G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes

 ,
,
Abstract

{kind=link}
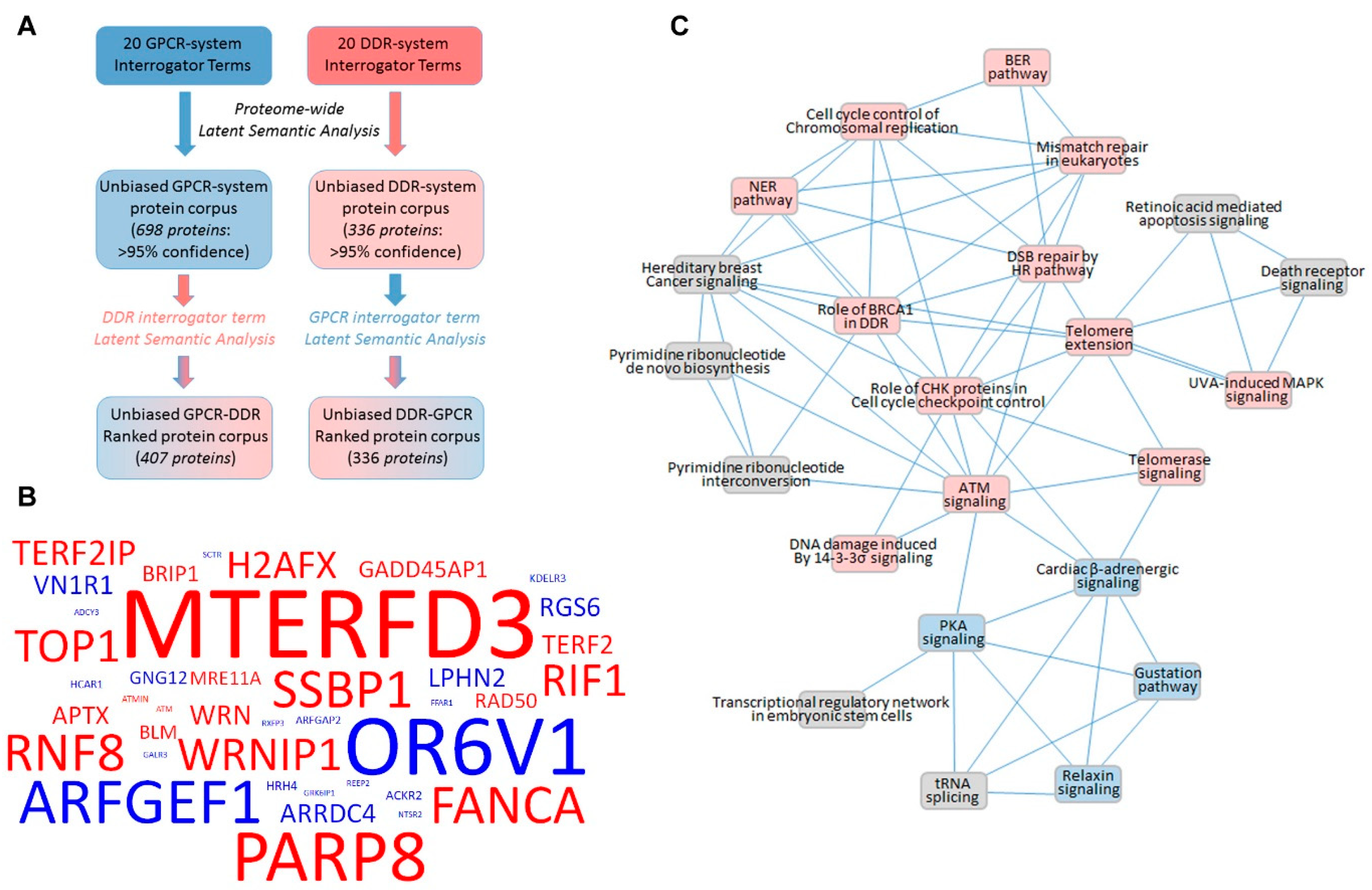{kind=link}
1. Introduction
2. Aging, Metabolic Functionality, DNA Stability, Damage and Repair
2.1. Metabolic Dysfunction, Oxidative Damage and Aging
2.2. Oxidative Aging and DNA Damage Responses
2.3. Metabolic-Clock Process Linked with DDR
3. G Protein-Coupled Receptor Systems: Intersections with DNA Damage and Repair Processes
3.1. GPCR Signaling Diversity
3.2. GPCR Functionality in the Context of Molecular Gerontology
3.3. GPCR Signaling Systems and DNA Damage Repair
3.3.1. Heptahelical GPCRs and DNA Damage
Lysophosphatidic Acid Receptor
Dopamine D2 Receptor
CXCR4 Receptor
Hydroxycarboxylic Acid (Lactate) Receptor
Melanocortin 1 Receptor
Angiotensin II Receptor
3.3.2. β-Arrestin Family Proteins
3.3.3. G Protein-Coupled Receptor Kinases and Associated Proteins
3.3.4. Regulator of G Protein Signaling Proteins
3.3.5. Non-Canonical GPCR-Interacting Proteins
Regulated in Development and DNA Damage Responses
Fanconi Anemia A Protein
Poly(ADP-ribose) Polymerase 1 Protein
Angiotensin II Type 2 Receptor-Interacting Protein
4. The GPCR-DDR Signaling Intersection and Its Potential Therapeutic Exploitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Van Dijk, G.; van Heijningen, S.; Reijne, A.C.; Nyakas, C.; van der Zee, E.A.; Eisel, U.L. Integrative neurobiology of metabolic diseases, neuroinflammation, and neurodegeneration. Front. Neurosci. 2015, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- World Report on Ageing and Health. Available online: http://www.who.int/ageing/events/world-report-2015-launch/en/ (accessed on 30 September 2015).
- Nkuipou-Kenfack, E.; Koeck, T.; Mischak, H.; Pich, A.; Schanstra, J.P.; Zürbig, P.; Schumacher, B. Proteome analysis in the assessment of ageing. Ageing Res. Rev. 2014, 18, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, W.; Martin, B.; Chapter, M.C.; Park, S.-S.; Wang, L.; Daimon, C.M.; Brenneman, R.; Maudsley, S. GIT2 Acts as a Potential Keystone Protein in Functional Hypothalamic Networks Associated with Age-Related Phenotypic Changes in Rats. PLoS ONE 2012, 7, e36975. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-R.; Li, K.; Lin, S.Y.; Hung, W.C. Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci. 2016, 17, 685. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Martin, B.; Janssens, J.; Etienne, H.; Jushaj, A.; van Gastel, J.; Willemsen, A.; Chen, H.; Gesty-Palmer, D.; Luttrell, L.M. Informatic deconvolution of biased GPCR signaling mechanisms from in vivo pharmacological experimentation. Methods 2016, 92, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Martin, B.; Gesty-Palmer, D.; Cheung, H.; Johnson, C.; Patel, S.; Becker, K.G.; Wood, W.H., 3rd; Zhang, Y.; Lehrmann, E.; et al. Delineation of a Conserved Arrestin-Biased Signaling Repertoire In Vivo. Mol. Pharmacol. 2015, 87, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ishida, M.; Tashiro, S.; Yoshizumi, M.; Kihara, Y. Role of DNA damage in cardiovascular disease. Circ. J. 2014, 8, 42–50. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Dogra, N.; Singh, S. The Mitochondrial Unfolded Protein Response: Role in Cellular Homeostasis and Disease. Curr. Mol. Med. 2017, 17, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H. The role of DNA-PK in aging and energy metabolism. FEBS J. 2018, 285, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Awate, S.; Brosh, R.M., Jr. Interactive Roles of DNA Helicases and Translocases with the Single-Stranded DNA Binding Protein RPA in Nucleic Acid Metabolism. Int. J. Mol. Sci. 2017, 18, 1233. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Honda, S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999, 13, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Sohal, R.S. Extension of lifespan by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Seroude, L.; Benzer, S. Extended life-span and stress resistance in the drosophila mutant methuselah. Science 1998, 282, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Klopp, R.G.; Weindruch, R.; Prolla, T.A. Gene expression profile of aging and its retardation by caloric restriction. Science 1999, 285, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.B.; Summer, W.; Cutler, R.G.; Martin, B.; Hyun, D.H.; Dixit, V.D.; Pearson, M.; Nassar, M.; Telljohann, R.; Maudsley, S.; et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 2007, 42, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Carlson, O.; Martin, B.; Stote, K.S.; Golden, E.; Maudsley, S.; Najjar, S.S.; Ferrucci, L.; Ingram, D.K.; Longo, D.L.; Rumpler, W.V.; et al. Impact of reduced meal frequency without caloric restriction on glucose regulation in healthy, normal-weight middle-aged men and women. Metabolism 2007, 56, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Huffman, D.M.; Muzumdar, R.H.; Bartke, A. The critical role of metabolic pathways in aging. Diabetes 2012, 61, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Terman, A. Catabolic insufficiency and aging. Ann. N. Y. Acad. Sci. 2006, 1067, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kühlbrandt, W. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 15301–15306. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2107, 2010, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Viña, J.; Borras, C.; Abdelaziz, K.M.; Garcia-Valles, R.; Gomez-Cabrera, M.C. The free radical theory of aging revisited: The cell signaling disruption theory of aging. Antioxid. Redox Signal 2013, 19, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, W.; Zhou, Y.; Park, S.S.; Wang, L.; Mitchell, N.; Stone, M.D.; Becker, K.G.; Martin, B.; Maudsley, S. Minimal Peroxide Exposure of Neuronal Cells Induces Multifaceted Adaptive Responses. PLoS ONE 2010, 5, e14352. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Seviour, E.G.; Lin, S.Y. The DNA damage response: Balancing the scale between cancer and ageing. Aging 2010, 2, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Ambekar, S.S.; Hattur, S.S.; Bule, P.B. DNA: Damage and Repair Mechanisms in Humans. Glob. J. Pharm. Pharm. Sci. 2017, 3, 555613. [Google Scholar]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Nickoloff, J.A. DNA Repair, Genetic Instability, and Cancer; World Scientific: River Edge, NJ, USA, 2007; pp. 119–156. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Mari, P.-O.; Florea, B.I.; Persengiev, S.P.; Verkaik, N.S.; Brüggenwirth, H.T.; Modesti, M.; Giglia-Mari, G.; Bezstarosti, K.; Demmers, J.A.; Luider, T.M.; et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. USA 2006, 103, 18597–18602. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [PubMed]
- Aubert, G.; Lansdorp, P.M. Telomeres and Aging. Physiol. Rev. 2008, 88, 557–579. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomeres. Curr. Opin. Cell Biol. 1991, 3, 444–451. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2018. pii:S0047-6374(18)30052-6. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.; Stier, A. Does oxidative stress shorten telomeres in vivo? A review. Biol. Lett. 2017, 13, 20170463. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.K.; Meeker, A. Telomeres and telomerase in prostate cancer development and therapy. Nat. Rev. Urol. 2017, 14, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef] [PubMed]
- Cattan, V.; Mercier, N.; Gardner, J.P.; Regnault, V.; Labat, C.; Maki-Jouppila, J.; Nzietchueng, R.; Benetos, A.; Kimura, M.; et al. Chronic oxidative stress induces a tissue-specific reduction in telomere length in CAST/Ei mice. Free Radic. Biol. Med. 2008, 44, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Kaul, Z.; Cesare, A.J.; Huschtscha, L.I.; Neumann, A.A.; Reddel, R.R. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012, 13, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Lidzbarsky, G.; Gutman, D.; Shekhidem, H.A.; Sharvit, L.; Atzmon, G. Genomic Instabilities, Cellular Senescence, and Aging: In Vitro, In Vivo and Aging-Like Human Syndromes. Front. Med. 2018, 5, 104. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Caux, F.; Dubosclard, E.; Lascols, O.; Buendia, B.; Chazouilleres, O.; Cohen, A.; Courvalin, J.C.; Laroche, L.; Capeau, J.; Vigouroux, C.; et al. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J. Clin. Endocrinol. Metab. 2003, 88, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, M.; Kamei, S.; Kinoshita, T.; Sanada, J.; Fushimi, Y.; Irie, S.; Hirata, Y.; Tanabe, A.; Hirukawa, H.; Kimura, T.; et al. Werner Syndrome and Diabetes Mellitus Accompanied by Adrenal Cortex Cancer. Intern. Med. 2017, 56, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Okamoto, M.; Yamada, K.; Yoshimasa, Y.; Kosaki, A.; Kono, S.; Inoue, G.; Maeda, I.; Kubota, M.; Hayashi, T.; et al. Insulin resistance in Werner’s syndrome. Mech. Ageing Dev. 1992, 63, 11–25. [Google Scholar] [CrossRef]
- Hayashi, A.; Takemoto, M.; Shoji, M.; Hattori, A.; Sugita, K.; Yokote, K. Pioglitazone improves fat tissue distribution and hyperglycemia in a case of cockayne syndrome with diabetes. Diabetes Care 2015, 38, e76. [Google Scholar] [CrossRef] [PubMed]
- Schalch, D.S.; McFarlin, D.E.; Barlow, M.H. An unusual form of diabetes mellitus in ataxia telangiectasia. N. Engl. J. Med. 1970, 282, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Bar, R.S.; Levis, W.R.; Rechler, M.M.; Harrison, L.C.; Siebert, C.; Podskalny, J.; Roth, J.; Muggeo, M. Extreme insulin resistance in ataxia telangiectasia: Defect in affinity of insulin receptors. N. Engl. J. Med. 1978, 298, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Arellanes-Licea, E.; Caldelas, I.; De Ita-Pérez, D.; Díaz-Muñoz, M. The circadian timing system: A recent addition in the physiological mechanisms underlying pathological and aging processes. Aging Dis. 2014, 5, 406–418. [Google Scholar] [PubMed]
- Kang, T.-H.; Reardon, J.T.; Kemp, M.; Sancar, A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. USA 2009, 106, 2864–2867. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Hogenesch, J.B.; Kay, S.A. Circadian rhythms from flies to human. Nature 2002, 417, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Hirayama, J.; Nishina, H. A Common Origin: Signaling Similarities in the Regulation of the Circadian Clock and DNA Damage Responses. Biol. Pharm. Bull. 2010, 33, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Taguchi, A.; Matsumura, T.; Nakabayashi, H.; Akiyama, M.; Yamamoto, K.; Fujimoto, R.; Suetomi, R.; Yanai, A.; Shinoda, K.; et al. Clock Gene Dysregulation Induced by Chronic ER Stress Disrupts β-cell Function. EBioMedicine 2017, 18, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Ingenwerth, M.; Reinbeck, A.L.; Stahr, A.; Partke, H.J.; Roden, M.; Burkart, V.; von Gall, C. Perturbation of the molecular clockwork in the SCN of non-obese diabetic mice prior to diabetes onset. Chronobiol. Int. 2016, 33, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Saini, C.; Petrenko, V.; Pulimeno, P.; Giovannoni, L.; Berney, T.; Hebrok, M.; Howald, C.; Dermitzakis, E.T.; Dibner, C. A functional circadian clock is required for proper insulin secretion by human pancreatic islet cells. Diabetes Obes. Metab. 2016, 18, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Kohsaka, A.; Bhawal, U.K.; Muragaki, Y. Potential Roles of Dec and Bmal1 Genes in Interconnecting Circadian Clock and Energy Metabolism. Int. J. Mol. Sci. 2018, 19, 781. [Google Scholar] [CrossRef] [PubMed]
- Krishnaiah, S.Y.; Wu, G.; Altman, B.J.; Growe, J.; Rhoades, S.D.; Coldren, F.; Venkataraman, A.; Olarerin-George, A.O.; Francey, L.J.; Mukherjee, S.; et al. Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab. 2017, 25, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Chang, H.C. Physiological links of circadian clock and biological clock of aging. Protein Cell 2017, 8, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Tevy, M.F.; Giebultowicz, J.; Pincus, Z.; Mazzoccoli, G.; Vinciguerra, M. Aging signaling pathways and circadian clock-dependent metabolic derangements. Trends Endocrinol. Metab. 2013, 24, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-Fat Diet Disrupts Behavioral and Molecular Circadian Rhythms in Mice. Cell Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Yanagihara, H.; Hayashi, Y.; Obi, Y.; Tsuruoka, S.; Takamura, T.; Kaneko, S.; Fujimura, A. Rhythmic messenger ribonucleic acid expression of clock genes and adipocytokines in mouse visceral adipose tissue. Endocrinology 2005, 146, 5631–5636. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Bee, L.; Marini, S.; Pontarin, G.; Ferraro, P.; Costa, R.; Albrecht, U.; Celotti, L. Nucleotide excision repair efficiency in quiescent human fibroblasts is modulated by circadian clock. Nucleic Acids Res. 2015, 43, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Im, J.-S.; Jung, B.-H.; Kim, S.-E.; Lee, K.-H.; Lee, J.-K. Per3, a circadian gene, is required for Chk2 activation in human cells. FEBS Lett. 2010, 584, 4731–4734. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.M.; Lee, G.; Bhin, J.; Oh, S.K.; Kim, K.; Pyo, K.E.; Lee, J.S.; Yim, H.Y.; Kim, K.I.; et al. DNA Damage-Induced RORα Is Crucial for p53 Stabilization and Increased Apoptosis. Mol. Cell 2011, 44, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Geyfman, M.; Kumar, V.; Liu, Q.; Ruiz, R.; Gordon, W.; Espitia, F.; Cam, E.; Millar, S.E.; Smyth, P.; Ihler, A. Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc. Natl. Acad. Sci. USA 2012, 109, 11758–11763. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; Boulton, S.J. Emerging links between the biological clock and the DNA damage response. Chromosoma 2007, 116, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, Y. From clock genes to telomeres in the regulation of the healthspan. Nutr. Rev. 2012, 70, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Khapre, R.V.; Samsa, W.E.; Kondratov, R.V. Circadian regulation of cell cycle: Molecular connections between aging and the circadian clock. Ann. Med. 2010, 42, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Lim, M.M.; Yang, G.; Bauer, A.Q.; Qi, L.; Lee, Y.; Roh, J.H.; Ortiz-Gonzalez, X.; Dearborn, J.T.; Culver, J.P.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Investig. 2013, 123, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Hood, S.; Amir, S. Neurodegeneration and the Circadian Clock. Front. Aging Neurosci. 2017, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Pagano, E.S.; Spinedi, E.; Gagliardino, J.J. At the Cutting Edge White Adipose Tissue and Circadian Rhythm Dysfunctions in Obesity: Pathogenesis and Available Therapies. Neuroendocrinology 2017, 104, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Ripperger, J.A.; Hoegger, D.C.; Bruegger, P.; Buch, T.; Birchler, T.; Mueller, A.; Albrecht, U.; Contaldo, C.; Brown, S.A. NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. USA 2013, 110, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; West, M.D.; Allsopp, R.C.; Davison, T.S.; Wu, Y.S.; Arrowsmith, C.H.; Poirier, G.G.; Benchimol, S. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. 1997, 16, 6018–6033. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.; Gupta, R.; Makwana, K.; Kondratov, R. Circadian clocks, diets and aging. Nutr. Heal. Aging 2017, 4, 101–112. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Nohara, K.; Park, N.; Park, Y.S.; Guillory, B.; Zhao, Z.; Garcia, J.M.; Koike, N.; Lee, C.C.; Takahashi, J.S. The Small Molecule Nobiletin Targets the Molecular Oscillator to Enhance Circadian Rhythms and Protect against Metabolic Syndrome. Cell Metab. 2016, 23, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Gloston, G.F.; Yoo, S.-H.; Chen, Z.J. Clock-Enhancing Small Molecules and Potential Applications in Chronic Diseases and Aging Front. Neurol. 2017, 8, 100. [Google Scholar]
- Vass, M.; Kooistra, A.J.; Yang, D.; Stevens, R.C.; Wang, M.W.; de Graaf, C. Chemical Diversity in the G Protein-Coupled Receptor Superfamily. Trends Pharmacol. Sci. 2018, 39, 494–512. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Martin, B.; Luttrell, L.M. The origins of diversity and specificity in g protein-coupled receptor signaling. J. Pharmacol. Exp. Ther. 2005, 314, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 2010, 62, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Kenakin, T.P. Refining efficacy: Allosterism and bias in G protein-coupled receptor signaling. Methods Mol. Biol. 2011, 756, 3–35. [Google Scholar] [PubMed]
- Maudsley, S.; Patel, S.A.; Park, S.S.; Luttrell, L.M.; Martin, B. Functional signaling biases in G protein-coupled receptors: Game Theory and receptor dynamics. Mini. Rev. Med. Chem. 2012, 12, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Maudsley, S.; Gesty-Palmer, D. Translating in vitro ligand bias into in vivo efficacy. Cell Signal 2018, 41, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Ward, R.; An, S.; Guo, X.X.; Li, W.; Xu, T.R. Biased signalling: The instinctive skill of the cell in the selection of appropriate signalling pathways. Biochem. J. 2015, 470, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Gent, J.P.; Findlay, J.B.; Donnelly, D. The relationship between the agonist-induced activation and desensitization of the human tachykinin NK2 receptor expressed in Xenopus oocytes. Br. J. Pharmacol. 1998, 124, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Dunn, H.; Ferguson, S.S. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br. J. Pharmacol. 2012, 165, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Sachs, B.D.; Caron, M.G.; Lefkowitz, R.J. Pharmacological blockade of a β(2)AR-β-arrestin-1 signaling cascade prevents the accumulation of DNA damage in a behavioral stress model. Cell Cycle 2013, 12, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Zhang, Z.; Wu, Y.; Kang, J.; Pei, G. Beta-arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-kappaB activation. EMBO J. 2005, 24, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Stäubert, C.; Schöneberg, T. GPCR Signaling From Intracellular Membranes—A Novel Concept. BioEssays 2017, 39, 1700200. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, A.M.; Halls, M.L. Compartmentalization of GPCR signalling controls unique cellular responses. Biochem. Soc. Trans. 2016, 44, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Nishimoto, S.K.; Quarles, L.D. GPRC6A: Jack of all metabolism (or master of none). Mol. Metab. 2016, 6, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Reimann, F.; Gribble, F.M. G protein-coupled receptors as new therapeutic targets for type 2 diabetes. Diabetologia 2016, 59, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Amisten, S.; Neville, M.; Hawkes, R.; Persaud, S.J.; Karpe, F.; Salehi, A. An atlas of G-protein coupled receptor expression and function in human subcutaneous adipose tissue. Pharmacol. Ther. 2015, 146, 61–93. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.D.; Ulven, T.; Milligan, G. The therapeutic potential of allosteric ligands for free fatty acid sensitive GPCRs. Curr. Top. Med. Chem. 2013, 13, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Van Gastel, J.; Janssens, J.; Etienne, H.; Azmi, A.; Maudsley, S. The synergistic GIT2-RXFP3 system in the brain and its importance in age-related disorders. Front. Aging Neurosci. 2016. [Google Scholar] [CrossRef]
- Janssens, J.; Etienne, H.; Idriss, S.; Azmi, A.; Martin, B.; Maudsley, S. Systems-Level G Protein-Coupled Receptor Therapy Across a Neurodegenerative Continuum by the GLP-1 Receptor System. Front. Endocrinol. 2014, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R.; Perona, J.S.; Sánchez-Dominguez, J.M.; Montero, E.; Cañizares, J.; Bressani, R.; Escribá, P.V.; Ruiz-Gutierrez, V. G protein-coupled receptor systems and their lipid environment in health disorders during aging. Biochim. Biophys. Acta 2007, 1768, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J.; Jang, I.S.; Lim, H.K.; Ha, K.S.; Park, S.C. Agonist-specific differential changes of cellular signal transduction pathways in senescent human diploid fibroblasts. Exp. Gerontol. 2002, 37, 871–883. [Google Scholar] [CrossRef]
- Yeo, E.J.; Park, S.C. Age-dependent agonist-specific dysregulation of membrane-mediated signal transduction: Emergence of the gate theory of aging. Mech. Ageing Dev. 2002, 123, 1563–1578. [Google Scholar] [CrossRef]
- Hakim, M.A.; Buchholz, J.N.; Behringer, E.J. Electrical dynamics of isolated cerebral and skeletal muscle endothelial tubes: Differential roles of G-protein-coupled receptors and K+ channels. Pharmacol. Res. Perspect. 2018, 6, e00391. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Huang, X.; Huang, L.; Yang, J.; Li, A.; Shen, K.; Wedegaertner, P.B.; Jiang, X. G protein-coupled receptor kinase 4-induced cellular senescence and its senescence-associated gene expression profiling. Exp. Cell Res. 2017, 360, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D. Healing and hurting: Molecular mechanisms, functions, and pathologies of cellular senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espin, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Niederländer, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pan, K.H.; Cohen, S.N. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc. Natl. Acad. Sci. USA 2003, 100, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, Z.; Xu, B.; Hu, H.; Wei, Z.; Liu, Q.; Zhang, X.; Ding, X.; Wang, Y.; Zhao, M.; et al. Chemokine receptor CXCR2 is transactivated by p53 and induces p38-mediated cellular senescence in response to DNA damage. Aging Cell 2013, 12, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Lee, H.J.; Heo, J.; Lim, J.; Kim, M.; Kim, M.K.; Nam, H.Y.; Hong, G.H.; Cho, Y.S.; Choi, S.J.; et al. Senescence-Associated MCP-1 Secretion Is Dependent on a Decline in BMI1 in Human Mesenchymal Stromal Cells. Antioxid Redox Signal 2016, 24, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Shaker, M.E.; Mehal, W.Z. Therapeutic Opportunities in Damage-Associated Molecular Pattern-Driven Metabolic Diseases. Antioxid Redox Signal 2015, 23, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.T.; Staples, C.J. Approaches for Identifying Novel Targets in Precision Medicine: Lessons from DNA Repair. Adv. Exp. Med. Biol. 2017, 1007, 1–16. [Google Scholar] [PubMed]
- Gold, M. Phase II clinical trials of anti-amyloid β antibodies: When is enough, enough? Alzheimers Dement. (NY). 2017, 3, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.; Lu, D.; Ni, B.; Chadwick, W.; Siddiqui, S.; Azmi, A.; Etienne, H.; Jushaj, A.; van Gastel, J.; Martin, B. Development of Precision Small-Molecule Proneurotrophic Therapies for Neurodegenerative Diseases. Vitam. Horm. 2017, 104, 263–311. [Google Scholar] [PubMed]
- Chadwick, W.; Mitchell, N.; Martin, B.; Maudsley, S. Therapeutic targeting of the endoplasmic reticulum in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015, 98, 541–555. [Google Scholar] [CrossRef] [PubMed]
- De Pascali, F.; Reiter, E. β-arrestins and biased signalling in gonadotropin receptors. Minerva Ginecol. 2018. [Google Scholar] [CrossRef]
- Luttrell, L.M. Minireview: More than just a hammer: Ligand “bias” and pharmaceutical discovery. Mol. Endocrinol. 2014, 28, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ‘small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Chadwick, W.; Janssens, J.; Premont, R.T.; Schmalzigaug, R.; Becker, K.G.; Lehrmann, E.; Wood, W.H.; Zhang, Y.; Siddiqui, S.; et al. GIT2 Acts as a Systems-Level Coordinator of Neurometabolic Activity and Pathophysiological Aging. Front. Endocrinol. 2015, 6, 191. [Google Scholar] [CrossRef] [PubMed]
- Han, J.D.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Ferguson, S.S. Minireview: Role of intracellular scaffolding proteins in the regulation of endocrine G protein-coupled receptor signaling. Mol. Endocrinol. 2015, 29, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Zamah, A.M.; Rahman, N.; Blitzer, J.T.; Luttrell, L.M.; Lefkowitz, R.J.; Hall, R.A. Platelet-derived growth factor receptor association with Na(+)/H(+) exchanger regulatory factor potentiates receptor activity. Mol. Cell Biol. 2000, 20, 8352–8363. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qiao, Y.; Li, Z. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 2018, 39, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, D.A.; Gosmanova, E.; Johnson, L.R.; Tigyi, G. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am. J. Physiol. Liver Physiol. 2003, 284, G821–G829. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.E.; Herr, D.R.; Chun, J. Lysophosphatidic acid (LPA) receptors: Signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010, 91, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Poppleton, H.; Yasuda, S.; Makarova, N.; Shinozuka, Y.; Wang, D.A.; Johnson, L.R.; Patel, T.B.; Tigyi, G. Optimal lysophosphatidic acid-induced DNA synthesis and cell migration but not survival require intact autophosphorylation sites of the epidermal growth factor receptor. J. Biol. Chem. 2004, 279, 47871–47880. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.C.; Li, M.; Asaithamby, A. New insights into the roles of ATM and DNA-PKcs in the cellular response to oxidative stress. Cancer Lett. 2012, 327, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Balogh, A.; Shimizu, Y.; Lee, S.C.; Norman, D.D.; Gangwar, R.; Bavaria, M.; Moon, C.; Shukla, P.; Rao, R.; Ray, R. The autotaxin-LPA2 GPCR axis is modulated by γ-irradiation and facilitates DNA damage repair. Cell Signal 2015, 27, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Szabó, E.; Fells, J.I.; Balogh, A.; Lim, K.G.; Fujiwara, Y.; Norman, D.D.; Lee, S.C.; Balazs, L.; Thomas, F.; et al. Combined mitigation of the gastrointestinal and hematopoietic acute radiation syndromes by an LPA2 receptor-specific nonlipid agonist. Chem. Biol. 2015, 22, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Likhite, N.; Jackson, C.A.; Liang, M.S.; Krzyzanowski, M.C.; Lei, P.; Wood, J.F.; Birkaya, B.; Michaels, K.L.; Andreadis, S.T.; Clark, S.D.; et al. The protein arginine methyltransferase PRMT5 promotes D2-like dopamine receptor signaling. Sci. Signal 2015, 8, ra115. [Google Scholar] [CrossRef] [PubMed]
- Yong, M.; Yu, T.; Tian, S.; Liu, S.; Xu, J.; Hu, J.; Hu, L. DR2 blocker thioridazine: A promising drug for ovarian cancer therapy. Oncol. Lett. 2017, 14, 8171–8177. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.; Kulbe, H.; Wong, D.; Chakravarty, P.; Balkwill, F. An antagonist of the chemokine receptor CXCR4 induces mitotic catastrophe in ovarian cancer cells. Mol. Cancer Ther. 2009, 8, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dépond, M.; He, L.; Foudi, A.; Kwarteng, E.O.; Lauret, E.; Plo, I.; Desterke, C.; Dessen, P.; Fujii, N.; et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci. Rep. 2016, 6, 37827. [Google Scholar] [CrossRef] [PubMed]
- Proia, P.; Di Liegro, C.M.; Schiera, G.; Fricano, A.; Di Liegro, I. Lactate as a Metabolite and a Regulator in the Central Nervous System. Int. J. Mol Sci. 2016, 17, 1450. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Nguyen, T.; Lynch, K.R.; Cheng, R.; Vanti, W.B.; Arkhitko, O.; Lewis, T.; Evans, J.F.; George, S.R.; O’Dowd, B.F. Discovery and mapping of ten novel G protein-coupled receptor genes. Gene 2001, 275, 83–91. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Lee, K.; Martin, B.; Maudsley, S.; Golden, E.; Cutler, R.G.; Mattson, M.P. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus 2009, 19, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Peng, X.; Xiang, W.; Han, J.; Li, K. The effect of resistance training on cognitive function in the older adults: A systematic review of randomized clinical trials. Aging Clin. Exp. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Ciszewski, W.M.; Kania, K.D. L- and D-lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun. Signal 2015, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Kania, K.D.; Ciszewski, W.M. Stimulation of lactate receptor (HCAR1) affects cellular DNA repair capacity. DNA Repair 2017, 52, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Kania, K.D.; Blauz, A.; Ciszewski, W.M. The lactate receptor (HCAR1/GPR81) contributes to doxorubicin chemoresistance via ABCB1 transporter up-regulation in human cervical cancer HeLa cells. J. Physiol. Pharmacol. 2017, 68, 555–564. [Google Scholar] [PubMed]
- Swope, V.B.; Abdel-Malek, Z.A. Significance of the Melanocortin 1 and Endothelin B Receptors in Melanocyte Homeostasis and Prevention of Sun-Induced Genotoxicity. Front Genet. 2016, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Kadekaro, A.L.; Chen, J.; Yang, J.; Chen, S.; Jameson, J.; Swope, V.B.; Cheng, T.; Kadakia, M.; Abdel-Malek, Z. Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol. Cancer Res. 2012, 10, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.S.; Maudsley, S.; An, S.S.; Santhanam, L.; Martin, B.; Faulkner, S.; et al. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.M.; Kuller, L.H.; Barinas-Mitchell, E.J.; McDade, E.M.; Klunk, W.E.; Cohen, A.D.; Mathis, C.A.; Dekosky, S.T.; Price, J.C.; Lopez, O.L. Arterial stiffness and β-amyloid progression in nondemented elderly adults. JAMA Neurol. 2014, 71, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, G.; Stopper, H.; Schinzel, R.; Ni, C.W.; Jo, H.; Schupp, N. Angiotensin II induces DNA damage via AT1 receptor and NADPH oxidase isoform Nox4. Mutagenesis 2012, 27, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.E.; Mistry, Y.; Hastings, R.; Poolman, T.; Niklason, L.; Williams, B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 2008, 102, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Yuri, K.; Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein–Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar]
- Fan, Y.; Huang, Z.; Long, C.; Ning, J.; Zhang, H.; Kuang, X.; Zhang, Q.; Shen, H. ID2 protects retinal pigment epithelium cells from oxidative damage through p-ERK1/2/ID2/NRF2. Arch. Biochem. Biophys. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature 2011, 477, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Ritov, G.; Richter-Levin, G.; Barki-Harrington, L. Selective increase in the association of the β2 adrenergic receptor, β Arrestin-1 and p53 with Mdm2 in the ventral hippocampus one month after underwater trauma. Behav. Brain Res. 2013, 240, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, C.; Garcia-Borron, J.C.; Jiménez-Cervantes, C.; Olivares, C. MC1R signaling. Intracellular partners and pathophysiological implications. Biochim. Biophys. Acta 2017, 1863, 2448–2461. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wang, L.; Zhang, J.; Dong, W.; Zhang, T.; Ni, Y.; Cao, H.; Wang, K.; Li, Y.; Wang, Y.; et al. ARRB1 enhances the chemosensitivity of lung cancer through the mediation of DNA damageresponse. Oncol. Rep. 2017, 37, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; Barak, L.S.; Zhang, J.; Caron, M.G. G-protein-coupled receptor regulation: Role of G-protein-coupled receptor kinases and arrestins. Can. J. Physiol. Pharmacol. 1996, 74, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Inglese, J.; Lefkowitz, R.J. Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 1995, 9, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Mayor, F. Jr. Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sainz, M.C.; Murga, C.; Kavelaars, A.; Jurado-Pueyo, M.; Krakstad, B.F.; Heijnen, C.J.; Mayor, F. Jr.; Aragay, A.M. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G. protein subunits. Mol. Biol. Cell 2006, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Zhu, S.; Rockey, D.C. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat. Med. 2005, 11, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Sallese, M.; Mariggio, S.; Collodel, G.; Moretti, E.; Piomboni, P.; Baccetti, B.; De Blasi, A. G protein-coupled receptor kinase GRK4. Molecular analysis of the four isoforms and ultrastructural localization in spermatozoa and germinal cells. J. Biol. Chem. 1997, 272, 10188–10195. [Google Scholar] [CrossRef] [PubMed]
- Virlon, B.; Firsov, D.; Cheval, L.; Reiter, E.; Troispoux, C.; Guillou, F.; Elalouf, J.M. Rat G protein-coupled receptor kinase GRK4: Identification, functional expression, and differential tissue distribution of two splice variants. Endocrinology 1998, 139, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Sallese, M.; Salvatore, L.; D’Urbano, E.; Sala, G.; Storto, M.; Launey, T.; Nicoletti, F.; Knopfel, T.; De Blasi, A. The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J. 2000, 14, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhu, H.; Yuan, M.; Fu, J.; Zhou, Y.; Ma, L. G-protein-coupled receptor kinase 5 phosphorylates p53 and inhibits DNA damage-induced apoptosis. J. Biol. Chem. 2010, 285, 12823–12830. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Cox, A.A.; Bartelli, N.; Rasul, I.; Festoff, B.W.; Premont, R.T.; Arendash, G.W. GRK5 deficiency leads to early Alzheimer-like pathology and working memory impairment. Neurobiol. Aging 2007, 28, 1873–1888. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rasul, I.; Liu, J.; Zhao, B.; Tang, R.; Premont, R.T.; Suo, W.Z. Augmented axonal defects and synaptic degenerative changes in female GRK5 deficient mice. Brain Res. Bull. 2009, 78, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Peng, W.; Zhang, Q.; Ding, X.; Suo, W.Z. GRK5 deficiency leads to susceptibility to intermittent hypoxia-induced cognitive impairment. Behav. Brain Res. 2016, 302, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Takagi, C.; Urasawa, K.; Yoshida, I.; Takagi, Y.; Kaneta, S.; Nakano, N.; Onozuka, H.; Kitabatake, A. Enhanced GRK5 expression in the hearts of cardiomyopathic hamsters, J2N-k. Biochem. Biophys. Res. Commun. 1999, 262, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Claing, A.; Vitale, N.; Perry, S.J.; Lefkowitz, R.J. The GIT family of ADP-ribosylation factor GTPase-activating proteins. Functional diversity of GIT2 through alternative splicing. J. Biol. Chem. 2000, 275, 22373–22380. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.J.; Schmalzigaug, R.; Roseman, J.T.; Xing, Y.; Claing, A. The GIT/PIX complex: An oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell Signal. 2004, 16, 1001–1011. [Google Scholar]
- Van Gastel, J.; Jushaj, A.; Boddaert, J.; Premont, R.T.; Luttrell, L.M.; Janssens, J.; Martin, B.; Maudsley, S. GIT2—A keystone in ageing and age-related disease. Ageing Res. Rev. 2018, 43, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Cai, H.; Park, S.S.; Siddiqui, S.; Premont, R.T.; Schmalzigaug, R.; Paramasivam, M.; Seidman, M.; Bodogai, I.; Biragyn, A.; et al. Nuclear GIT2 Is an ATM Substrate and Promotes DNA Repair. Mol. Cell. Biol. 2015, 35, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.; Lustig, A.; Carter, A.; Sankar, M.; Daimon, C.M.; Premont, R.T.; Etienne, H.; van Gastel, J.; Azmi, A.; Janssens, J.; et al. Genomic deletion of GIT2 induces a premature age-related thymic dysfunction and systemic immune system disruption. Aging 2017, 9, 706–740. [Google Scholar] [CrossRef] [PubMed]
- Van Gastel, J.; Hendrickx, J.; Leysen, H.; Luttrell, L.M.; Lee, M.-H.M.; Azmi, A.; Janssens, J.; Maudsley, S. The RXFP3-GIT2 signaling system represents a potential multidimensional therapeutic target in age-related disorders. FASEB J. 2018, 32, 1. [Google Scholar]
- Roux, B.T.; Cottrell, G.S. G protein-coupled receptors: What a difference a ‘partner’ makes. Int. J. Mol. Sci. 2014, 15, 1112–1142. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Fisher, R.A. Introduction: G Protein-coupled Receptors and RGS Proteins. Prog. Mol. Biol. Transl. Sci. 2015, 133, 1–11. [Google Scholar] [PubMed]
- Berman, D.M.; Wilkie, T.M.; Gilman, A.G. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 1996, 86, 445–452. [Google Scholar] [CrossRef]
- Wiechec, E.; Overgaard, J.; Hansen, L.L. A fragile site within the HPC1 region at 1q25.3 affecting RGS16, RGSL1, and RGSL2 in human breast carcinomas. Genes Chromosomes Cancer 2008, 47, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, S.; Lu, Y.; Xie, Z.; Druey, K.M. p53 negatively regulates RGS13 protein expression in immune cells. J. Biol Chem. 2011, 286, 22219–22226. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, J.; Maity, B.; Mayuzumi, D.; Fisher, R.A. Regulator of G protein signaling 6 mediates doxorubicin-induced ATM and p53 activation by a reactive oxygen species-dependent mechanism. Cancer Res. 2011, 71, 6310–6319. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Maity, B.; Huang, J.; Gao, Z.; Stewart, A.; Weiss, R.M.; Anderson, M.E.; Fisher, R.A. G-protein inactivator RGS6 mediates myocardial cell apoptosis and cardiomyopathy caused by doxorubicin. Cancer Res. 2013, 73, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Maity, B.; Stewart, A.; O’Malley, Y.; Askeland, R.W.; Sugg, S.L.; Fisher, R.A. Regulator of G protein signaling 6 is a novel suppressor of breast tumor initiation and progression. Carcinogenesis 2013, 34, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, B.; Swaney, S.; Neubig, R.R. FBXO44-Mediated Degradation of RGS2 Protein Uniquely Depends on a Cullin 4B/DDB1 Complex. PLoS ONE 2015, 10, e0123581. [Google Scholar] [CrossRef] [PubMed]
- Iovine, B.; Iannella, M.L.; Bevilacqua, M.A. Damage-specific DNA binding protein 1 (DDB1): A protein with a wide range of functions. Int. J. Biochem. Cell Biol. 2011, 43, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. mTOR: On target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013, 19, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Michel, G.; Matthes, H.W.; Hachet-Haas, M.; El Baghdadi, K.; de Mey, J.; Pepperkok, R.; Simpson, J.C.; Galzi, J.L.; Lecat, S. Plasma membrane translocation of REDD1 governed by GPCRs contributes to mTORC1 activation. J. Cell Sci. 2014, 127, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Deans, A.J. The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Palovcak, A.; Liu, W.; Yuan, F.; Zhang, Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009, 668, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Larder, R.; Karali, D.; Nelson, N.; Brown, P. Fanconi anemia A is a nucleocytoplasmic shuttling molecule required for gonadotropin-releasing hormone (GnRH) transduction of the GnRH receptor. Endocrinology 2006, 147, 5676–5689. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Nara, K.; Yoshikawa, H.; Suzuki, N. Txk, a member of the non-receptor tyrosine kinase of the Tec family, forms a complex with poly(ADP-ribose) polymerase 1 and elongation factor 1alpha and regulates interferon-gamma gene transcription in Th1 cells. Clin. Exp. Immunol. 2007, 147, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Ahel, I.; Ahel, D.; Matsusaka, T.; Clark, A.J.; Pines, J.; Boulton, S.J.; West, S.C. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008, 451, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Reinemund, J.; Seidel, K.; Steckelings, U.M.; Zaade, D.; Klare, S.; Rompe, F.; Katerbaum, M.; Schacherl, J.; Li, Y.; Menk, M. Poly(ADP-ribose) polymerase-1 (PARP-1) transcriptionally regulates angiotensin AT2 receptor (AT2R) and AT2R binding protein (ATBP) genes. Biochem Pharmacol. 2009, 77, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Ahel, D.; Horejsí, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Grube, K.; Bürkle, A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. USA 1992, 89, 11759–11763. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6, 8897. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Nho, K.; Risacher, S.L.; Inlow, M.; Swaminathan, S.; Yoder, K.K.; Shen, L.; West, J.D.; McDonald, B.C.; Tallman, E.F.; et al. PARP1 gene variation and microglial activity on [11C]PBR28 PET in older adults at risk for Alzheimer’s disease. Multimodal Brain Image Anal. 2013, 8159, 150–158. [Google Scholar]
- Adamczyk, A.; Jeśko, H.; Strosznajder, R.P. Alzheimer’s disease related peptides affected cholinergic receptor mediated poly(ADP-ribose) polymerase activity in the hippocampus. Folia Neuropathol. 2005, 43, 139–142. [Google Scholar] [PubMed]
- Kunieda, T.; Minamino, T.; Nishi, J.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Min, L.J.; Mogi, M.; Iwanami, J.; Li, J.M.; Sakata, A.; Fujita, T.; Tsukuda, K.; Iwai, M.; Horiuchi, M.; et al. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc. Res. 2007, 76, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Min, L.J.; Mogi, M.; Iwanami, J.; Li, J.M.; Sakata, A.; Fujita, T.; Tsukuda, K.; Iwai, M.; Horiuchi, M. Angiotensin II type 2 receptor deletion enhances vascular senescence by methyl methanesulfonate sensitive 2 inhibition. Hypertension 2008, 51, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Daviet, L.; Lehtonen, J.Y.; Tamura, K.; Griese, D.P.; Horiuchi, M.; Dzau, V.J. Cloning and characterization of ATRAP, a novel protein that interacts with the angiotensin II type 1 receptor. J. Biol. Chem. 1999, 274, 17058–17062. [Google Scholar] [CrossRef] [PubMed]
- Min, L.J.; Mogi, M.; Tamura, K.; Iwanami, J.; Sakata, A.; Fujita, T.; Tsukuda, K.; Jing, F.; Iwai, M.; Horiuchi, M. Angiotensin II type 1 receptor-associated protein prevents vascular smooth muscle cell senescence via inactivation of calcineurin/nuclear factor of activated T cells pathway. J. Mol. Cell Cardiol. 2009, 47, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Nouet, S.; Amzallag, N.; Li, J.M.; Louis, S.; Seitz, I.; Cui, T.X.; Alleaume, A.M.; Di Benedetto, M.; Boden, C.; Masson, M.; et al. Trans-inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein. ATIP J. Biol. Chem. 2004, 279, 28989–38997. [Google Scholar] [CrossRef] [PubMed]
- Min, L.J.; Mogi, M.; Iwanami, J.; Jing, F.; Tsukuda, K.; Ohshima, K.; Horiuchi, M. Angiotensin II type 2 receptor-interacting protein prevents vascular senescence. J. Am. Soc. Hypertens. 2012, 6, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Devanarayan, V.; Martin, B.; Geerts, H.; Brain Health Modeling Initiative (BHMI). Intelligent and effective informatic deconvolution of “Big Data” and its future impact on the quantitative nature of neurodegenerative disease therapy. Alzheimers Dement. 2018, 14, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Martin, B.; Daimon, C.M.; Maudsley, S. Effective use of latent semantic indexing and computational linguistics in biological and biomedical applications. Front. Physiol. 2013, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Cashion, A.; Stanfill, A.; Thomas, F.; Xu, L.; Sutter, T.; Eason, J.; Ensell, M.; Homayouni, R. Expression levels of obesity-related genes are associated with weight change in kidney transplant recipients. PLoS ONE 2013, 8, e59962. [Google Scholar] [CrossRef] [PubMed]
- Leff, P. The two-state model of receptor activation. Trends Pharmacol. Sci. 1995, 16, 89–97. [Google Scholar] [CrossRef]
- De Lean, A.; Stadel, J.M.; Lefkowitz, R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980, 255, 7108–7117. [Google Scholar] [PubMed]
- Samama, P.; Cotecchia, S.; Costa, T.; Lefkowitz, R.J. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993, 268, 4625–4636. [Google Scholar] [PubMed]
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol. Pharmacol. 2015, 88, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Kiran, A.; Saini, D.K.G. protein Signaling, Journeys Beyond the Plasma Membrane. J. Indian Inst. Sci. 2017, 97, 95–108. [Google Scholar] [CrossRef]
- Irannejad, R.; von Zastrow, M. GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 2014, 27, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Jong, Y.I.; Harmon, S.K.; O’Malley, K.L. GPCR signaling from within the cell. Br. J. Pharmacol. 2017, 1–10. [Google Scholar]
- Jong, Y.I.; Harmon, S.K.; O’Malley, K.L. Intracellular GPCRs Play Key Roles in Synaptic Plasticity. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Römpler, H.; Yu, H.T.; Arnold, A.; Orth, A.; Schöneberg, T. Functional consequences of naturally occurring DRY motif variants in the mammalian chemoattractant receptor GPR33. Genomics 2006, 87, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Wilbanks, A.M.; Laporte, S.A.; Bohn, L.M.; Barak, L.S.; Caron, M.G. Apparent Loss-of-Function Mutant GPCRs Revealed as Constitutively Desensitized Receptors. Biochemistry 2002, 41, 11981–11989. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leysen, H.; Van Gastel, J.; Hendrickx, J.O.; Santos-Otte, P.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. Int. J. Mol. Sci. 2018, 19, 2919. https://doi.org/10.3390/ijms19102919
Leysen H, Van Gastel J, Hendrickx JO, Santos-Otte P, Martin B, Maudsley S. G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. International Journal of Molecular Sciences. 2018; 19(10):2919. https://doi.org/10.3390/ijms19102919
Chicago/Turabian StyleLeysen, Hanne, Jaana Van Gastel, Jhana O. Hendrickx, Paula Santos-Otte, Bronwen Martin, and Stuart Maudsley. 2018. "G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes" International Journal of Molecular Sciences 19, no. 10: 2919. https://doi.org/10.3390/ijms19102919
APA StyleLeysen, H., Van Gastel, J., Hendrickx, J. O., Santos-Otte, P., Martin, B., & Maudsley, S. (2018). G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. International Journal of Molecular Sciences, 19(10), 2919. https://doi.org/10.3390/ijms19102919