Preservation Method and Phosphate Buffered Saline Washing Affect the Acute Myeloid Leukemia Proteome

 , ,
, ,  and
and 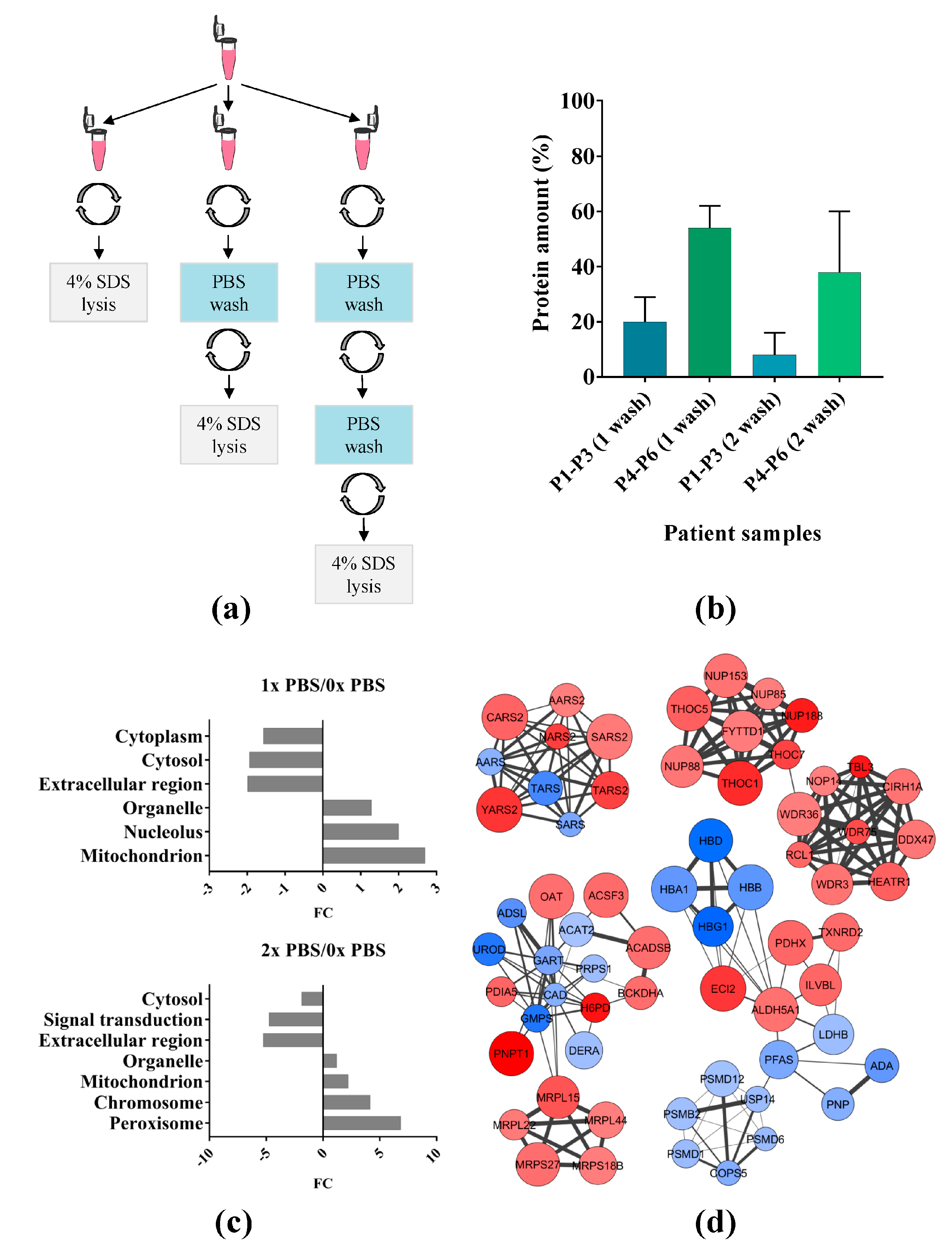{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Effect of Phosphate Buffered Saline (PBS) Wash on Primary Acute Myeloid Leukemia (AML) Cells
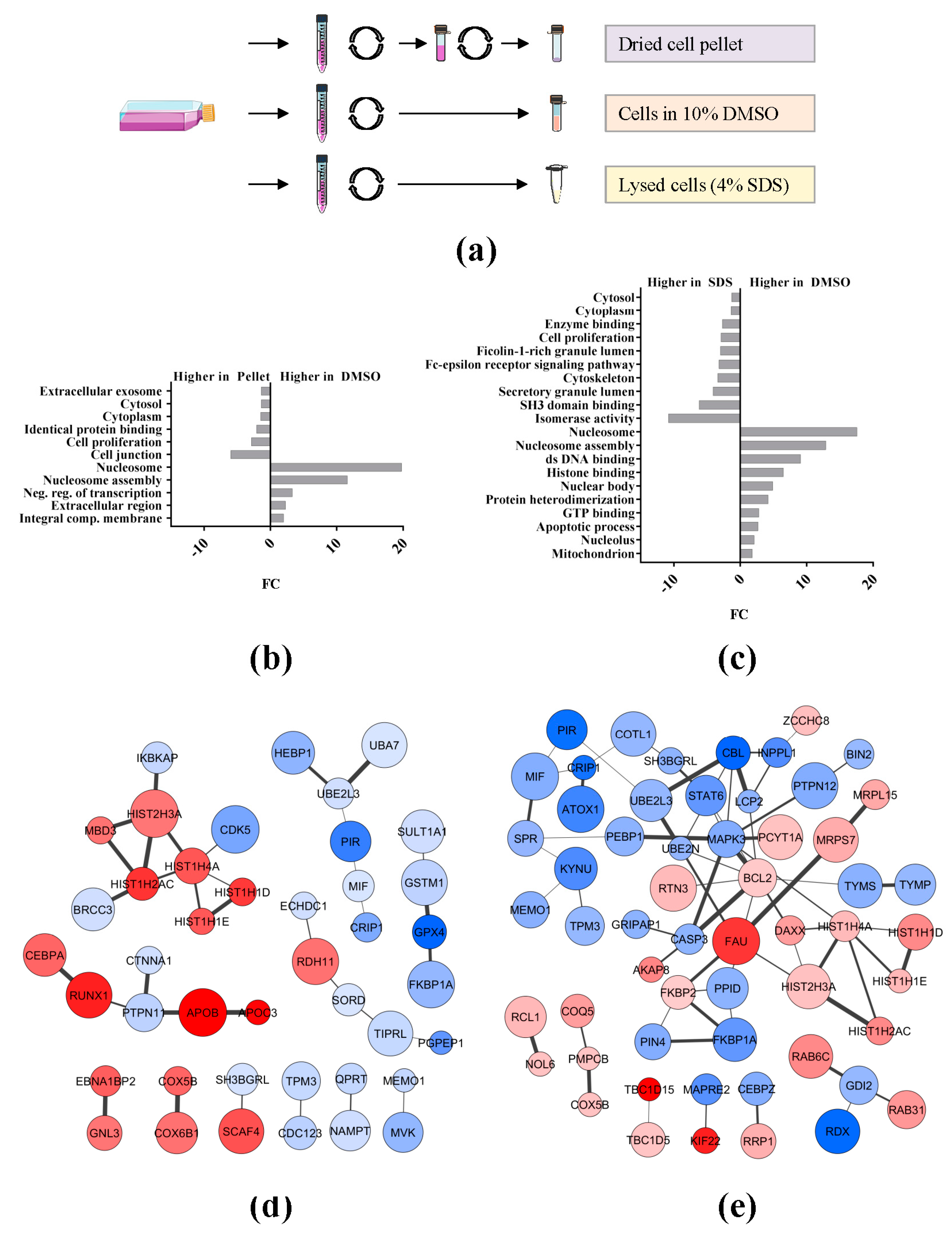
2.2. Effect of Sample Preservation for the Study of the AML Proteome
3. Discussion
3.1. PBS Wash
3.2. Preservation Conditions
4. Materials and Methods
4.1. Primary AML Cells
4.2. AML Cell Lines
4.3. Sample Preparation for Mass Spectrometry (MS) Analysis
4.4. Bioinformatic Analysis of Proteomic Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| AML | Acute myeloid leukemia |
| DMSO | Dimethyl sulfoxide |
| FA | Formic acid |
| FASP | Filter-aided sample preparation |
| FBS | Fetal bovine serum |
| FCS | Fetal calf serum |
| FC | Fold change |
| GO | Gene ontology |
| LC | Liquid chromatography |
| MS | Mass spectrometry |
| PBS | Phosphate buffered saline |
| RBC | Red blood cell |
| SD | Standard deviation |
| SDS | Sodium dodecyl sulfate |
| SILAC | Stable isotope labeling with amino acids in cell culture |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Böchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Malm, J.; Marko-Varga, G. Clinical protein science developments for patient monitoring in hospital central laboratories. Clin. Transl. Med. 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Farag, Y.; Selheim, F.; Berven, F.; Bruserud, Ø.; Hernandez-Valladares, M. Freezing effects on the acute myeloid leukemia cell proteome and phosphoproteome revealed using optimal quantitative workflows. J. Proteom. 2016, 145, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Tubaon, R.M.; Haddad, P.R.; Quirino, J.P. Sample clean-up strategies for ESI mass spectrometry applications in bottom-up proteomics: Trends from 2012 to 2016. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass spectrometry applied to bottom-up proteomics: Entering the high-throughput era for hypothesis testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Vaudel, M.; Sickmann, A.; Martens, L. Introduction to opportunities and pitfalls in functional mass spectrometry based proteomics. Biochim. Biophys. Acta 2014, 1844 1 Pt A, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Verheggen, K.; Raeder, H.; Berven, F.S.; Martens, L.; Barsnes, H.; Vaudel, M. Anatomy and evolution of database search engines-a central component of mass spectrometry based proteomic workflows. Mass Spectrom. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Tabb, D.L.; Impens, F.; Staes, A.; Timmerman, E.; Martens, L.; Laukens, K. Quality control in mass spectrometry-based proteomics. Mass Spectrom. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Valkenborg, D.; Martens, L.; Laukens, K. Computational quality control tools for mass spectrometry proteomics. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Aasebø, E.; Opsahl, J.A.; Bjørlykke, Y.; Myhr, K.M.; Kroksveen, A.C.; Berven, F.S. Effects of blood contamination and the rostro-caudal gradient on the human cerebrospinal fluid proteome. PLoS ONE 2014, 9, e90429. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, B.T.; Øyan, A.M.; Marzolf, B.; Hovland, R.; Gausdal, G.; Døskeland, S.O.; Dimitrov, K.; Golden, A.; Kalland, K.H.; Hood, L.; et al. Analysis of acute myelogenous leukemia: Preparation of samples for genomic and proteomic analyses. J. Hematother. Stem Cell Res. 2002, 11, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In vitro culture of aml cells—The present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ryningen, A.; Ersvaer, E.; Øyan, A.M.; Kalland, K.H.; Vintermyr, O.K.; Gjertsen, B.T.; Bruserud, Ø. Stress-induced in vitro apoptosis of native human acute myelogenous leukemia (AML) cells shows a wide variation between patients and is associated with low BCL-2:Bax ratio and low levels of heat shock protein 70 and 90. Leuk. Res. 2006, 30, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Wangen, R.; Aasebø, E.; Trentani, A.; Døskeland, S.O.; Bruserud, Ø.; Selheim, F.; Hernandez-Valladares, M. The enrichment analysis of DMSO vs. pellet samples showed approximately the same results for biological processes and cellular components as for the DMSO vs. SDS samples. Unpublished work. 2017. [Google Scholar]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Akkök, C.A.; Liseth, K.; Hervig, T.; Ryningen, A.; Bruserud, Ø.; Ersvaer, E. Use of different DMSO concentrations for cryopreservation of autologous peripheral blood stem cell grafts does not have any major impact on levels of leukocyte- and platelet-derived soluble mediators. Cytotherapy 2009, 11, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.J.; Hovland, R.; Øyan, A.M.; Kalland, K.H.; Ryningen, A.; Gjertsen, B.T.; Bruserud, Ø. Release of angiopoietin-1 by primary human acute myelogenous leukemia cells is associated with mutations of nucleophosmin, increased by bone marrow stromal cells and possibly antagonized by high systemic angiopoietin-2 levels. Leukemia 2008, 22, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Tiziani, S.; Kang, Y.; Harjanto, R.; Axelrod, J.; Piermarocchi, C.; Roberts, W.; Paternostro, G. Metabolomics of the tumor microenvironment in pediatric acute lymphoblastic leukemia. PLoS ONE 2013, 8, e82859. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, Ø.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics quality control: Quality control software for maxquant results. J. Proteome Res. 2016, 15, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Arntzen, M.Ø.; Koehler, C.J.; Barsnes, H.; Berven, F.S.; Treumann, A.; Thiede, B. IsobariQ: Software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. J. Proteome Res. 2011, 10, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Schölz, C.; Lyon, D.; Refsgaard, J.C.; Jensen, L.J.; Choudhary, C.; Weinert, B.T. Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nat. Methods 2015, 12, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wangen, R.; Aasebø, E.; Trentani, A.; Døskeland, S.-O.; Bruserud, Ø.; Selheim, F.; Hernandez-Valladares, M. Preservation Method and Phosphate Buffered Saline Washing Affect the Acute Myeloid Leukemia Proteome. Int. J. Mol. Sci. 2018, 19, 296. https://doi.org/10.3390/ijms19010296
Wangen R, Aasebø E, Trentani A, Døskeland S-O, Bruserud Ø, Selheim F, Hernandez-Valladares M. Preservation Method and Phosphate Buffered Saline Washing Affect the Acute Myeloid Leukemia Proteome. International Journal of Molecular Sciences. 2018; 19(1):296. https://doi.org/10.3390/ijms19010296
Chicago/Turabian StyleWangen, Rebecca, Elise Aasebø, Andrea Trentani, Stein-Ove Døskeland, Øystein Bruserud, Frode Selheim, and Maria Hernandez-Valladares. 2018. "Preservation Method and Phosphate Buffered Saline Washing Affect the Acute Myeloid Leukemia Proteome" International Journal of Molecular Sciences 19, no. 1: 296. https://doi.org/10.3390/ijms19010296
APA StyleWangen, R., Aasebø, E., Trentani, A., Døskeland, S.-O., Bruserud, Ø., Selheim, F., & Hernandez-Valladares, M. (2018). Preservation Method and Phosphate Buffered Saline Washing Affect the Acute Myeloid Leukemia Proteome. International Journal of Molecular Sciences, 19(1), 296. https://doi.org/10.3390/ijms19010296