The Many Faces of Rap1 GTPase




Abstract
1. Introduction
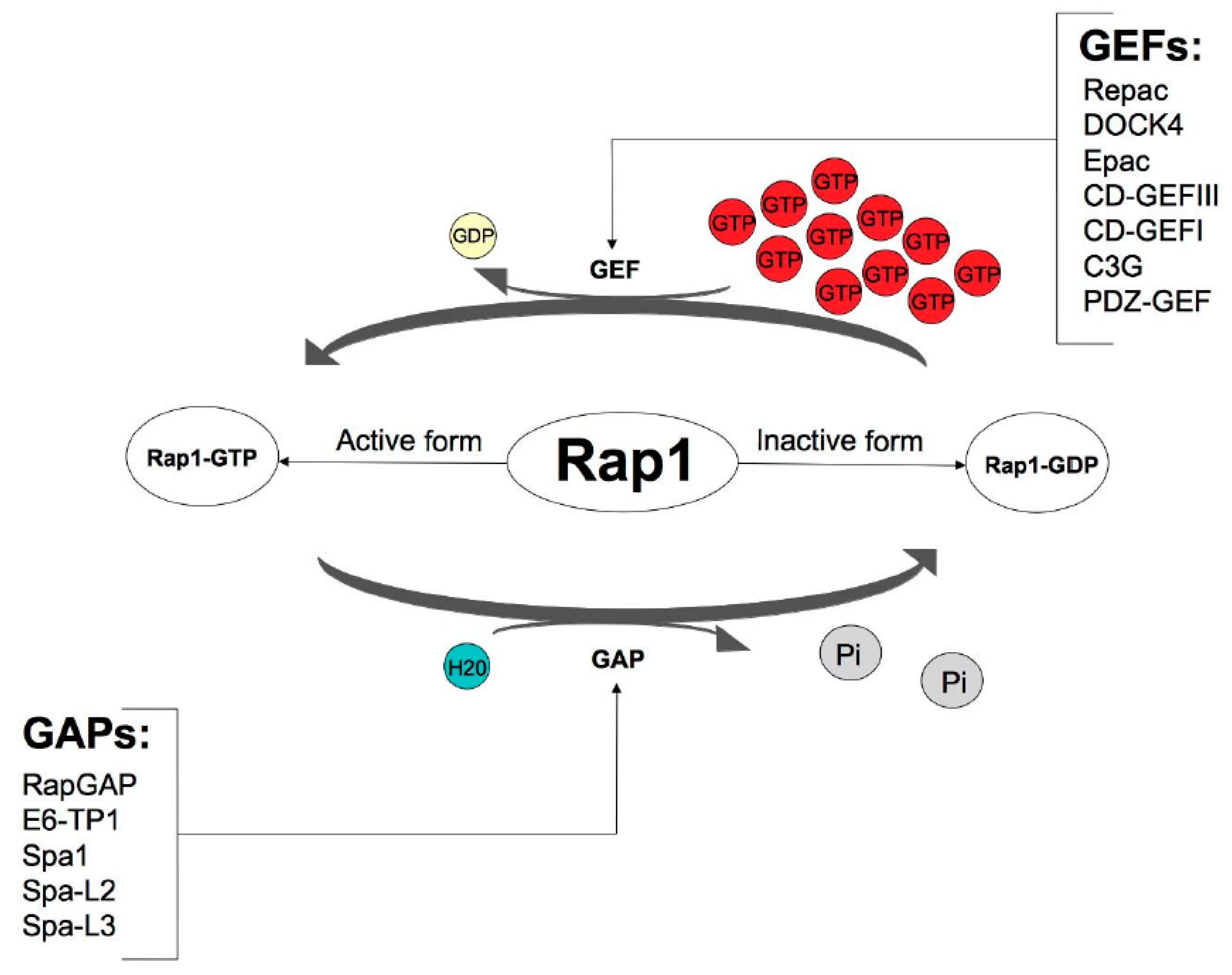
2. Activation of Rap1
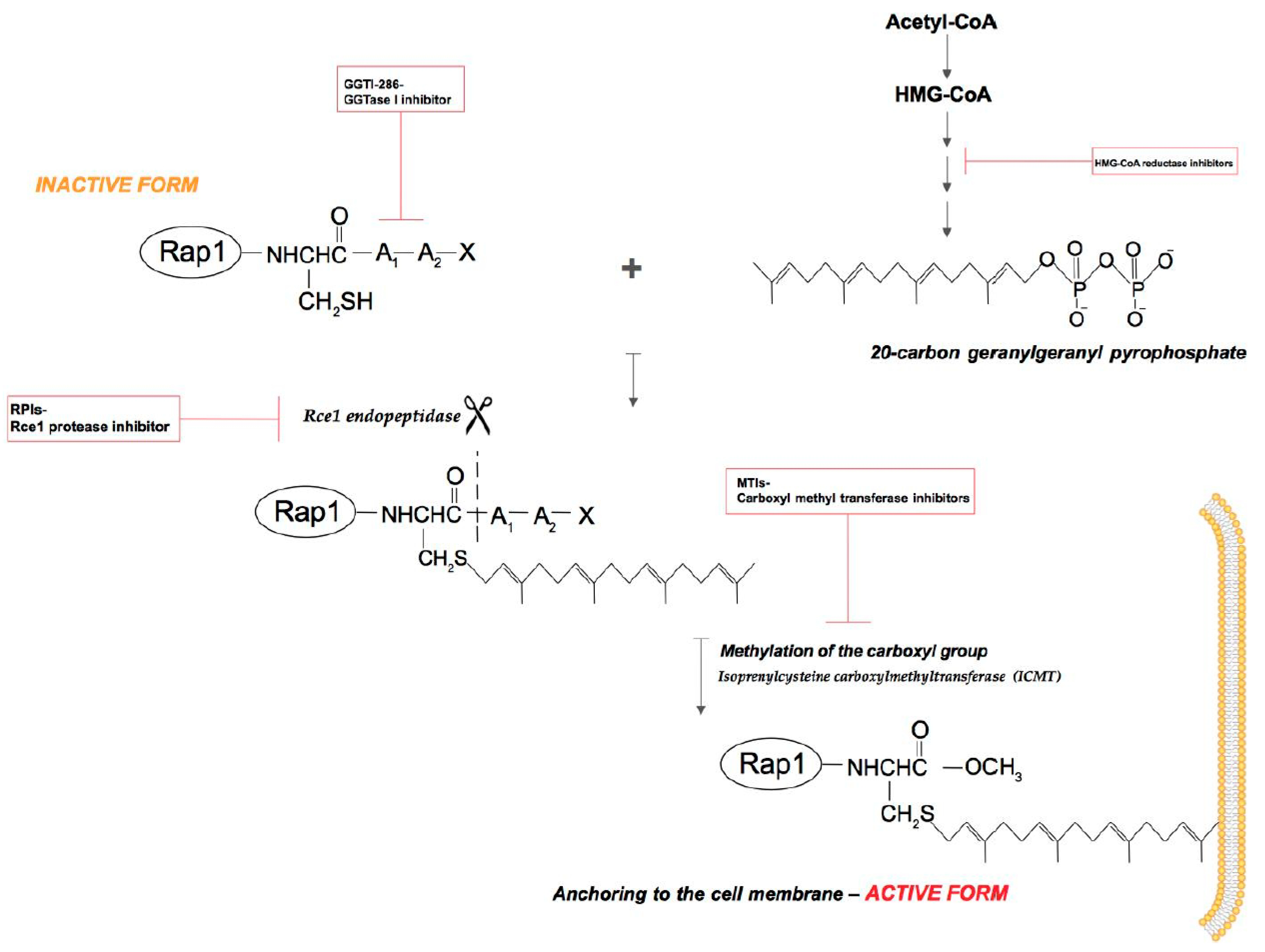
2.1. Isoprenylation
2.2. Other Post-Translational Modifications of Rap1
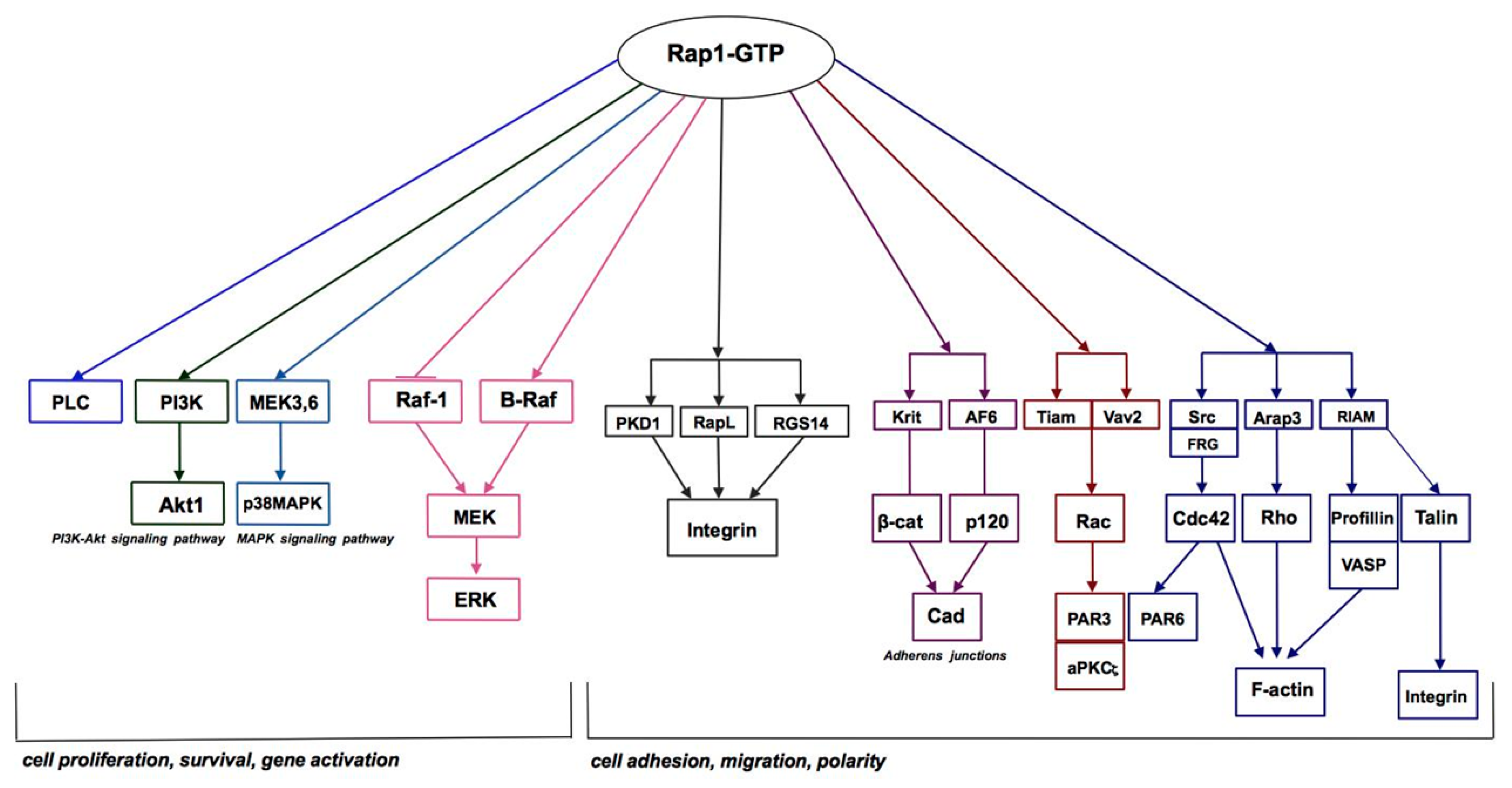
3. Rap1 Protein is A Link Between Cadherins and Integrins
4. Rap1 in Skeletal Muscle Differentiation
Rap1 Degradation in Skeletal Muscle Cells
5. Rap1 in the Inflammatory Response
6. Rap1 in Neoplastic Transformation
7. Rap1 in Genetic Disorders
8. Summary and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | Fluorouracil |
| AF6 | Afadin |
| Akt1 | RAC-alpha protein serine/threonine kinase |
| aPKCζ | Atypical protein kinase C zeta |
| Arap3 | Arf-GAP with Rho-domain, ANK repeat and PH domain-containing protein 3 |
| BCR | B cell antigen receptor |
| B-Raf | B-Raf proto-oncogene protein serine/threonine kinase, MAPKKK |
| C3G | Rap guanine nucleotide exchange factor 1 |
| Ca2+ | Calcium ion |
| CAAX | Cys-aliphatic residue-aliphatic residue-X |
| CAD | Cadherin 1, type 1, E-cadherin |
| CalDAG-GEF | Calcium/diacylglycerol guanine nucleotide exchange factor |
| cAMP | 3’,5’ cyclic adenosine monophosphate |
| CAMs | Cellular adhesion molecules |
| Cdc42 | Cell division control protein 42 |
| CD-GEFII | Ras guanyl-releasing protein 2 |
| CD-GEFIII | Ras guanyl-releasing protein 3 |
| CFC | Cardio-Facio-Cutaneous syndrome |
| CLM | Chronic myelogenous leukemia |
| CS | Costello syndrome |
| C3G | Rap guanine nucleotide exchange factor 1 |
| DAG | Diacylglycerol |
| DOCK4 | Dedicator of cytokinesis protein 4 |
| E6TP1 | Signal-induced proliferation-associated 1 like protein 1 |
| eIF3-F | Eukaryotic translation initiation factor 3 subunit F |
| EMT | Epithelial-mesenchymal transition |
| ERK1/2 | Extracellular signal-regulated kinase 1/2, MAPK |
| F-actin | Filamentous actin |
| FOXO1 | Forkhead box protein O1 |
| FOXO3 | Forkhead box protein O3 |
| FPP | Farnesyl pyrophosphate |
| FRG | Ferm, RhoGEF and pleckstrin domain protein 2 |
| FTase | Farnesyl transferase |
| GAPs | GTPase-activating proteins |
| G-CSF | Granulocyte colony stimulating agent |
| GEFs | Guanine nucleotide exchange factors |
| GGPP | Geranylgeranyl diphosphate |
| GGTase I | Geranylgeranyl transferase-1 |
| GGTase II | Rab geranylgeranyl transferase, geranylgeranyl transferase-2 |
| GM-CSF | High-affinity receptor for human granulocyte/macrophage colony-stimulating factor |
| GPCRs | G protein-coupled receptors, serpentine receptors |
| GTPase | Guanosine triphosphatase |
| HMG-CoAR | 3-hydroxy-3-methylglutaryl coenzyme A reductase |
| ICAM-1 | Intercellular adhesion molecule 1 |
| ICMT | Isoprenylcysteine carboxyl methyltransferase |
| IGF-1 | Insulin like growth factor |
| JAM-A | Junctional adhesion molecule-A |
| JNK | C-Jun N-terminal kinase |
| KDM6A | (K)-specific demethylase 6A |
| KMT2D | Lysine-specific methyltransferase 2D |
| Krit | Krev interaction trapped protein 1 |
| KS | Kabuki syndrome |
| KSR | Kinase Suppressor of Ras |
| LFA-1 | Lymphocyte function-associated antigen 1 |
| LS | Legius syndrome |
| MAFbx | Muscle-specific F-box |
| MAPK | Mitogen-activated protein kinase |
| MDCK-f3 | Mesenchymal Ras-transformed Madin-Darby canine kidney cells |
| MEK | Mitogen-activated protein kinase, MAPKK |
| MEK3,6 | Mitogen-activated protein kinase 3/6, MAPKK |
| MET | Mesenchymal-epithelial transition |
| miR-203 | Short non-coding RNA molecule |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NF1 | Neurofibromatosis type 1 |
| NO | Nitric oxide |
| Nox2 | NADH oxidase 2 |
| NS | Noonan syndrome |
| NSML | Noonan syndrome with multiple lentigins syndrome |
| p120ctn | Catenin (cadherin-associated protein), delta 1 |
| p38MAPK | P38 MAP kinase |
| PAR3 | Partitioning defective protein 3 |
| PAR6 | Partitioning defective protein 6 |
| PECAM-1 | Platelet endothelial cell adhesion molecule—also known as CD31—cluster of differentiation 31 |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha/beta/delta |
| PKA | Protein kinase A |
| PKD1 | Protein kinase D1 |
| PLC | Phosphatidylinositol phospholipase C |
| PLC-β | Phospholipase C beta |
| PLC-γ | Phospholipase C gamma |
| PDZ-GEF | Rap guanine nucleotide exchange factor 2 |
| Rac1/RAC | Rac1-Ras-related C3 botulinum toxin substrate 1 |
| Raf-1 | B-Raf proto-oncogene protein serine/threonine kinase, MAPKKK |
| RapL | Ras associated domain-containing protein 5 |
| RapGAP1 | Rap1 GTPase activating protein 1 |
| Repac-RAPGEF5 | Rap guanine nucleotide exchange factor 5 |
| RGS14 | Regulator of G-protein signaling 14 |
| RIAM | Amyloid beta A4 precursor protein-binding family B member 1- interacting protein |
| Rho | Ras homolog gene family, member A |
| Spa1 | Signal-induced proliferation-associated gene 1 |
| Spa-L2 | Signal-induced proliferation-associated 1 like protein 2 |
| Spa-L3 | Signal-induced proliferation-associated 1 like protein 3 |
| Src | Sarcoma family protein tyrosine kinases |
| TEM | Transendothelial migration |
| THP-1 | Human monocytic cell line 1 |
| TIAM | T-lymphoma invasion and metastasis-inducing protein 1 |
| TLN | Talin |
| TLRs | Toll-Like Receptors |
| TPA | 12-O-tetradecanoylphorbol 13-acetate |
| TSC | Tuberous sclerosis |
| TSC1-TSC2 | Hamartin-tuberin complex |
| TSH | Thyrotropin |
| VASP | Vasodilator-stimulated phosphoprotein |
| Vav2 | Vav guanine nucleotide exchange factor 2 |
| VCAM-1 | Vascular cell adhesion protein 1 |
| α4β1 (VLA-4) | Very late antigen-4 |
| β-cat | Catenin beta 1 |
References
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Cichowski, K. An expanding role for RAS GTPase-activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Brock, J.E.; Ji, K.; Mattingly, R.R. Ras and Rap1: A tale of two GTPases. Sem. Cancer Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, H.; Sugimoto, Y.; Matsuzaki, T.; Ikawa, Y.; Noda, M. A ras-related gene with transformation suppressor activity. Cell 1989, 56, 77–84. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Wittchen, E.S.; Aghajanian, A.; Burridge, K. Isoform-specific differences between RAP1A and RAP1B in the formation of endothelial cell junctions. Small GTPases 2011, 2, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Ikegami, T.; Nagadoi, A.; Kamatari, Y.O.; Park, S.-Y.; Tame, J.R.H.; Unzai, S. The structure and conformational switching of Rap1B. Biochem. Biophys. Res. Commun. 2015, 462, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Klose, A.; Ahmadian, M.R.; Schuelke, M.; Scheffzek, K.; Hoffmeyer, S.; Gewies, A.; Schmitz, F.; Kaufmann, D.; Peters, H.; Wittinghofer, A.; et al. Selective disactivation of neurofibromin GAP activity in neurofibromatosis type 1. Hum. Mol. Genet. 1998, 7, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Audagnotto, M.; Dal Peraro, M. Protein post-translational modifications: In silico prediction tools and molecular modeling. Comput. Struct. Biotechnol. J. 2017, 15, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Karbstein, K. Role of GTPases in ribosome assembly. Biopolymers 2007, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Fierke, C.A.; Casey, P.J. The biochemistry of farnesyltransferase and geranylgeranyltransferase I. In Farnesyltransferase Inhibitors in Cancer Therapy. Cancer Drug Discovery and Development; Sebti, S.M., Hamilton, A.D., Eds.; Humana Press: Totowa, NJ, USA, 2001; pp. 21–36. [Google Scholar]
- Tsukamoto, N.; Hattori, M.; Yang, H.; Bos, J.L. Rap1 GTPase-activating protein SPA-1 negatively regulates cell adhesion. J. Biol. Chem. 1999, 274, 18463–18469. [Google Scholar] [CrossRef] [PubMed]
- Franke, B.; Akkerman, J.W.; Bos, J.L. Rapid Ca2+mediated activation of Rap1 in human platelets. EMBO J. 1997, 16, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Matsuda, M.; Anafi, M.; Pawson, T.; Pessin, J.E. Insulin regulates the dynamic balance between Ras and Rap1 signaling by coordinating the assembly states of the Grb2-SOS and CrkII-C3G complexes. EMBO J. 1998, 17, 2554–2565. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.; Freeman, G.J.; Berezovskaya, A.; Barber, D.W.; Nadler, L.M. Maintenance of human T cell anergy: Blocking of IL-2 gene transcription by activated Rap1. Science 1997, 278, 124–128. [Google Scholar] [CrossRef] [PubMed]
- M’Rabet, M.; Coffer, P.; Zwartkruis, F.; Franke, B.; Segal, W.; Koenderman, L.; Bos, L. Activation of the small GTPase Rap1 in human neutrophils. Blood 1998, 92, 2133–2140. [Google Scholar] [PubMed]
- Scheffzek, K.; Ahmadin, M.R.; Wittinghofer, A. GTPase-activating proteins: Helping hands to complement an active site. Trends Biochem. Sci. 1998, 23, 257–262. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is RAP1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Vossler, M.R.; Yao, H.; York, R.D.; Pan, M.G.; Rim, C.S.; Stork, P.J.S. cAMP activates MAP kinase and Elk-1 through a B- Raf- and Rap1-dependent pathway. Cell 1997, 89, 73–82. [Google Scholar] [CrossRef]
- Burgering, B.M.; Bos, J.L. Regulation of Ras-mediated signaling: More than one way to skin a cat. Trends Biochem. Sci. 1995, 20, 18–22. [Google Scholar] [CrossRef]
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, 678–693. [Google Scholar]
- Byrne, D.P.; Vonderach, M.; Ferries, S.; Brownridge, P.J.; Eyers, C.E.; Eyers, P.A. cAMP-dependent protein kinase (PKA) complexes probed by complementary differential scanning fluorimetry and ion mobility-mass spectrometry. Biochem. J. 2016, 473, 3159–3175. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Arthur, W.T.; Quilliam, L.A.; Cooper, J.A. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J. Cell Biol. 2004, 167, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Ogita, H.; Kawakatsu, T.; Fukuhara, T.; Yamada, T.; Sato, T.; Shimizu, K.; Nakamura, T.; Matsuda, M.; Takai, Y. Involvement of the c-Src-Crk-C3G-Rap1 signaling pathway in the nectin-induced activation of Cdc42 and formation of adherens junctions. J. Biol. Chem. 2005, 280, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Ogita, H.; Kawakatsu, T.; Inagaki, M.; Takai, Y. Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene 2006, 25, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Novick, P. Regulation of membrane traffic by Rab GEF and GAP cascades. Small GTPases 2016, 7, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, J.R.; Bergmeier, W.; Zhang, Y.; Piffath, C.L.; Liang, Y.; Wagner, D.D.; Housman, D.E.; Graybiel, A.M. CalDAG-GEfI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 2004, 10, 982–986. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.J.; Ingham, R.J.; Bos, J.L.; Kurosaki, T.; Gold, M.R. Activation of the Rap1 GTPase by the B cell antigen receptor. J. Biol. Chem. 1998, 273, 29218–29223. [Google Scholar] [CrossRef] [PubMed]
- Tsygankova, O.M.; Saavedra, A.; Rebhun, J.F.; Quilliam, L.A.; Meinkoth, J.L. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol. Cell. Biol. 2001, 21, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, K.M.T.; Zwartkruis, F.J.T.; de Rooij, J.; Akkerman, J.W.N.; Bos, J.L. The small GTPase Rap1 is activated by turbulence and is involved in integrin αIIbβ3-mediated cell adhesion in human megakaryocytes. J. Biol. Chem. 2003, 278, 22412–22417. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Ye, W. Regulation of vascular endothelial junction stability and remodeling through Rap1-Rasip1 signaling. Cell Adh. Migr. 2014, 8, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M. Regulation of angiogenesis by small GTPase Rap1. Vasc. Pharmacol. 2010, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M. Distinct functions for Rap1 signaling in vascular morphogenesis and dysfunction. Exp. Cell Res. 2013, 319, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M. Rap1 in endothelial biology. Curr. Opin. Hematol. 2017, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Post, A.; Pannekoek, W.-J.; Ponsioen, B.; Vliem, M.J.; Bos, J.L. Rap1 spatially controls ArhGAP29 to inhibit Rho signaling during endothelial barrier regulation. Mol. Cell. Biol. 2015, 35, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Pannekoek, W.-J.; Post, A.; Bos, J.L. Rap1 signaling in endothelial barrier control. Cell Adh. Migr. 2014, 8, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.J.; Gastonguay, A.J.; Lorimer, E.L.; Kuhnmuench, J.R.; Li, R.; Fields, A.P.; Williams, C.L. Splice variants of SmgGDS control small GTPase prenylation and membrane localization. J. Biol. Chem. 2010, 285, 35255–35266. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, A.; Bełtowski, J. Izoprenylacja białek. Postępy Biochem. 2004, 50, 316–329. [Google Scholar]
- Erhardt, A.; David, M.D.; Erhardt, G.R.A.; Schrader, J.W. Distinct mechanisms determine the patterns of differential activation of H-Ras, N-Ras, K-Ras 4B, and M-Ras by receptors for growth factors, or antigen. Mol. Cell. Biol. 2004, 24, 6311–6323. [Google Scholar] [CrossRef] [PubMed]
- Ochocki, J.D.; Distefano, M.D. Prenytransferase inhibitors: Treating human ailments from cancer to parasitic infections. Medchemcomm 2013, 4, 476–492. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.V.; Robison, W.G.; Bettelheim, F.; Lin, L.R.; Reddy, V.N.; Zigler, J. Role of small GTP-binding proteins in lovastatin-induced cataracts. Invest. Ophthalmol. Vis. Sci. 1997, 38, 2313–2321. [Google Scholar] [PubMed]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Stork, P.J.S. Phosphorylation of Rap1 by cAMP-dependent protein kinase (PKA) creates a binding site for KSR to sustain ERK activation by cAMP. J. Biol. Chem. 2017, 292, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Dillon, T.J.; Liu, C.; Kariya, Y.; Wang, Z.; Stork, P.J.S. Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J. Biol. Chem. 2013, 288, 27712–27723. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Prokop, J.W.; Lorimer, E.; Ntantie, E.; Williams, C.L. Differences in the phosphorylation-dependent regulation of prenylation of Rap1A and Rap1B. J. Mol. Biol. 2016, 428, 4929–4945. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Zipursky, S.L. Cell-Cell adhesion and communication. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 2011, 124, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 1999, 24, 73–76. [Google Scholar] [CrossRef]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 5, 701–706. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in the cancer progression. Biochim. Biophys. Acta 2013, 12, 3481–3498. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion in tumor invasion and metastasis: Loss of the glue is not enough. Biochim. Biophys. Acta 2001, 1552, 39–45. [Google Scholar] [CrossRef]
- Price, L.S.; Hajdo-Milasinovic, A.; Zhao, J.; Zwartkruis, F.J.T.; Collard, J.G.; Bos, J.L. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J. Biol. Chem. 2004, 279, 35127–35132. [Google Scholar] [CrossRef] [PubMed]
- Balzac, F.; Avolio, M.; Degani, S.; Kaverina, I.; Torti, M.; Silengo, L.; Small, J.V.; Retta, S.F. E-cadherin endocytosis regulates the activity of Rap1: A traffic light GTPase at the crossroads between cadherin and integrin function. J. Cell Sci. 2005, 118, 4765–4783. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Garry, D.J. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006, 20, 1692–1708. [Google Scholar] [CrossRef] [PubMed]
- Pizon, V.; Cifuentes-Diaz, C.; Mege, R.M.; Baldacci, G.; Rieger, F. Expression and localization of Rap1 proteins during myogenic differentiation. Eur. J. Cell Biol. 1996, 69, 224–235. [Google Scholar] [PubMed]
- Pizon, V.; Mechali, F.; Baldacci, G. RAP1A GTP/GDP cycles determine the intracellular location of the late endocytic compartments and contribute to myogenic differentiation. Exp. Cell Res. 1999, 246, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Pizon, V.; Desjardins, M.; Bucci, C.; Parton, R.G.; Zerial, M. Association of Rap1 and Rap1b proteins with late endocytic/phagocytic compartments and Rap2a with the Golgi complex. J. Cell Sci. 1994, 107, 1661–1670. [Google Scholar] [PubMed]
- Pizon, V.; Baldacci, G. Rap1A protein interferes with various MAP kinase activating pathways in skeletal myogenic cells. Oncogene 2000, 19, 6074–6081. [Google Scholar] [CrossRef] [PubMed]
- Meresse, S.; Gorvel, J.P.; Chavrier, P. The rab7 GTPase resides on a vesicular compartment connected to lysosomes. J. Cell Sci. 1995, 108, 3349–3358. [Google Scholar] [PubMed]
- Sin, J.; Andres, A.M.; Taylor, D.J.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.E.; Zhang, X.; Bleicher, K.B.; Dysart, G.; Loughlin, A.F.; Schaefer, W.H.; Umbenhauer, D.R. Statins induce apoptosis in rat and human myotube cultures by inhibiting protein geranylgeranylation but not ubiquinone. Toxicol. Appl. Pharmacol. 2004, 200, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Crick, D.C.; Andres, D.A.; Danesi, R.; Macchia, M.; Waechte, C.J. Geranylgeraniol overcomes the block of cell proliferation by lovastatin in C6 glioma cells. J. Neurochem. 1998, 70, 2387–2405. [Google Scholar] [CrossRef]
- Jaśkiewicz, A.; Pająk, B.; Litwiniuk, A.; Urbańska, K.; Orzechowski, A. Geranylgeraniol prevents statin-dependent myotoxicity in C2C12 muscle cells through RAP1 GTPase prenylation and cytoprotective autophagy. Oxid. Med. Cell. Longev. 2018, 2018, 6463807. [Google Scholar] [CrossRef] [PubMed]
- Siddals, K.W.; Marshman, E.; Westwood, M.; Gibson, J.M. Abrogation of insulin-like growth factor-I (IGF-I) and insulin action by mevalonic acid depletion: Synergy between protein prenylation and receptor glycosylation pathways. J. Biol. Chem. 2004, 279, 38353–38359. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy in autoimmunity: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 5, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2003, 501, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Reedquist, K.A.; Ross, E.; Koop, E.A.; Wolthuis, R.M.F.; Zwartkruis, F.J.T.; van Kooyk, Y.; Salmon, M.; Buckley, C.D.; Bos, J.L. The small GTPase Rap1, Mediates Cd31-induced integrin adhesion. J. Cell Biol. 2000, 148, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P. Regulation of B-cell responses by Toll-like receptors. Immulology 2012, 136, 370–379. [Google Scholar]
- Schaefer, T.M.; Desouza, K.; Fahey, J.V.; Beagley, K.W.; Wira, C.R. Toll-like receptor (TLR) expression and TLR-mediated cytokine/chemokine production by human uterine epithelial cells. Immunology 2004, 112, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.J.; Mitroulis, I.; Wiessner, J.R.; Zheng, Y.Y.; Siegert, G.; Sperandio, M.; Chavakis, T. A novel pathway of rapid TLR-triggered activation of integrin-dependent leukocyte adhesion that requires Rap1 GTPase. Mol. Biol. Cell 2014, 25, 2948–2955. [Google Scholar] [CrossRef] [PubMed]
- Dorn, A.; Zoellner, A.; Follo, M.; Martin, S.; Weber, F.; Marks, R.; Melchinger, W.; Zeiser, R.; Fisch, P.; Scheele, J.S. Rap1a deficiency modifies cytokine responses and MAPK-signaling in vitro and impairs the in vivo inflammatory response. Cell Immunol. 2012, 276, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, B.A.; Nelson, W.J. Alterations in the establishment and maintenance of epithelial cell polarity as a basis of disease processes. J. Clin. Invest. 1990, 85, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Nelson, C.M.; Myers, C.A.; Bissell, M.J. Rap1 integrates tissue polarity, lumen formation, and tumorigenic potential in human breast epithelial cells. Cancer Res. 2007, 67, 4759–4766. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.; Olson, M.F. Targeting Ras and Rho GTPases as opportunities for cancer therapeutics. Curr. Opin. Gentet. Dev. 2005, 15, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Labbe, D.P.; Hardy, S.; Tremblay, M.L. Protein tyrosine phosphatases in cancer: Friends and foes! Prog. Mol. Biol. Transl. Sci. 2012, 106, 253–306. [Google Scholar] [PubMed]
- Coxon, F.P.; Helfrich, M.H.; Larijani, B.; Muzylak, M.; Dunford, J.E.; Marshall, D.; McKinnon, A.D.; Nesbitt, S.A.; Horton, M.A.; Seabra, M.C.; et al. Identification of a novel phosphonocarboxylate inhibitor of Rab geranylgeranyl transferase that specifically prevents Rab prenylation in osteoclasts and macrophages. J. Biol. Chem. 2001, 276, 48213–48222. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.A.; McLeod, S.J.; Dukowski, J.; Austin, P.; Lee, C.C.; Millen-Martin, B.; Kubes, P.; McCaffert, D.M.; Gold, M.R.; Roskelley, C.D. Preventing the activation or cycling of the Rap1 GTPase alterts adhesion and cytoskeletal dynamics and blocks metastatic melanoma cell extravasation into the lungs. Cancer Res. 2010, 70, 4590–4601. [Google Scholar] [CrossRef] [PubMed]
- McSherry, E.A.; Brennan, K.; Hudson, L.; Hill, A.D.; Hopkins, A.M. Breast cancer cell migration is regulated through junctional adhesion molecule-A-mediated activation of Rap2 GTPase. Breast Cancer Res. 2011, 13, R31. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.L.; Kelly, P.; Casey, P.J. Activation of Rap1 promotes prostate cancer metastasis. Cancer Res. 2009, 69, 4962–4968. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Bian, C.; Wang, H.; Huang, S.; Wu, D. MiR-203 down-regulates Rap1A and suppresses cell proliferation, adhesion and invasion in prostate cancer. J. Exp. Clin. Cancer Res. 2015, 34, 8. [Google Scholar] [CrossRef] [PubMed]
- Pieters, T.; van Roy, F.; van Hengel, J. Functions of p120ctn isoforms in cell-cell adhesion and intracellular signaling. Front. Biosci. 2012, 17, 1669–1694. [Google Scholar] [CrossRef]
- Kumper, S.; Ridley, A. p120ctn and P-cadherin but not E-cadherin regulate cell motility and invasion of DU145 prostate cancer cells. PLoS ONE 2010, 5, e11801. [Google Scholar] [CrossRef] [PubMed]
- Ishida, D.; Kometani, K.; Yang, H.; Kakugawa, K.; Masuda, K.; Iwai, K.; Suzuki, M.; Itohara, S.; Nakahata, T.; Hiai, H.; et al. Myeloproliferative stem cell disorders by deregulated Rap1 activation in SPA-1-deficient mice. Cancer Cell 2003, 4, 55–65. [Google Scholar] [CrossRef]
- Zhang, L.; Chenwei, L.; Mahmood, R.; van Golen, K.; Greenson, J.; Li, G.; D’Silva, N.J.; Li, X.; Burant, C.F.; Logsdon, C.D.; et al. Identification of a putative tumor suppressor gene Rap1GAP in pancreatic cancer. Cancer Res. 2006, 66, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Soucek, T.; Pusch, O.; Wienecke, R.; DeClue, J.; Hengstschlager, M. Role of the tuberous sclerosis gene-2 product in cell cycle control. J. Biol. Chem. 1997, 272, 29301–29308. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.H.; Shoarinejad, F.; Jin, F.; Golemis, E.A.; Yeung, R.S. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J. Biol. Chem. 1997, 272, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.; Bailey, C.L.; Fueger, P.T.; Newgard, C.B.; Casey, P.J.; Kimple, M.E. Rap1 promotes multiple pancreatic islet cell functions and signals through mammalian target of rapamycin complex1. J. Biol. Chem. 2010, 285, 15777–15785. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Schill, L.; Schoyer, L.; Andresen, B.S.; Bakker, A.; Bayrak-Toydemir, P.; Burkitt-Wright, E.; Chatfield, K.; Elefteriou, F.; Elgersma, Y.; et al. The Fourth International Symposium on Genetic Disorders of the Ras/MAPK pathway. Am. J. Med. Genet. 2016, 170, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Bogershausen, N.; Tsai, I.C.; Pohl, E.; Kiper, P.O.; Beleggia, F.; Percin, E.F.; Keupp, K.; Matchan, A.; Milz, E.; Alanay, Y.; et al. RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. J. Clin. Investig. 2015, 125, 3585–3599. [Google Scholar] [CrossRef] [PubMed]
- Bogershausen, N.; Wollnik, B. Unmasking Kabuki syndrome. Clin. Genet. 2013, 83, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Boettner, B.; Van Aelst, L. Control of cell adhesion dynamics by Rap1 signaling. Curr. Opin. Cell Biol. 2009, 21, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Boettner, B.; Govek, E.; Cross, J.; Van Aelst, L. The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc. Natl. Acad. Sci. USA 2000, 97, 9064–9069. [Google Scholar] [CrossRef] [PubMed]
- Krugmann, S.; Andrews, S.; Stephens, L.; Hawkins, P. ARAP3 is essential for formation of lamellipodia after growth factor stimulation. J. Cell Sci. 2006, 119, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.W.; Li, Z.; Brown, M.; Sacks, D. IQGAP1 Binds Rap1 and modulates its activity. J. Biol. Chem. 2007, 282, 20752–20762. [Google Scholar] [CrossRef] [PubMed]
- Glading, A.; Han, J.; Stockton, A.; Ginsberg, M. KRIT-1/CCM1 is a RapGlading. J. Cell Biol. 2007, 179, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, K.; Maeda, A.; Shimonaka, M.; Kinashi, T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 2003, 4, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.; Bos, J. Specificity in Ras and Rap signaling. J. Biol. Chem. 2009, 284, 10995–10999. [Google Scholar] [CrossRef] [PubMed]
- Smolen, G.; Schott, B.; Stewart, R.; Diederichs, S.; Muir, B.; Provencher, H.; Look, A.; Sgroi, D.; Peterson, R.; Haber, D. A Rap GTPase interactor, RADIL, mediates migration of neural crest precursors. Genes Dev. 2007, 21, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Sakisaka, T.; Hisata, S.; Baba, T. RA-RhoGAP, Rap-activated Rho GTPase-activating protein implicated in neurite outgrowth through Rho. J. Biol. Chem. 2005, 280, 33026–33034. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Lim, C.J.; Puzon-McLaughlin, W.; Shattil, S.J.; Ginsberg, M.H. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J. Biol. Chem. 2009, 284, 5119–5127. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.R.; Puzon-McLaughlin, W.; Bobkov, A.A.; Ginsberg, M.H. Recognition of the Ras subfamily of GTP-binding proteins. Structure 2017, 24, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, D.L.; Ribeiro-Neto, F. Mitogenic and oncogenic properties of the small G protein Rap1b. Proc. Natl. Acad. Sci. USA 1998, 95, 7475–7479. [Google Scholar] [CrossRef] [PubMed]
- Apicelli, A.J.; Uhlmann, E.J.; Baldwin, R.L.; Ding, H.; Nagy, A.; Guha, A.; Gutmann, D.H. Role of the Rap1 GTPase in astrocyte growth regulation. Glia 2003, 42, 225–234. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, N.J.; Mitra, R.S.; Zhang, Z.; Kurnit, D.M.; Babcock, C.R.; Polverini, P.J.; Carey, T.E. Rap1, a small GTP-binding protein is upregulated during arrest of proliferation in human keratinocytes. J. Cell Physiol. 2003, 196, 532–540. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rap1 GTPase Effector | Cell Type | Effect | References |
|---|---|---|---|
| Afadin/AF-6 | Epithelial cells | Adherens junction formation | [95,96] |
| Arap3 | Endothelial cells | Regulation of lamellipodia formation | [97] |
| IQGAP1 | Epithelial cells | Regulation of cytoskeleton fomation | [98] |
| Krit-1/CCM1 | Arterial/Venous endhothelial cells | Maintaining the integrity of endothelial junctions | [99] |
| Mst1 RAPL | Leukocytes | Integrin α1β2 activation | [100] |
| Phg2 serine/threonine protein kinase | Dictyostelium discoideum cells | Regulation of chemotaxis and cell adhesion | [98] |
| PKD1 serine/threonine protein kinase D1 | T cells | Promotion of cell adhesion | [101] |
| Rac Cdc42 | MDCK | Actin cytoskeleton assembly | [27] |
| Radil | Neural crest (NC) cells | Promotion of integrin-mediated cell adhesion and migration | [102] |
| RA-RhoGAP | Neuronal cells | Regulation of neurite outgrowth | [103] |
| RIAM | Hematopoetic cells | Talin dependent integrins activation | [104] |
| Rasip1/Rain | Endothelial cells | Maintaining proper endothelial barrier functioning | [105] |
| Raf-1 serine/threonine protein kinase (the Rap1 GTPase inhibitory effect) B-Raf serine/threonine protein kinase (the Rap1 GTPase activating effect) | PC12 cells | Neuronal differentiation | [21] |
| Tiam1 Vav2 | Fibroblasts | Promotion of cell spreading | [26] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaśkiewicz, A.; Pająk, B.; Orzechowski, A. The Many Faces of Rap1 GTPase. Int. J. Mol. Sci. 2018, 19, 2848. https://doi.org/10.3390/ijms19102848
Jaśkiewicz A, Pająk B, Orzechowski A. The Many Faces of Rap1 GTPase. International Journal of Molecular Sciences. 2018; 19(10):2848. https://doi.org/10.3390/ijms19102848
Chicago/Turabian StyleJaśkiewicz, Anna, Beata Pająk, and Arkadiusz Orzechowski. 2018. "The Many Faces of Rap1 GTPase" International Journal of Molecular Sciences 19, no. 10: 2848. https://doi.org/10.3390/ijms19102848
APA StyleJaśkiewicz, A., Pająk, B., & Orzechowski, A. (2018). The Many Faces of Rap1 GTPase. International Journal of Molecular Sciences, 19(10), 2848. https://doi.org/10.3390/ijms19102848