubMed]
- Gouw, A.A.; van der Flier, W.M.; Fazekas, F.; van Straaten, E.C.W.; Pantoni, L.; Poggesi, A.; Inzitari, D.; Erkinjuntti, T.; Wahlund, L.O.; Waldemar, G.; et al. Progression of White Matter Hyperintensities and Incidence of New Lacunes Over a 3-Year Period: The Leukoaraiosis and Disability Study. Stroke 2008, 39, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Erten-Lyons, D.; Woltjer, R.; Kaye, J.; Mattek, N.; Dodge, H.H.; Green, S.; Tran, H.; Howieson, D.B.; Wild, K.; Silbert, L.C. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology 2013, 81, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.R.L.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Tang, D.G.; Tokumoto, Y.M.; Apperly, J.A.; Lloyd, A.C.; Raff, M.C. Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science (80-.) 2001, 291, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Kujuro, Y.; Suzuki, N.; Kondo, T. Esophageal cancer-related gene 4 is a secreted inducer of cell senescence expressed by aged CNS precursor cells. Proc. Natl. Acad. Sci. USA 2010, 107, 8259–8264. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain 1999, 122, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Balaratnam, M.; Vora, A.; Reynolds, R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol. Appl. Neurobiol. 2007, 33, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-I.; Yoo, K.H.; Hussaini, S.M.Q.; Jeon, B.T.; Welby, J.; Gan, H.; Scarisbrick, I.A.; Zhang, Z.; Baker, D.J.; van Deursen, J.M.; et al. The progeroid gene BubR1 regulates axon myelination and motor function. Aging (Albany NY) 2016, 8, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xiao, M. Oligodendrocytes and Alzheimer’s disease. Int. J. Neurosci. 2016, 126, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [PubMed]
- Panossian, L.; Fenik, P.; Zhu, Y.; Zhan, G.; McBurney, M.W.; Veasey, S. SIRT1 regulation of wakefulness and senescence-like phenotype in wake neurons. J. Neurosci. 2011, 31, 4025–4036. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Korsakova, E.; Allen, D.; Lee, P.; Fu, K.; Vargas, B.S.; Cinkornpumin, J.; Salas, C.; Park, J.C.; Germanguz, I.; et al. Loss of MECP2 Leads to Activation of P53 and Neuronal Senescence. Stem Cell Rep. 2018, 10, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Duncan, T.; Valenzuela, M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res. Ther. 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Ferron, S.; Mira, H.; Franco, S.; Cano-Jaimez, M.; Bellmunt, E.; Ramírez, C.; Fariñas, I.; Blasco, M.A. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 2004, 131, 4059–4070. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Jin, W.-L.; Lok, K.-H.; Wang, Y.; Yin, M.; Wang, Z.-J. Amyloid-β1–42 oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013, 4, e924. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Q.; Wang, Z.; Liu, S.; Hu, Y.; Yin, M.; Lu, Y. N-Stearoyl-L-Tyrosine Inhibits the Senescence of Neural Stem/Progenitor Cells Induced by Aβ 1-42 via the CB2 Receptor. Stem Cells Int. 2016, 2016, 1–9. [Google Scholar]
- Bose, R.; Moors, M.; Tofighi, R.; Cascante, A.; Hermanson, O.; Ceccatelli, S. Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis. 2010, 1, e92. [Google Scholar] [CrossRef] [PubMed]
- Ferron, S.R.; Marques-Torrejon, M.A.; Mira, H.; Flores, I.; Taylor, K.; Blasco, M.A.; Farinas, I. Telomere Shortening in Neural Stem Cells Disrupts Neuronal Differentiation and Neuritogenesis. J. Neurosci. 2009, 29, 14394–14407. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Magini, A.; Polchi, A.; Tozzi, A.; Tancini, B.; Tantucci, M.; Urbanelli, L.; Borsello, T.; Calabresi, P.; Emiliani, C. Abnormal cortical lysosomal β-hexosaminidase and β-galactosidase activity at post-synaptic sites during Alzheimer’s disease progression. Int. J. Biochem. Cell Biol. 2015, 58, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Tiribuzi, R.; Orlacchio, A.A.; Crispoltoni, L.; Maiotti, M.; Zampolini, M.; De Angelis, M.; Mecocci, P.; Cecchetti, R.; Bernardi, G.; Datti, A.; et al. Lysosomal β-galactosidase and β-hexosaminidase activities correlate with clinical stages of dementia associated with Alzheimer’s disease and type 2 diabetes mellitus. J. Alzheimer’s Dis. 2011, 24, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Tiribuzi, R.; Crispoltoni, L.; Porcellati, S.; Di Lullo, M.; Florenzano, F.; Pirro, M.; Bagaglia, F.; Kawarai, T.; Zampolini, M.; Orlacchio, A.A.; et al. MiR128 up-regulation correlates with impaired amyloid β(1–42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol. Aging 2014, 35, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.; Rosado, M.; Davies, P. Mitotic mechanisms in Alzheimer’s disease? J. Cell Biol. 1996, 132, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z. The last neuronal division: A unifying hypothesis for the pathogenesis of Alzheimer’s disease. J. Cell. Mol. Med. 2005, 9, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Hortobágyi, T.; Soriano, S. The neuronal cell cycle as a mechanism of pathogenesis in Alzheimer’s. Aging (Albany NY) 2009, 1, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Watanabe, K.; Broude, E.V.; Fang, J.; Poole, J.C.; Kalinichenko, T.V.; Roninson, I.B. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 4291–4296. [Google Scholar] [CrossRef] [PubMed]
- Engidawork, E.; Gulesserian, T.; Seidl, R.; Cairns, N.; Lubec, G. Expression of apoptosis related proteins in brains of patients with Alzheimer’s disease. Neurosci. Lett. 2001, 303, 79–82. [Google Scholar] [CrossRef]
- Yates, S.C.; Zafar, A.; Rabai, E.M.; Foxall, J.B.; Nagy, S.; Morrison, K.E.; Clarke, C.; Esiri, M.M.; Christie, S.; Smith, A.D.; et al. The Effects of Two Polymorphisms on p21cip1 Function and Their Association with Alzheimer’s Disease in a Population of European Descent. PLoS ONE 2015, 10, e0114050. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Holzer, M.; Gärtner, U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J. Neural Transm. 1998, 105, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Bartolomé, F.; Alquézar, C.; Antequera, D.; Muñoz, Ú.; Carro, E.; Martín-Requero, Á. Altered cell cycle-related gene expression in brain and lymphocytes from a transgenic mouse model of Alzheimer’s disease [amyloid precursor protein/presenilin 1 (PS1)]. Eur. J. Neurosci. 2012, 36, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Delobel, P.; Lavenir, I.; Ghetti, B.; Holzer, M.; Goedert, M. Cell-Cycle Markers in a Transgenic Mouse Model of Human Tauopathy. Am. J. Pathol. 2006, 168, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Wang, S.; Song, J.; Jia, J. Combination of p53(ser15) and p21/p21(thr145) in peripheral blood lymphocytes as potential Alzheimer’s disease biomarkers. Neurosci. Lett. 2012, 516, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Alquézar, C.; Bermejo-Pareja, F.; Bialopiotrowicz, E.; Wojda, U.; Martín-Requero, Á. Downregulation of extracellular signal-regulated kinase 1/2 activity by calmodulin KII modulates p21 Cip1 levels and survival of immortalized lymphocytes from Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Bialopiotrowicz, E.; Kuzniewska, B.; Kachamakova-Trojanowska, N.; Barcikowska, M.; Kuznicki, J.; Wojda, U. Cell cycle regulation distinguishes lymphocytes from sporadic and familial Alzheimer’s disease patients. Neurobiol. Aging 2011, 32, 2319-e13. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, T.; Marksteiner, J.; Defrancesco, M.; Deisenhammer, E.A.; Kemmler, G.; Humpel, C. Two Blood Monocytic Biomarkers (CCL15 and p21) Combined with the Mini-Mental State Examination Discriminate Alzheimer’s Disease Patients from Healthy Subjects. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Rödel, L.; Gärtner, U.; Holzer, M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. Neuroreport 1996, 7, 3047–3049. [Google Scholar] [PubMed]
- McShea, A.; Harris, P.L.; Webster, K.R.; Wahl, A.F.; Smith, M.A. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 1933–1939. [Google Scholar] [PubMed]
- Luth, I.H.J.; Holzer, M.; Gertz, H.-J.; Arendt, T. Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21 ras and p16. Brain Res. 2000, 852, 45–55. [Google Scholar] [CrossRef]
- Monte, S.M. De; Sohn, Y.K.; Wands, J.R. Correlates of p53- and Fas (CD95) -mediated apoptosis in Alzheimer’s disease. J. Neurol. Sci. 1997, 152, 73–83. [Google Scholar] [CrossRef]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1997, 232, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Ohyagi, Y.; Asahara, H.; Chui, D.-H.; Tsuruta, Y.; Sakae, N.; Miyoshi, K.; Yamada, T.; Kikuchi, H.; Taniwaki, T.; Murai, H.; et al. Intracellular Aβ42 activates p53 promoter: A pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 2005, 19, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.-J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimers Dis. 2001, 3, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, C.; Sheng, X.; Zhang, X.; Wang, B.; Zhang, G. Peripheral expression of MAPK pathways in Alzheimer’s and Parkinson’s diseases. J. Clin. Neurosci. 2014, 21, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Lin, Y.-G.; Ciallella, J.R.; Flood, D.G.; Scott, R.W. Activation of c-Jun N-Terminal Kinase and p38 in an Alzheimer’s Disease Model Is Associated with Amyloid Deposition. J. Neurosci. 2002, 22, 3376–3385. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.A.; Wood, P.L.; Ryan, R.; Graff-Radford, N.R.; Pilapil, C.; Robitaille, Y.; Quirion, R. Cytokine indices in Alzheimer’s temporal cortex: No changes in mature IL-1 beta or IL-1RA but increases in the associated acute phase proteins IL-6, alpha 2-macroglobulin and C-reactive protein. Brain Res. 1993, 629, 245–252. [Google Scholar] [CrossRef]
- Cacabelos, R.; Alvarez, X.A.; Fernández-Novoa, L.; Franco, A.; Mangues, R.; Pellicer, A.; Nishimura, T. Brain interleukin-1 beta in Alzheimer’s disease and vascular dementia. Methods Find. Exp. Clin. Pharmacol. 1994, 16, 141–151. [Google Scholar] [PubMed]
- Luterman, J.D.; Haroutunian, V.; Yemul, S.; Ho, L.; Purohit, D.; Aisen, P.S.; Mohs, R.; Pasinetti, G.M. Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch. Neurol. 2000, 57, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Blum-Degen, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Liljeroth, A.-M.; Minthon, L.; Tarkowski, A.; Wallin, A.; Blennow, K. Cerebral pattern of pro-and anti-inflammatory cytokines in dementias. Brain Res. Bull. 2003, 61, 255–260. [Google Scholar] [CrossRef]
- Gezen-Ak, D.; Dursun, E.; Hanağası, H.; Bilgiç, B.; Lohman, E.; Araz, Ö.S.; Atasoy, İ.L.; Alaylıoğlu, M.; Önal, B.; Gürvit, H.; et al. BDNF, TNFα, HSP90, CFH, and IL-10 Serum Levels in Patients with Early or Late Onset Alzheimer’s Disease or Mild Cognitive Impairment. J. Alzheimer’s Dis. 2013, 37, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, İ.L.; Alaylıoğlu, M.; Araz, S.; Önal, B.; et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Leake, A.; Morris, C.; Whateley, J. Brain matrix metalloproteinase 1 levels are elevated in Alzheimer’s disease. Neurosci. Lett. 2000, 291, 201–203. [Google Scholar] [CrossRef]
- Bjerke, M.; Zetterberg, H.; Edman, Å.; Blennow, K.; Wallin, A.; Andreasson, U. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Horstmann, S.; Budig, L.; Gardner, H.; Koziol, J.; Deuschle, M.; Schilling, C.; Wagner, S. Matrix metalloproteinases in peripheral blood and cerebrospinal fluid in patients with Alzheimer’s disease. Int. Psychogeriatr. C Int. Psychogeriatr. Assoc. 2018, 226, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Asahina, M.; Hattori, T. Selective distribution of matrix metalloproteinase-3 (MMP-3) in Alzheimer’s disease brain. Acta Neuropathol. 2000, 99, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s Disease: Perspective of DNA Methylation. Mol. Neurobiol. 2018, 55, 1026–1044. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide12 DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; Cruchaga, C. The epigenetic landscape of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.R.T.; Santos, A.C.F.; Farfel, J.M.; Grinberg, L.T.; Ferretti, R.E.L.; Campos, A.H.J.F.M.; Cunha, I.W.; Begnami, M.D.; Rocha, R.M.; Carraro, D.M.; et al. Repair of Oxidative DNA Damage, Cell-Cycle Regulation and Neuronal Death May Influence the Clinical Manifestation of Alzheimer’s Disease. PLoS ONE 2014, 9, e99897. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Francois, M.; Hecker, J.; Faunt, J.; Fenech, M.F.; Leifert, W.R. γH2AX is increased in peripheral blood lymphocytes of Alzheimer’s disease patients in the South Australian Neurodegeneration, Nutrition and DNA Damage (SAND) study of aging. Mutat. Res.-Genet. Toxicol. Environ. Mutagen. 2018, 829–830, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Hernández, N.; Sproul, A.; Yu, W.H. Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Morishima-Kawashima, M.; Nixon, R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006361. [Google Scholar] [CrossRef] [PubMed]
- Zare-Shahabadi, A.; Masliah, E.; Johnson, G.V.W.; Rezaei, N. Autophagy in Alzheimer’s disease. Rev. Neurosci. 2015, 26, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Kim, D.H. Alzheimer’s disease genes and autophagy. Brain Res. 2016, 1649, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxid. (Basel, Switz.) 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Lee, C.J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Myung, N.-H.; Zhu, X.; Kruman, I.I.; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H.-G. Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age (Omaha) 2008, 30, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia and Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2004, 77, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, C.; Oliveira, A.F.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Fernandes, A.; Brites, D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci. 2014, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key aging-associated alterations in primary microglia response to beta-amyloid stimulation. Front. Aging Neurosci. 2017, 9, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Eitan, E.; Hutchison, E.R.; Mattson, M.P. Telomere shortening in neurological disorders: An abundance of unanswered questions. Trends Neurosci. 2014, 37, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Boccardi, V.; Pelini, L.; Ercolani, S.; Ruggiero, C.; Mecocci, P. From cellular senescence to Alzheimer’s disease: The role of telomere shortening. Ageing Res. Rev. 2015, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Forero, D.A.; González-Giraldo, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G.; Kritchevsky, S. Meta-analysis of Telomere Length in Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Hinterberger, M.; Fischer, P.; Huber, K.; Krugluger, W.; Zehetmayer, S. Leukocyte telomere length is linked to vascular risk factors not to Alzheimer’s disease in the VITA study. J. Neural Transm. 2017, 124, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Jordan-Sciutto, K.L.; Dorsey, R.; Chalovich, E.M.; Hammond, R.R.; Achim, C.L. Expression patterns of retinoblastoma protein in Parkinson disease. J. Neuropathol. Exp. Neurol. 2003, 62, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, G.U.; Breunig, J.J.; Depboylu, C.; Rouaux, C.; Michel, P.P.; Alvarez-Fischer, D.; Boutillier, A.-L.; DeGregori, J.; Oertel, W.H.; Rakic, P.; et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 3585–3590. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D.J. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-Motor Symptoms in Patients with Parkinson’s Disease—Correlations with Inflammatory Cytokines in Serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef] [PubMed]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Serum levels of interleukin-6 are elevated in patients with Parkinson’s disease and correlate with physical performance. Neurosci. Lett. 2010, 468, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.W.; Schuh, A.F.S.; Saute, J.; Townsend, R.; Fricke, D.; Leke, R.; Souza, D.O.; Portela, L.V.; Chaves, M.L.F.; Rieder, C.R.M. Interleukin-6 serum levels in patients with parkinson’s disease. Neurochem. Res. 2009, 34, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Choi, D.-H.; Kim, Y.-J.; Kim, Y.-G.; Joh, T.H.; Beal, M.F.; Kim, Y.-S. Role of Matrix Metalloproteinase 3-mediated α-Synuclein Cleavage in Dopaminergic Cell Death. J. Biol. Chem. 2011, 286, 14168–14177. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Gill, J.S.; Bansal, P.K.; Deshmukh, R. Neuroinflammation—A major cause for striatal dopaminergic degeneration in Parkinson’s disease. J. Neurol. Sci. 2017, 381, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Pitcairn, C.; Wani, W.Y.; Mazzulli, J.R. Dysregulation of the autophagic-lysosomal pathway in Gaucher and Parkinson’s disease. Neurobiol. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Ríos, P.; Madero-Pérez, J.; Fernández, B.; Hilfiker, S. Targeting the Autophagy/Lysosomal Degradation Pathway in Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Duchen, M.R. Crosstalk between Lysosomes and Mitochondria in Parkinson’s Disease. Front. Cell Dev. Biol. Cell Dev. Biol 2017, 5, 110. [Google Scholar] [CrossRef] [PubMed]
- Watfa, G.; Dragonas, C.; Brosche, T.; Dittrich, R.; Sieber, C.C.; Alecu, C.; Benetos, A.; Nzietchueng, R. Study of telomere length and different markers of oxidative stress in patients with Parkinson’s disease. J. Nutr. Health Aging 2011, 15, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Guan, J.Z.; Koyanagi, M.; Higuchi, Y.; Makino, N. Aging-Associated Alteration of Telomere Length and Subtelomeric Status in Female Patients with Parkinson’s Disease. J. Neurogenet. 2012, 26, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.Z.; Maeda, T.; Sugano, M.; Oyama, J.; Higuchi, Y.; Suzuki, T.; Makino, N. A percentage analysis of the telomere length in Parkinson’s disease patients. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Eerola, J.; Kananen, L.; Manninen, K.; Hellstrom, O.; Tienari, P.J.; Hovatta, I. No Evidence for Shorter Leukocyte Telomere Length in Parkinson’s Disease Patients. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65A, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Degerman, S.; Domellöf, M.; Landfors, M.; Linder, J.; Lundin, M.; Haraldsson, S.; Elgh, E.; Roos, G.; Forsgren, L. Long leukocyte telomere length at diagnosis is a risk factor for dementia progression in idiopathic parkinsonism. PLoS ONE 2014, 9, e113387. [Google Scholar] [CrossRef] [PubMed]
- Forero, D.A.; González-Giraldo, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G. Telomere length in Parkinson’s disease: A meta-analysis HHS Public Access. Exp. Gerontol. 2016, 75, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Ebers, G.C. Environmental factors and multiple sclerosis. Lancet Neurol. 2008, 7, 268–277. [Google Scholar] [CrossRef]
- Pardo, G.; Jones, D.E. The sequence of disease-modifying therapies in relapsing multiple sclerosis: Safety and immunologic considerations. J. Neurol. 2017, 264, 2351–2374. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Pham-Dinh, D.; Reynolds, R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp. Neurol. 2006, 197, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Dukes, S.; Patel, R.; Nicholas, R.; Vora, A.; Reynolds, R. Substantial Archaeocortical Atrophy and Neuronal Loss in Multiple Sclerosis. Brain Pathol. 2009, 19, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The Immunopathology of Multiple Sclerosis: An Overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. The role of oxidative stress in the pathogenesis of multiple sclerosis: The need for effective antioxidant therapy. J. Neurol. 2004, 251, 261–268. [Google Scholar] [PubMed]
- Mahad, D.; Lassmann, H.; Turnbull, D. Mitochondria and disease progression in multiple sclerosis. Neuropathol. Appl. Neurobiol. 2008, 34, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, R.; Quaglini, S.; Tavazzi, E.; Amato, M.P.; Paolicelli, D.; Zipoli, V.; Romani, A.; Tortorella, C.; Portaccio, E.; D’Onghia, M.; et al. Immunomodulatory therapies delay disease progression in multiple sclerosis. Mult. Scler. J. 2016, 22, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Karamita, M.; Nicholas, R.; Kokoti, L.; Rizou, S.; Mitsikostas, D.D.; Gorgoulis, V.; Probert, L.; Papadopoulos, D. Cellular Senescence Correlates with Demyelination, Brain Atrophy and Motor Impairment in a Model of Multiple Sclerosis (P2.405). Neurology 2018, 90 no 15 supplement. [Google Scholar]
- Bø, L.; Vedeler, C.A.; Nyland, H.I.; Trapp, B.D.; Mørk, S.J. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J. Neuropathol. Exp. Neurol. 2003, 62, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Filippi, M.; Gallo, P. Cortical lesions in multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Serafini, B.; Aloisi, F.; Reynolds, R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 2010, 68, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W.; Bö, L.; Mörk, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Cummings, J. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin. Transl. Sci. 2018, 11, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Marchant, N.L.; Reed, B.R.; DeCarli, C.S.; Madison, C.M.; Weiner, M.W.; Chui, H.C.; Jagust, W.J. Cerebrovascular disease, β-amyloid, and cognition in aging. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.M.; Scheltens, P.; Visser, P.J.; Verfaillie, S.C.J.; Zwan, M.D.; et al. Prevalence of Amyloid PET Positivity in Dementia Syndromes. JAMA 2015, 313, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.J.; Visser, P.J.; Aalten, P.; Aarsland, D.; Alcolea, D.; et al. Prevalence of Cerebral Amyloid Pathology in Persons Without Dementia. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Tap, W.D.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Antonescu, C.R.; Landa, J.; Qin, L.-X.; Rathbone, D.D.; Condy, M.M.; et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013, 31, 2024–2028. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2010, 2, 627–631. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D.P.; Kobayashi, A.; Taketo, M.M.; McMahon, A.P.; Humphreys, B.D. Wnt4/ -Catenin Signaling in Medullary Kidney Myofibroblasts. J. Am. Soc. Nephrol. 2013, 24, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Byun, H.-O.; Jee, B.A.; Cho, H.; Seo, Y.-H.; Kim, Y.-S.; Park, M.H.; Chung, H.-Y.; Woo, H.G.; Yoon, G. Implications of time-series gene expression profiles of replicative senescence. Aging Cell 2013, 12, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.-S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Schaffrath, C.; Van de Leur, E.; Haas, U.; Tihaa, L.; Meurer, S.K.; Nevzorova, Y.A.; Liedtke, C.; Weiskirchen, R. The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 902–914. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Fernández, R.; Schulzer, M.; Kuramoto, L.; Cragg, J.; Ramachandiran, N.; Au, W.L.; Mak, E.; McKenzie, J.; McCormick, S.; Sossi, V.; et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann. Neurol. 2011, 69, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-Term Metformin Usage and Cognitive Function among Older Adults with Diabetes. J. Alzheimer’s Dis. 2014, 41, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Kuan, Y.-C.; Huang, K.-W.; Lin, C.-L.; Hu, C.-J.; Kao, C.-H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [PubMed]
- As, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R.V.; Shen, Z.; Cook, L.S.; Doweyko, A.M.; Pitt, S.; et al. 2-Aminothiazole as a Novel Kinase Inhibitor Template. Structure−Activity Relationship Studies toward the Discovery of N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a potent pan-Src Kinase Inhibitor. J. Med. Chem. 2006, 49, 6819–6832. [Google Scholar]
- Han, L.; Schuringa, J.J.; Mulder, A.; Vellenga, E. Dasatinib impairs long-term expansion of leukemic progenitors in a subset of acute myeloid leukemia cases. Ann. Hematol. 2010, 89, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs 2017, 77, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Ay, M.; Charli, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Quercetin. In Nutraceuticals; Gupta, R.C., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 447–452. [Google Scholar]
- Jaffe, R.; Mani, J. Polyphenolics Evoke Healing Responses: Clinical Evidence and Role of Predictive Biomarkers. In Polyphenols in Human Health and Disease; Watson, R.R., Preedy, V.R., Sherma, Z., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 695–705. [Google Scholar]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Wang, W.; Sun, C.; Mao, L.; Ma, P.; Liu, F.; Yang, J.; Gao, Y. The biological activities, chemical stability, metabolism and delivery systems of quercetin: A review. Trends Food Sci. Technol. 2016, 56, 21–38. [Google Scholar] [CrossRef]
- Shankar, G.M.; Antony, J.; Anto, R.J. Quercetin and Tryptanthrin: Two Broad Spectrum Anticancer Agents for Future Chemotherapeutic Interventions. In The Enzymes; Bathaie, S.Z., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 37, pp. 43–72. [Google Scholar]
- Schnekenburger, M.; Diederich, M. Nutritional Epigenetic Regulators in the Field of Cancer: New Avenues for Chemopreventive Approaches. In Epigenetic Cancer Therapy; Gray, S.G., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 393–425. [Google Scholar]
- Hubbard, G.P.; Stevens, J.M.; Cicmil, M.; Sage, T.; Jordan, P.A.; Williams, C.M.; Lovegrove, J.A.; Gibbins, J.M. Quercetin inhibits collagen-stimulated platelet activation through inhibition of multiple components of the glycoprotein VI signaling pathway. J. Thromb. Haemost. 2003, 1, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, G.P.; Wolffram, S.; Lovegrove, J.A.; Gibbins, J.M. Ingestion of quercetin inhibits platelet aggregation and essential components of the collagen-stimulated platelet activation pathway in humans. J. Thromb. Haemost. 2004, 2, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.; Spencer, J.P.; Rice-Evans, C. Flavonoids: Antioxidants or signalling molecules? Free Radic. Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Iannitti, R.; Palumbo, R. Quercetin: A Pleiotropic Kinase Inhibitor Against Cancer BT—Advances in Nutrition and Cancer; Zappia, V., Panico, S., Russo, G.L., Budillon, A., Della Ragione, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 185–205. [Google Scholar]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Andò, S. The G Protein-coupled Receptor GPR30 Mediates c-fos up-regulation by 17β-Estradiol and Phytoestrogens in Breast Cancer Cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Garland, W.; Benezra, R.; Chaudhary, J. Targeting Protein–Protein Interactions to Treat Cancer—Recent Progress and Future Directions. In Annual Reports in Medicinal Chemistry; Desai, M.C., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 48, pp. 227–245. [Google Scholar]
- Kim, H.-N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. 2017, 99, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Del Gaizo Moore, V.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Invest. 2007, 117, 112–121. [Google Scholar] [CrossRef] [PubMed]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.-X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. Fisetin Acts on Multiple Pathways to Reduce the Impact of Age and Disease on CNS Function. Front. Biosci. (Schol. Ed.) 2015, 7, 58–82. [Google Scholar] [CrossRef] [PubMed]
- Naeimi, A.F.; Alizadeh, M. Antioxidant properties of the flavonoid fisetin: An updated review of in vivo and in vitro studies. Trends Food Sci. Technol. 2017, 70, 34–44. [Google Scholar] [CrossRef]
- Mathers, J.C.; Strathdee, G.; Relton, C.L. Induction of Epigenetic Alterations by Dietary and Other Environmental Factors. In Advances in Genetics; Herceg, Z., Ushijima, T., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 71, pp. 3–39. [Google Scholar]
- Kim, J.; Lee, K.W.; Lee, H.J. Polyphenols Suppress and Modulate Inflammation: Possible Roles in Health and Disease. In Polyphenols in Human Health and Disease; Academic Press: San Diego, CA, USA, 2014; pp. 393–408. [Google Scholar]
- Esselen, M.; Barth, S.W. Food-Borne Topoisomerase Inhibitors: Risk or Benefit. In Advances in Molecular Toxicology; Fishbein, J.C., Heilman, J.M., Eds.; Elsevier: New York, NY, USA, 2014; Volume 8, pp. 123–171. [Google Scholar]
- Adhami, V.M.; Syed, D.N.; Khan, N.; Mukhtar, H. Dietary flavonoid fisetin: A novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem. Pharmacol. 2012, 84, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chang, D.J.; Baratte, B.; Meijer, L.; Schulze-Gahmen, U. Crystal Structure of a Human Cyclin-Dependent Kinase 6 Complex with a Flavonol Inhibitor, Fisetin. J. Med. Chem. 2005, 48, 737–743. [Google Scholar] [CrossRef] [PubMed]
- WEBB, M.R.; EBELER, S.E. Comparative analysis of topoisomerase IB inhibition and DNA intercalation by flavonoids and similar compounds: Structural determinates of activity. Biochem. J. 2004, 384, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anticancer Agents Med. Chem. 2013, 13, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Tyagi, A.K.; Deshmukh-Taskar, P.; Hinojosa, M.; Prasad, S.; Aggarwal, B.B. Downregulation of tumor necrosis factor and other proinflammatory biomarkers by polyphenols. Arch. Biochem. Biophys. 2014, 559, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A Dietary Antioxidant for Health Promotion. Antioxid. Redox Signal. 2013, 19, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY) 2016, 8, 2915–2926. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Son, D.J.; Gu, S.M.; Woo, J.R.; Ham, Y.W.; Lee, H.P.; Kim, W.J.; Jung, J.K.; Hong, J.T. Piperlongumine inhibits lung tumor growth via inhibition of nuclear factor kappa B signaling pathway. Sci. Rep. 2016, 6, 26357. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Park, H.; Kim, H.P. Effects of flavonoids on senescence-associated secretory phenotype formation from bleomycin-induced senescence in BJ fibroblasts. Biochem. Pharmacol. 2015, 96, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Pospelova, T.V.; Leontieva, O.V.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Blagosklonny, M.V. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle 2012, 11, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Arriola Apelo, S.I.; Lamming, D.W. Rapamycin: An InhibiTOR of Aging Emerges From the Soil of Easter Island. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, D.; Pan, Z.; Xu, D.; Zhou, Y.; Wu, Y.; Cai, N.; Tang, Q.; Wang, C.; Yan, M.; et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016, 7, e2441. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.R. Targeting aging with metformin (tame). Innov. Aging 2017, 743. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.-M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.J.; Liu, S.; Demaria, M.; Cong, Y.-S.; Kapahi, P.; et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Guarente, L. Small-Molecule Allosteric Activators of Sirtuins. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Pitozzi, V.; Mocali, A.; Laurenzana, A.; Giannoni, E.; Cifola, I.; Battaglia, C.; Chiarugi, P.; Dolara, P.; Giovannelli, L. Chronic Resveratrol Treatment Ameliorates Cell Adhesion and Mitigates the Inflammatory Phenotype in Senescent Human Fibroblasts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science (80-.) 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Mouraret, N.; Marcos, E.; Abid, S.; Gary-Bobo, G.; Saker, M.; Houssaini, A.; Dubois-Rande, J.-L.; Boyer, L.; Boczkowski, J.; Derumeaux, G.; et al. Activation of Lung p53 by Nutlin-3a Prevents andeverses Experimental Pulmonary Hypertension. Circulation 2013, 127, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, R.C.; Watts, A.C.; Murphy, R.J.; Snelling, S.J.; Carr, A.J.; Hulley, P.A. Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: In vivo and in vitro evidence. Ann. Rheum. Dis. 2014, 73, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Mallette, F.A.; Mukhopadhyay, U.K.; Moores, A.; Ferbeyre, G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol. Biol. Cell. 2006, 17, 1583–1592. [Google Scholar] [CrossRef] [PubMed]


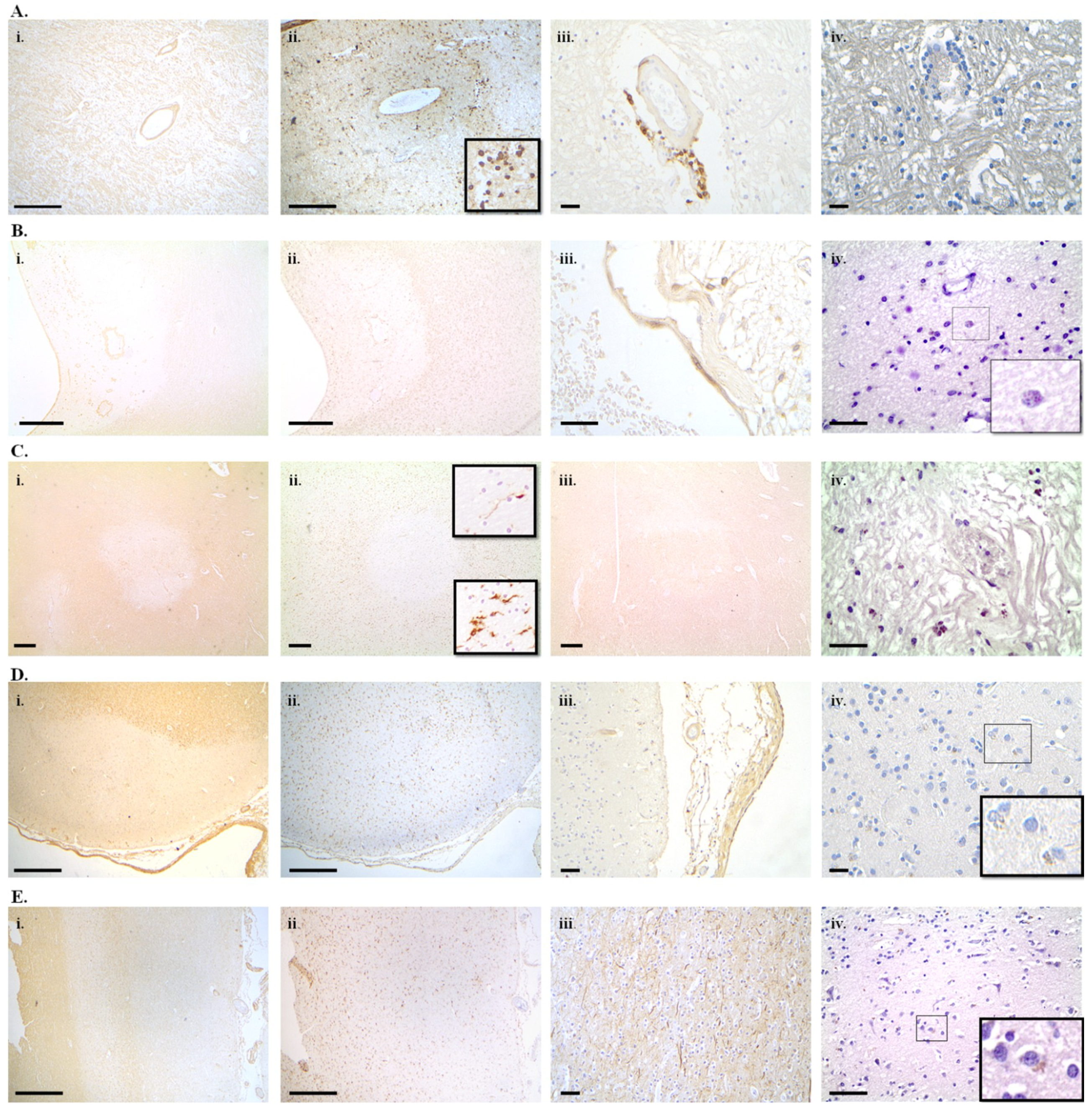{kind=link}
| Compound | MoA | Effect | Current Indication | Classification | References |
|---|---|---|---|---|---|
| Dasatinib (Sprycel) | Tyrosine kinase inhibitor, Inhibitor of Eph receptors | Reduced proliferation of senescent cells in vitro; Alleviated ageing phenotypes in treated animals | Philadelphia chromosome-CML and ALL | senolytic | [307,308,309,310] |
| Quercetin | Modulator of NF-κB, PI3K/Akt, estrogen receptor, mTOR, PIKδ kinase inhibitor, Potent antioxidant | Kills senescent human endothelial cells and murine bone marrow mesenchymal stem cells | experimental | senolytic | [311,312,313,314,315,316,317,318,319,320,321,322,323] |
| Navitoclax (ABT-263) | BCL-2 inhibitor (PPI) | Reduced survival of HUVEC, human lung fibroblasts and murine embryonic fibroblasts and mesenchymal stem cells in vitro | experimental | Senolytic | [324,325,326,327,328,329,330] |
| ABT-737 | BCL-XL inhibitor (PPI) | Reduced viability of senescent cell in vitro and in vivo | experimental | senolytic | [331,332,333,334] |
| A1331852 and A1155463 | BCL-XL inhibitor | Reduced viability of senescent cell in vitro. | experimental | senolytic | [335] |
| Fisetin | Interacts with: topoisomerases, cyclin-dependent kinases, NF-kB, PPAR, PARP1, PI3K/Akt/mTOR, antioxidant | Delay of age-related CNS complications in vivo | experimental | senomorphic | [335,336,337,338,339,340,341,342,343,344,345,346] |
| Piperlongumine | NF-kB modulator | Induces apoptosis in aged cells | experimental | senolytic | [347,348] |
| Geldanamycin | HSP90 inhibitor, down-regulation of PI3K/Akt | Induces death of senescent cells in vitro | experimental | senolytic | [349,350] |
| Tanespimycin (17-AAG) | HSP90 inhibitor, down-regulation of PI3K/Akt | Induces death of senescent cells in vitro | experimental | senolytic | [349,350] |
| Panobinostat (Farydak) | non-selective histone deacetylase inhibitor | Synergistic effect with taxol in inducing death of senescent cells, in vivo | multiple myeloma | senolytic | [351] |
| Apigenin and Kaempferol | Interference with the NF-kB p65 subunit and IkB | Inhibited components of SASP such as in IL-6, CXCL and GM-CSF in vivo. | experimental | senomorphics | [352] |
| Rapamycin (Rapamune) | mTOR kinase inhibitor | suppression of replicative senescence of rodent embryonic cells, lifespan extension in model systems | Lymphangio-myomatosis, coronary stent clot prevention, prevention of transplant rejection | senomorphic | [353,354,355,356] |
| Ruxolitinib (Jakafi) | JAK inhibitor | decreased systemic inflammation in aged animals and improved age-related dysfunctions and alleviating frailty | Polycytemia vera, Myelofibrosis | senomorphic | [357] |
| Metformin (GlucophageTM) | Inhibition of phosphorylation of IkB kinase | Increases lifespan, inhibits SASP, Prevents senescence in a disk degeneration model | Diabetes mellitus type 2 | senolytic | [358,359,360,361] |
| Cortisol and corticosterone | Prevented senescence of human fibroblasts in vitro | Inflammation, allergy | senomorphic | [362] | |
| Resveratrol and derivatives | SIRT1 and IkB inhibitor | Attenuates SASP in human fibroblasts in vitro | Dietary supplement | Senomorphic/senolytic/senescence modulator | [363,364,365] |
| Loperamide | Opioid receptor agonist and Ca++ channel blocker | Prevented senescence of primary MEFs in vitro | antidiarrheal | senomorphic | [349] |
| Niguldipine | Ca++ channel blocker, a1 adrenoreceptor antagonist | Prevented senescence of primary MEFs in vitro | experimental | senomorphic | [349] |
| Nutlin3a | p53 stabilizer | Prevented or inverted pulmonary hypertension in an in vivo model | experimental | Inducer of senescence | [366,367,368] |
| Dexamethasone | SIRT1 inhibition and p53/p21WAF/CIP1 activation | Increased percentage of senescent tenocytes in vitro and in vivo | Anti-edematous, anti-inflammatory | Inducer of senescence | [369] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kritsilis, M.; V. Rizou, S.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. https://doi.org/10.3390/ijms19102937
Kritsilis M, V. Rizou S, Koutsoudaki PN, Evangelou K, Gorgoulis VG, Papadopoulos D. Ageing, Cellular Senescence and Neurodegenerative Disease. International Journal of Molecular Sciences. 2018; 19(10):2937. https://doi.org/10.3390/ijms19102937
Chicago/Turabian StyleKritsilis, Marios, Sophia V. Rizou, Paraskevi N. Koutsoudaki, Konstantinos Evangelou, Vassilis G. Gorgoulis, and Dimitrios Papadopoulos. 2018. "Ageing, Cellular Senescence and Neurodegenerative Disease" International Journal of Molecular Sciences 19, no. 10: 2937. https://doi.org/10.3390/ijms19102937
APA StyleKritsilis, M., V. Rizou, S., Koutsoudaki, P. N., Evangelou, K., Gorgoulis, V. G., & Papadopoulos, D. (2018). Ageing, Cellular Senescence and Neurodegenerative Disease. International Journal of Molecular Sciences, 19(10), 2937. https://doi.org/10.3390/ijms19102937