BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy

 ,
, 
Abstract
:
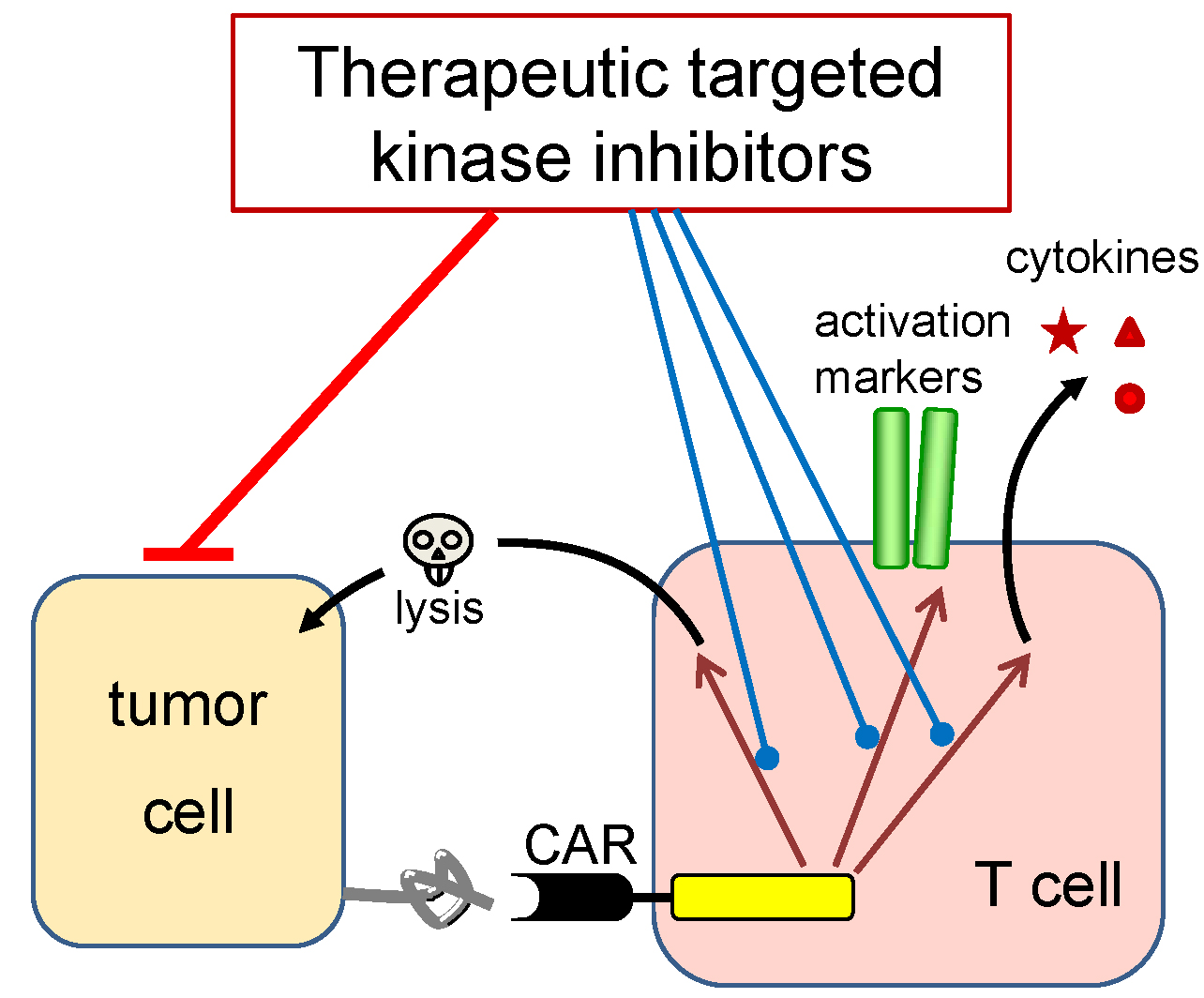
1. Introduction
2. Results
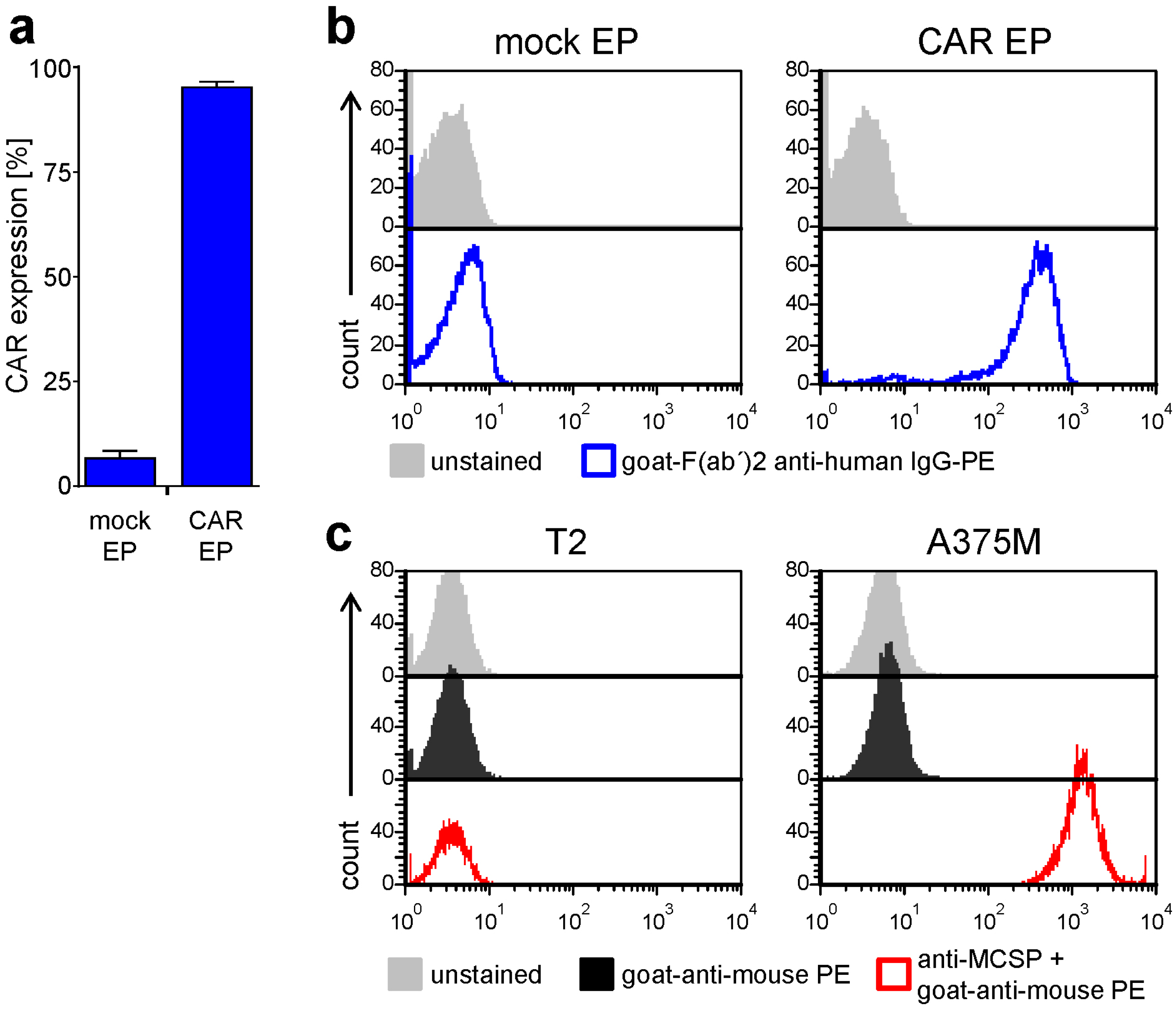
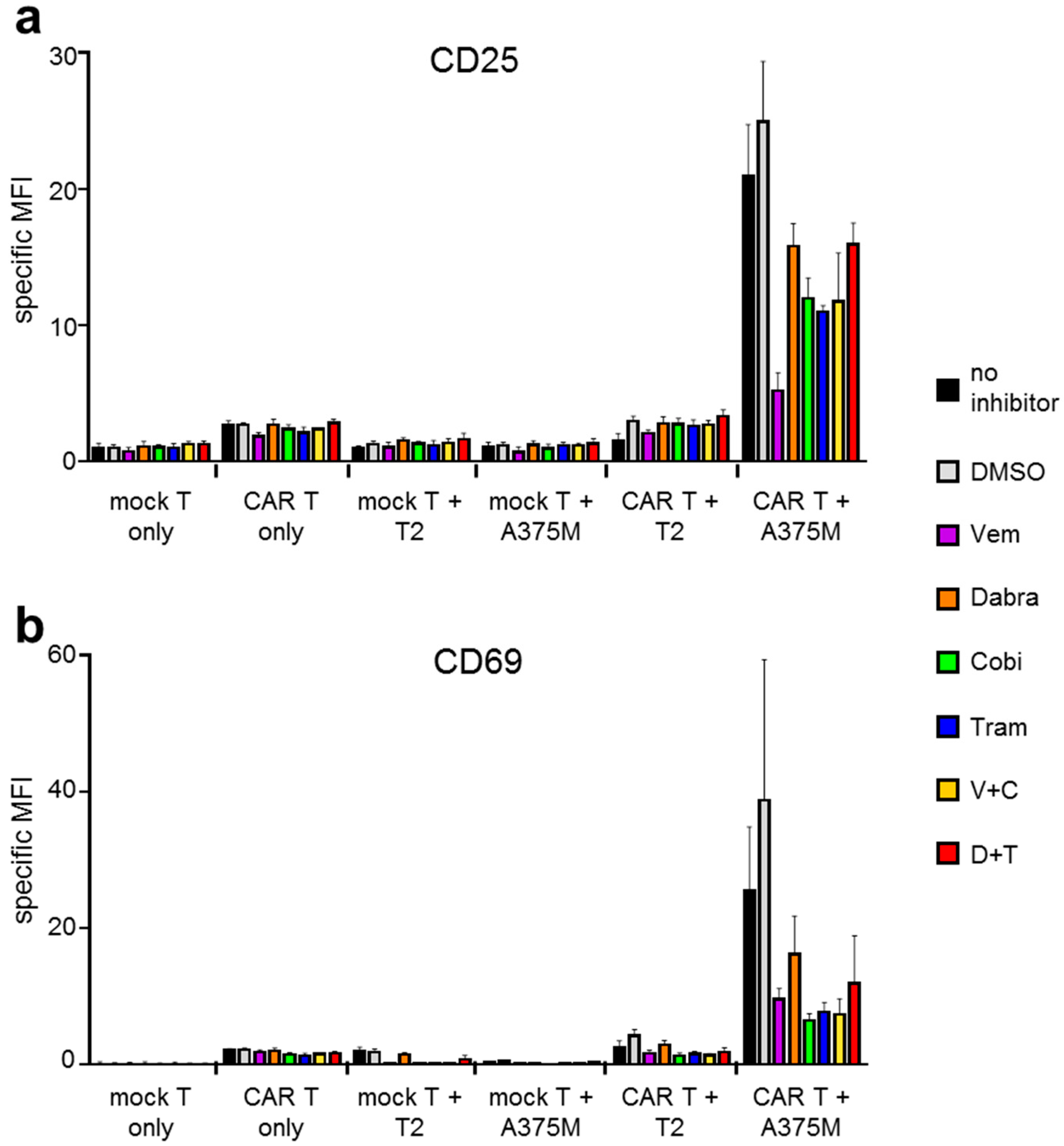
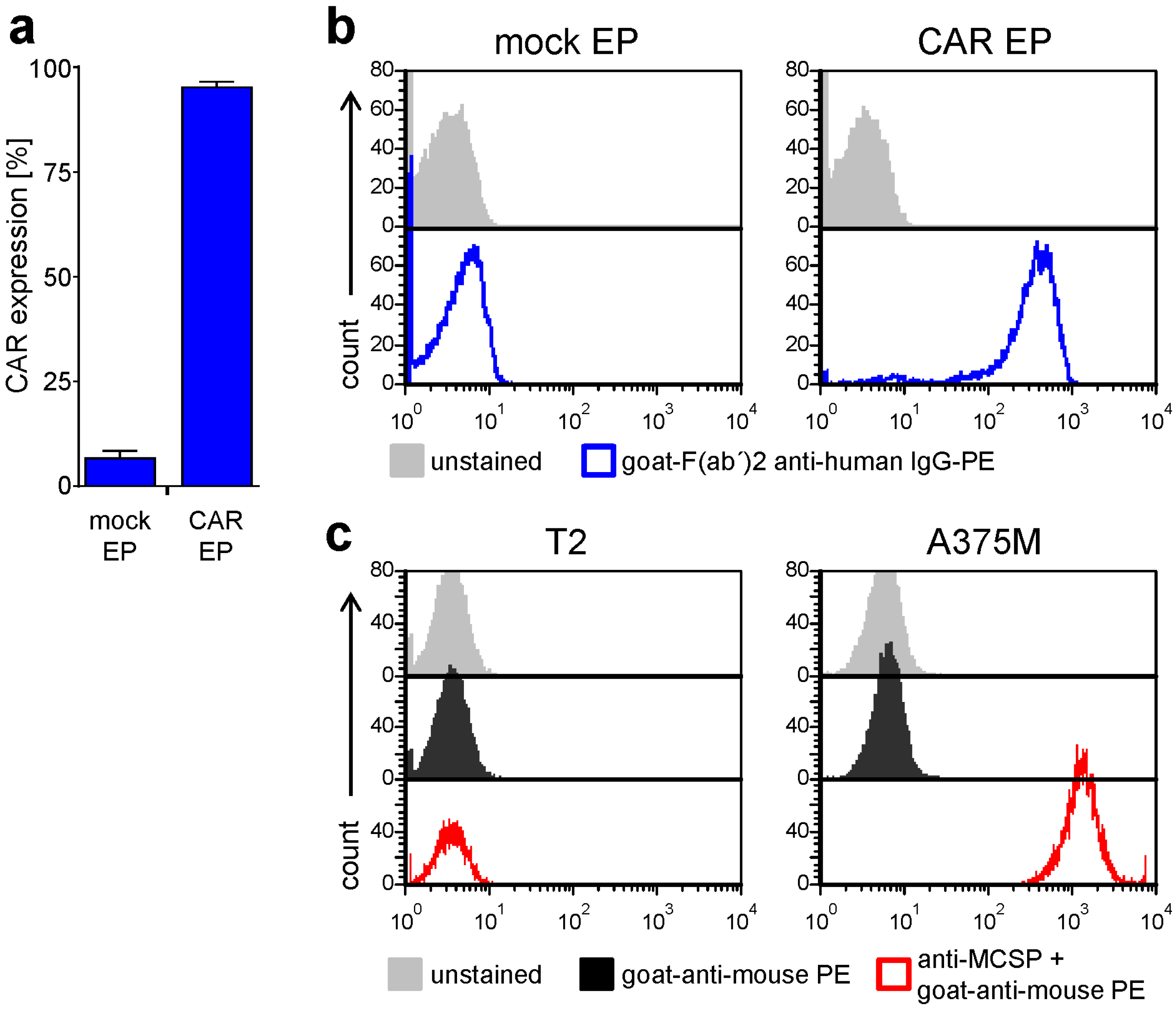
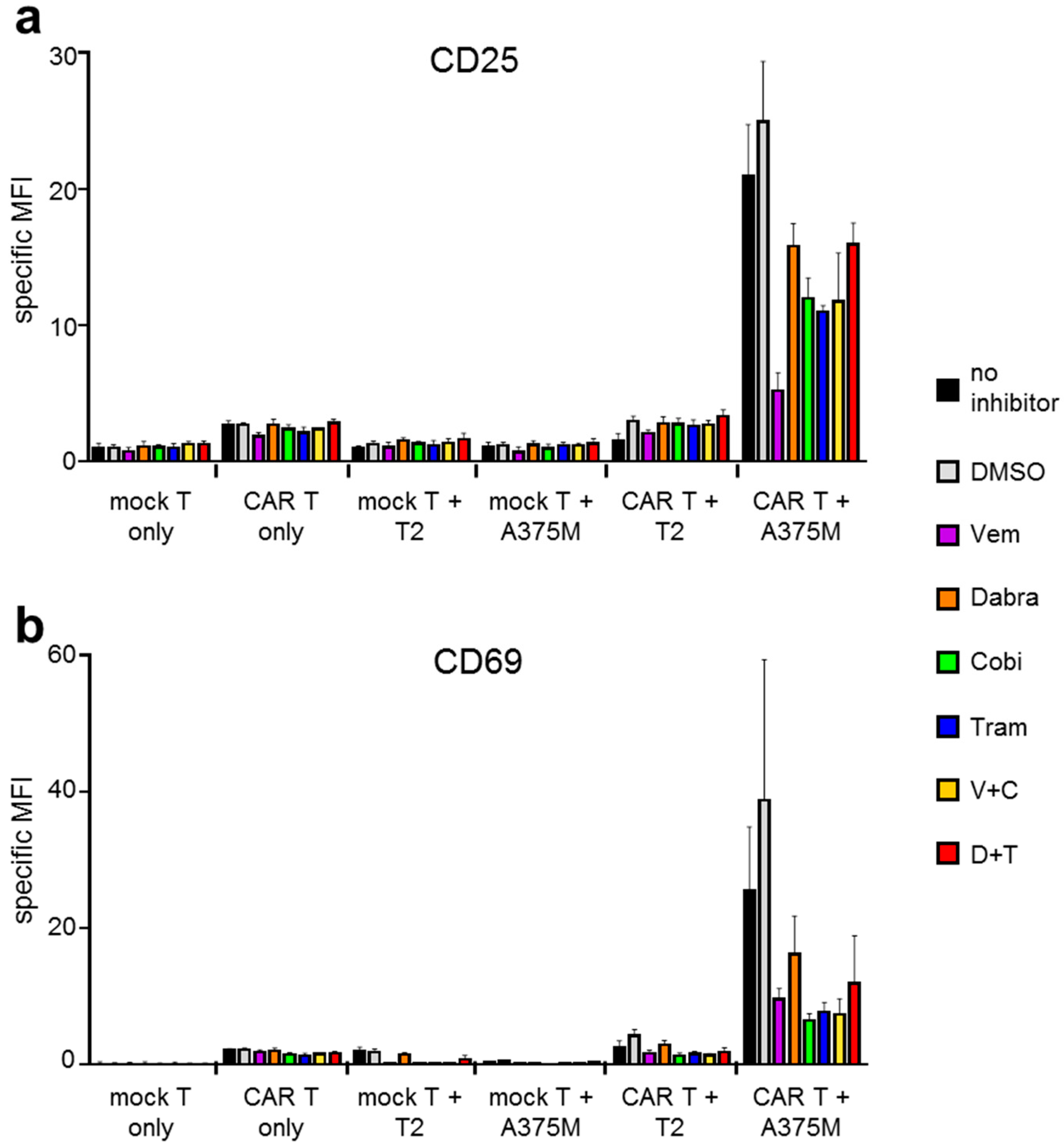
2.1. Antigen-Specific Activation of CAR-T Cells Is Differentially Affected by BRAFi/MEKi Treatment
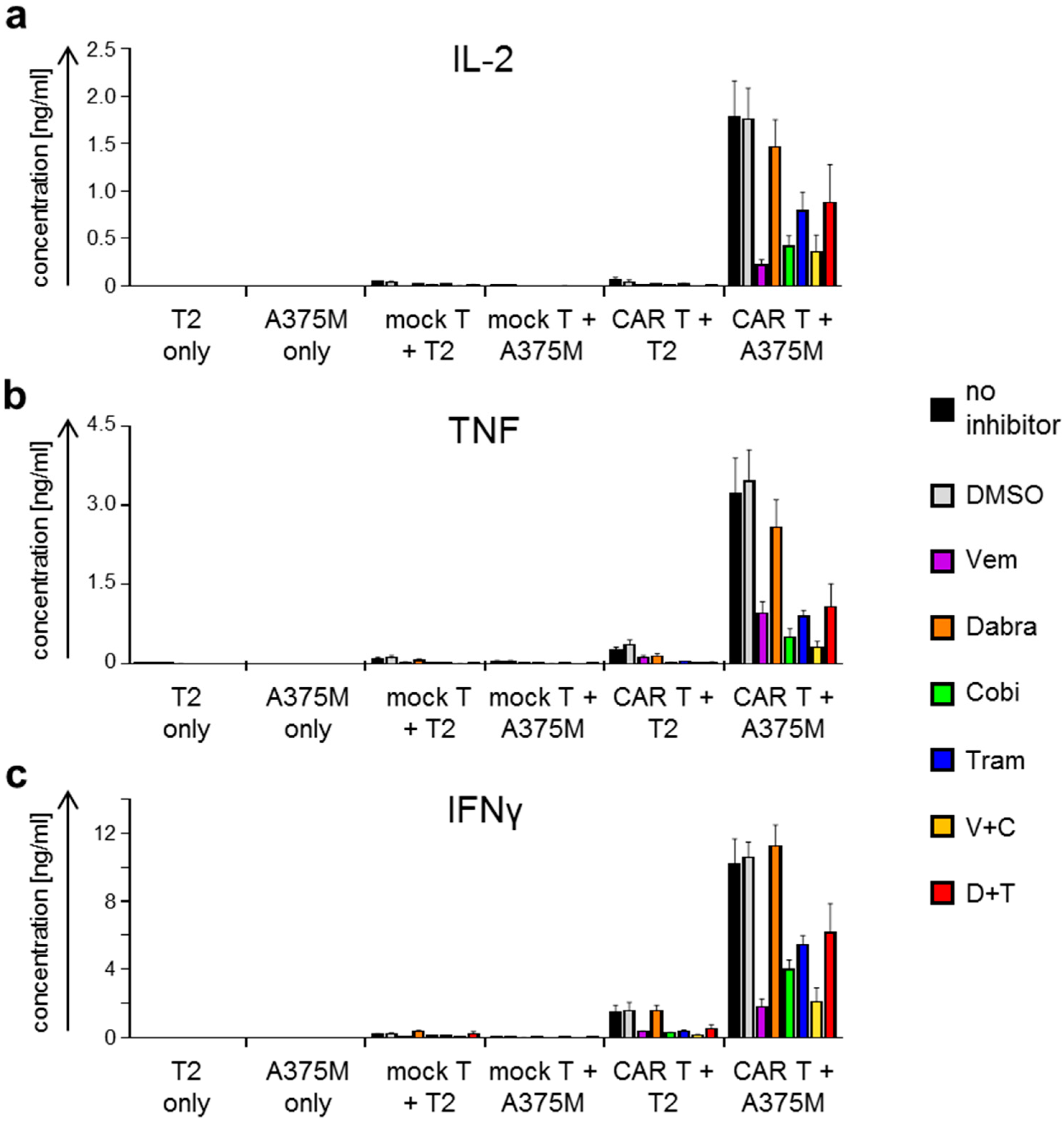
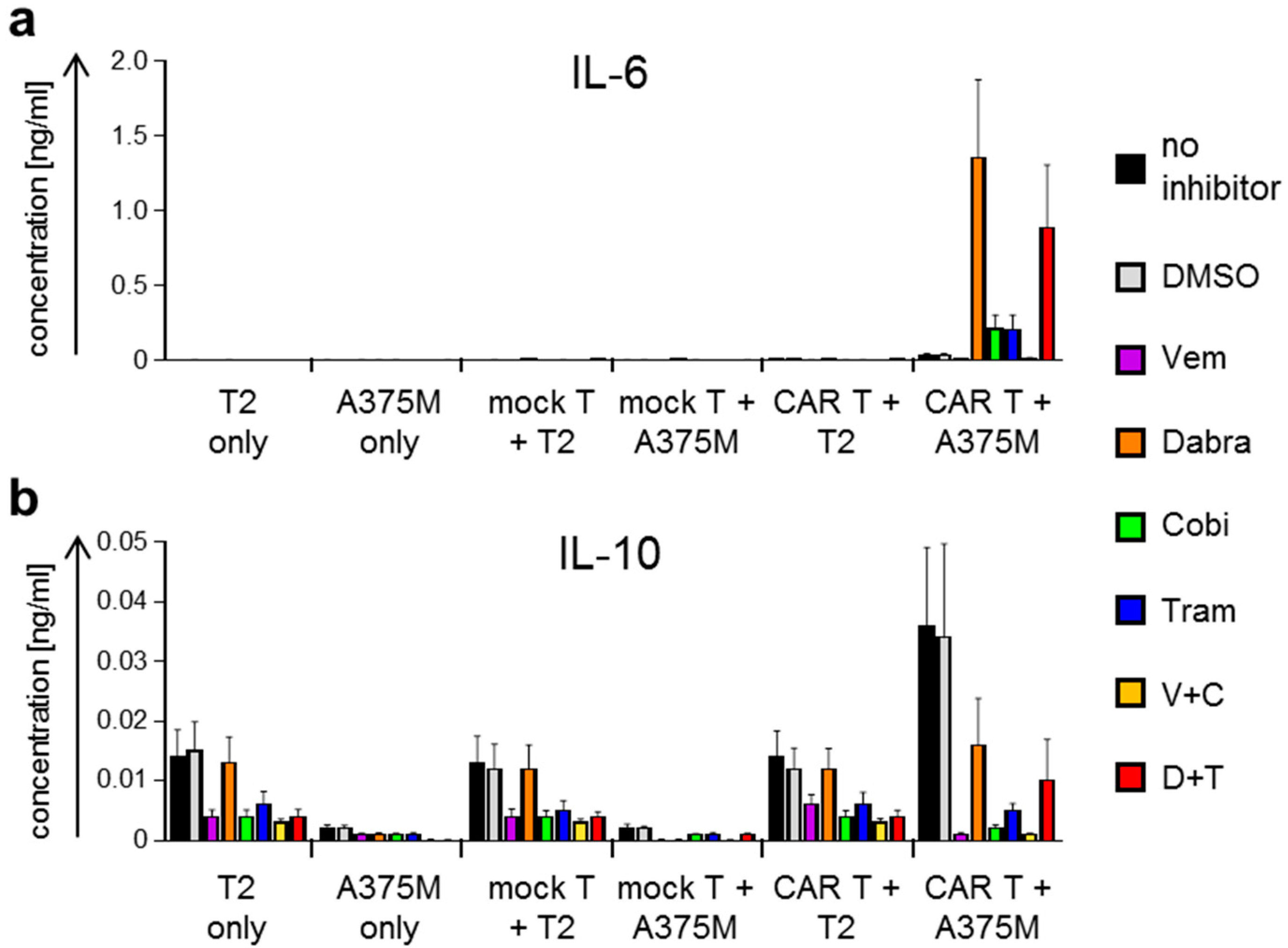
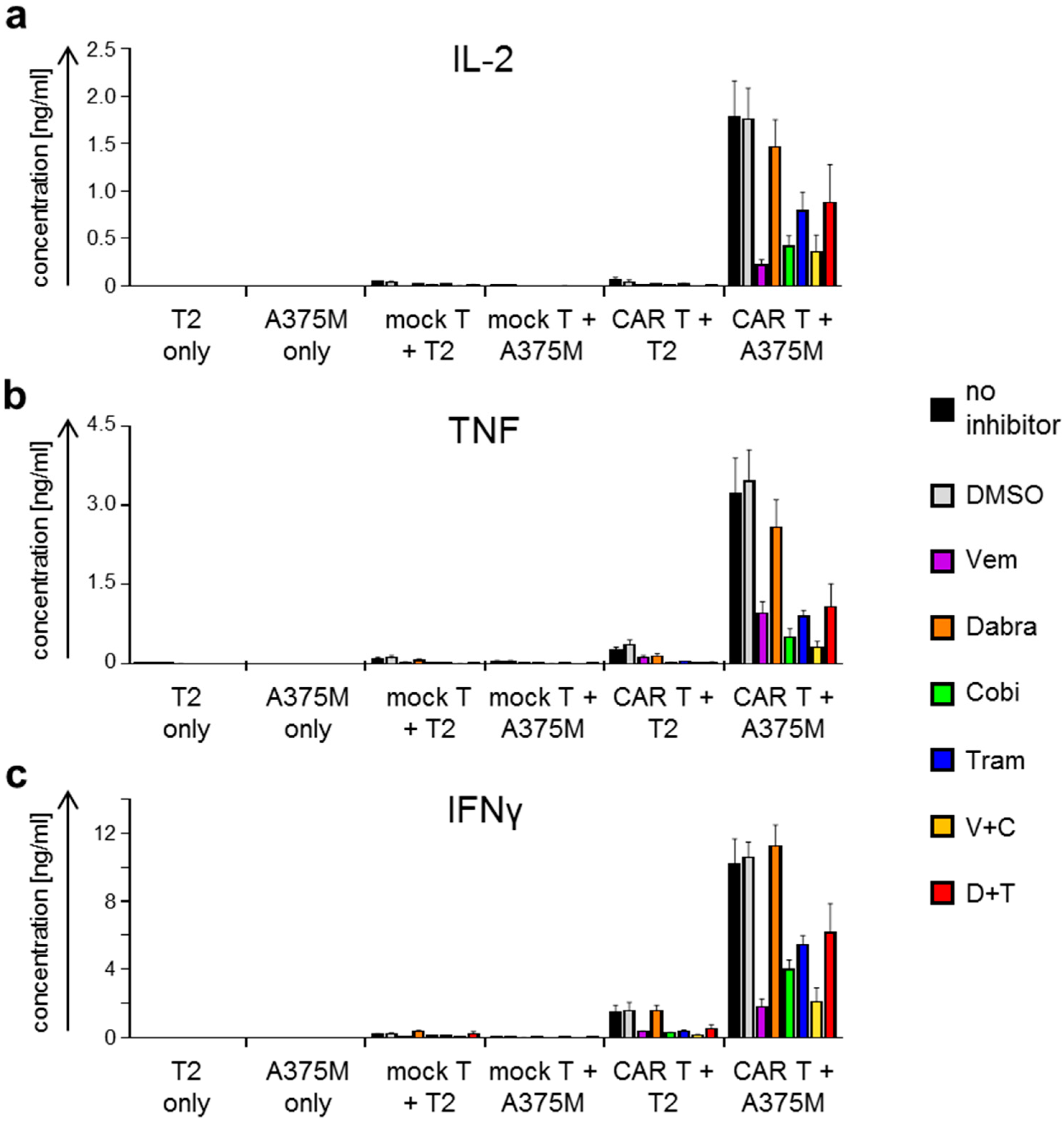
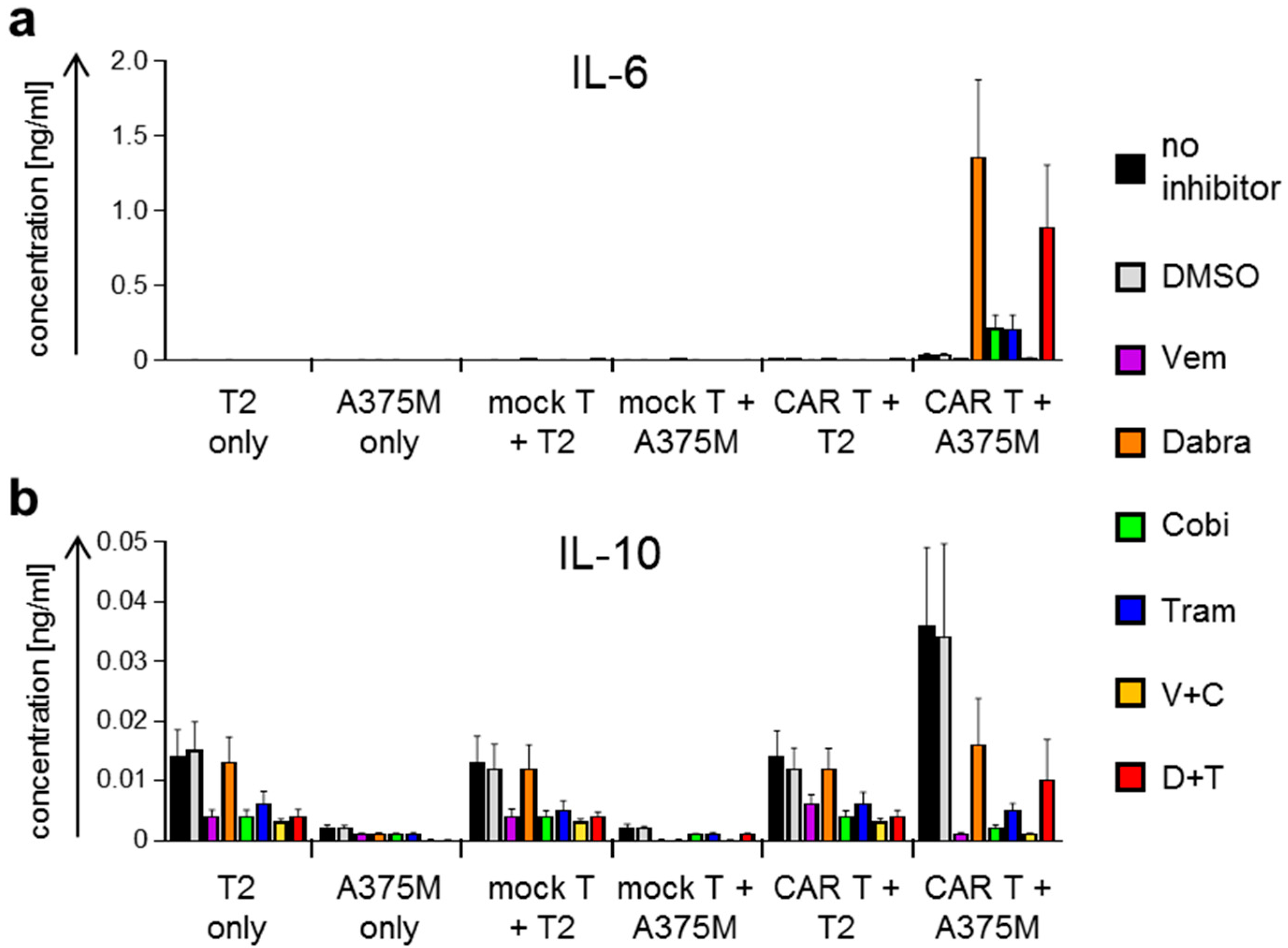
2.2. Antigen-Specific Cytokine Secretion by CAR-T Cells Is Differentially Affected by BRAFi/MEKi Treatment
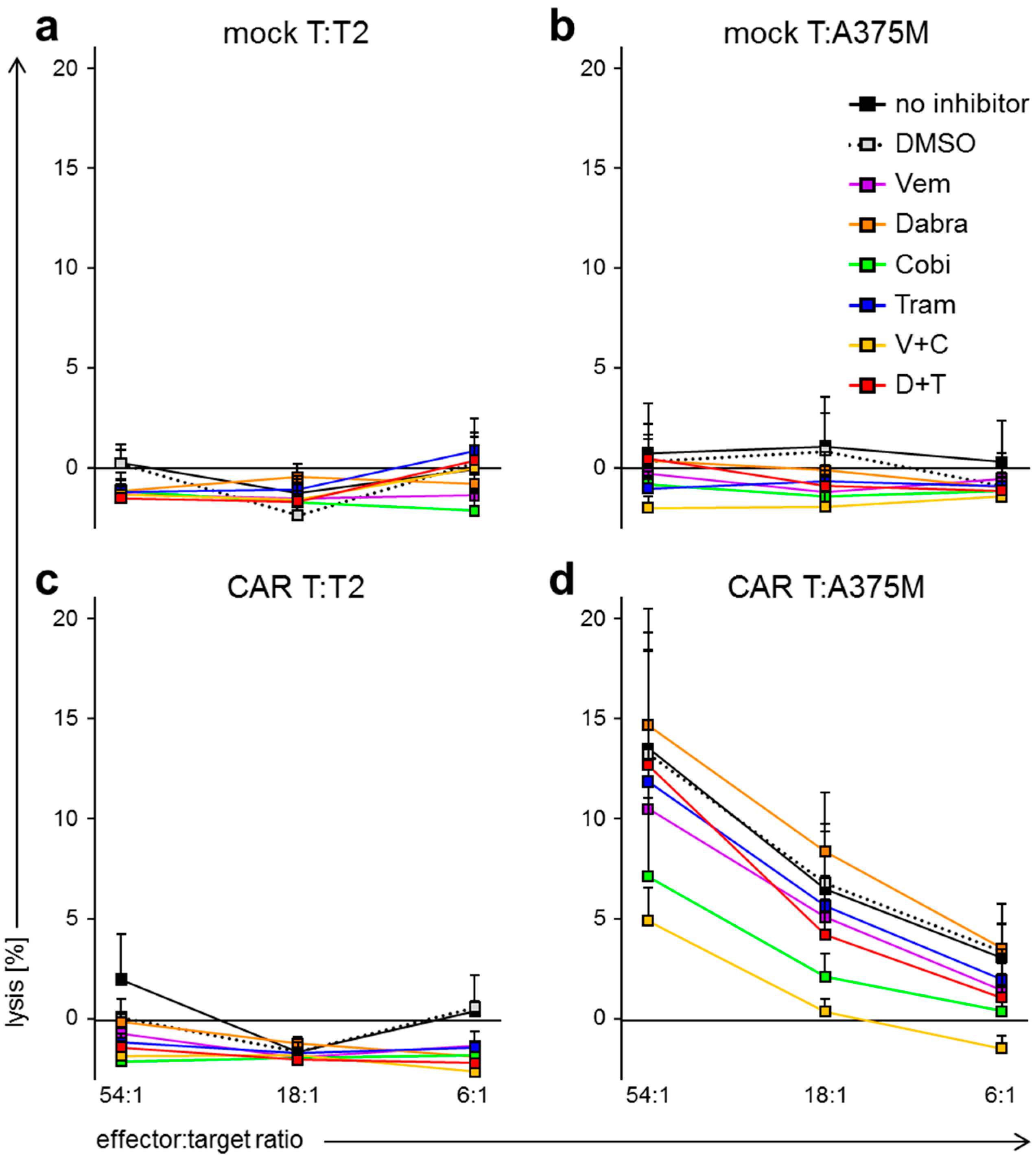
2.3. BRAFi/MEKi Treatment Differentially Affects Antigen-Specific Lytic Capacity of CAR-Transfected T Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture Media
4.2. BRAF and MEK Inhibitors
4.3. Cell Lines
4.4. T-Cell Isolation
4.5. RNA Transfection
4.6. Staining of CSPG4-Specific CAR on Transfected T Cells and CSPG4 on Target Cells
4.7. Staining of T-Cell Activation Markers
4.8. Cytokine Secretion Assay
4.9. Chromium Release Assay
4.10. Figure Preparation and Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CAR | chimeric antigen receptor |
| CSPG4 | chondroitin sulfate proteoglycan 4 |
| BRAF | v-Raf murine sarcoma viral oncogene homolog B |
| MEK | Mitogen-activated protein kinase kinase |
| Dabra | Dabrafenib |
| Tram | Trametinib |
| Vem | Vemurafenib |
| Cobi | Cobimetinib |
| ERK | extracellular signal-regulated kinases |
| MAPK | Mitogen-activated protein kinase |
| NRAS | Neuroblastoma RAS viral oncogene homolog |
| BRAFi | BRAF kinase inhibitor |
| MEKi | MEK inhibitor |
| FDA | Food and Drug Administration |
| PD1 | Programmed cell death protein 1 |
| MCSP | Melanoma-associated chondroitin sulfate proteoglycan |
| HMW-MAA | high molecular weight-melanoma associated antigen |
| RNA | Ribonucleic acid |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| IFN | Interferon |
| DMSO | Dimethyl sulfoxide |
| CRS | Cytokine release syndrome |
| MACS | Magnetic-activated cell sorting |
| PBMC | peripheral blood mononuclear cell |
References
- Ades, F.; Metzger-Filho, O. Targeting the Cellular Signaling: BRAF Inhibition and beyond for the Treatment of Metastatic Malignant Melanoma. Dermatol. Res. Pract. 2012, 2012, 259170. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Niault, T.S.; Baccarini, M. Targets of Raf in tumorigenesis. Carcinogenesis 2010, 31, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Ekedahl, H.; Cirenajwis, H.; Harbst, K.; Carneiro, A.; Nielsen, K.; Olsson, H.; Lundgren, L.; Ingvar, C.; Jonsson, G. The clinical significance of BRAF and NRAS mutations in a clinic-based metastatic melanoma cohort. Br. J. Dermatol. 2013, 169, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Lin, J.; Hocker, T.L.; Tsao, H. Genetics of melanoma tumorigenesis. Br. J. Dermatol. 2008, 158, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Jeong, J.H.; Asara, J.M.; Yuan, Y.Y.; Granter, S.R.; Chin, L.; Cantley, L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell 2009, 33, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Vence, L.; Falchook, G.; Radvanyi, L.G.; Liu, C.; Goodman, V.; Legos, J.J.; Blackman, S.; Scarmadio, A.; Kurzrock, R.; et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin. Cancer Res. 2012, 18, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Jarkowski, A., III; Khushalani, N.I. BRAF and beyond: Tailoring strategies for the individual melanoma patient. J. Carcinog. 2014, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Larkin, J. Vemurafenib: A new treatment for BRAF-V600 mutated advanced melanoma. Cancer Manag. Res. 2012, 4, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Cabanillas, M.E.; Lazar, A.J.; Williams, M.D.; Sanders, D.L.; Ilagan, J.L.; Nolop, K.; Lee, R.J.; Sherman, S.I. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid 2013, 23, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Boespflug, A.; Thomas, L. Cobimetinib and vemurafenib for the treatment of melanoma. Expert Opin. Pharmacother. 2016, 17, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Cooper, Z.A.; Reuben, A.; Austin-Breneman, J.; Wargo, J.A. Does It MEK a Difference? Understanding Immune Effects of Targeted Therapy. Clin. Cancer Res. 2015, 21, 3102–3104. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del, V.M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Rooprai, H.K.; Davies, D.; Levine, J.M.; Butt, A.M.; Pilkington, G.J. The NG2 chondroitin sulfate proteoglycan: Role in malignant progression of human brain tumours. Int. J. Dev. Neurosci. 1999, 17, 421–435. [Google Scholar] [CrossRef]
- Shoshan, Y.; Nishiyama, A.; Chang, A.; Mork, S.; Barnett, G.H.; Cowell, J.K.; Trapp, B.D.; Staugaitis, S.M. Expression of oligodendrocyte progenitor cell antigens by gliomas: Implications for the histogenesis of brain tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 10361–10366. [Google Scholar] [CrossRef] [PubMed]
- Malas, S.; Harrasser, M.; Lacy, K.E.; Karagiannis, S.N. Antibody therapies for melanoma: New and emerging opportunities to activate immunity (Review). Oncol. Rep. 2014, 32, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Fraser, C.K.; Dotti, G.; Yvon, E.S.; Brown, M.P. BRAF and MEK inhibition variably affect GD2-specific chimeric antigen receptor (CAR) T-cell function in vitro. J. Immunother. 2015, 38, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.; Gill, J.; Kamra, S.; Chen, L.; Ahuja, A. Indirect treatment comparison of dabrafenib plus trametinib versus vemurafenib plus cobimetinib in previously untreated metastatic melanoma patients. J. Hematol. Oncol. 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Adelmann, C.H.; Ching, G.; Du, L.; Saporito, R.C.; Bansal, V.; Pence, L.J.; Liang, R.; Lee, W.; Tsai, K.Y. Comparative profiles of BRAF inhibitors: The paradox index as a predictor of clinical toxicity. Oncotarget 2016, 7, 30453–30460. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Furqan, M.; Mukhi, N.; Ravella, P.; Liu, D. MEK and the inhibitors: From bench to bedside. J. Hematol. Oncol. 2013, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Jyothi, P.S.; Chandrasekar, B.; Nandi, D. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine 2010, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, H. Agents to reduce cytokine storm. F1000Research 2016, 5, 2909. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Kloss, C.C.; Gunset, G.; Sadelain, M. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS ONE 2013, 8, e61338. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Bouhassira, D.C.; Park, J.H.; Curran, K.J.; Smith, E.L.; Pegram, H.J.; Brentjens, R. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int. J. Hematol. 2014, 99, 361–371. [Google Scholar] [CrossRef] [PubMed]
- DeFrancesco, L. CAR-T cell therapy seeks strategies to harness cytokine storm. Nat. Biotechnol. 2014, 32, 604. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, J.H.; Gomez-Eerland, R.; van de Wiel, B.; Hulshoff, L.; van den Broek, D.; Bins, A.; Tan, H.L.; Harper, J.V.; Hassan, N.J.; Jakobsen, B.K.; et al. Case Report of a Fatal Serious Adverse Event upon Administration of T Cells Transduced with a MART-1-specific T-cell Receptor. Mol. Ther. 2015, 23, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. 1997, 6, 929–955. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Qureshi, A.A.; Niu, T.; Kraft, P.; Guo, Q.; Hunter, D.J.; Han, J. Interleukin and interleukin receptor gene polymorphisms and susceptibility to melanoma. Melanoma Res. 2008, 18, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015, 5, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Butler, M.; Lutzky, J.; Lawrence, D.P.; Robert, C.; Miller, W.; Linette, G.P.; Ascierto, P.A.; Kuzel, T.; Algazi, A.P.; et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J. Clin. Oncol. 2015, 33, 3003. [Google Scholar]
- Kozlowski, J.M.; Hart, I.R.; Fidler, I.J.; Hanna, N. A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J. Natl. Cancer Inst. 1984, 72, 913–917. [Google Scholar] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. CD28 cosignalling does not affect the activation threshold in a chimeric antigen receptor-redirected T-cell attack. Gene Ther. 2011, 18, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef] [PubMed]
- Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. Electroporation of mRNA as Universal Technology Platform to Transfect a Variety of Primary Cells with Antigens and Functional Proteins. Methods Mol. Biol. 2017, 1499, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dorrie, J.; Muller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kampgen, E.; Schuler, G. A new way to generate cytolytic tumor-specific T cells: Electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase Inhibitor | Target | Final Concentration |
|---|---|---|
| Vemurafenib (Vem, V) 1 | BRAFV600E | 60 µM |
| Dabrafenib (Dabra, D) 2 | BRAFV600 | 1 µM |
| Trametinib (Tram, T) 3 | MEK1/2 | 30 nM |
| Cobimetinib (Cobi, C) 4 | MEK1/2 | 0.5 µM |
| V + C | BRAFV600E + MEK1/2 | 60 µM + 0.5 µM |
| D + T | BRAFV600 + MEK1/2 | 1 µM + 30 nM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dörrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int. J. Mol. Sci. 2018, 19, 289. https://doi.org/10.3390/ijms19010289
Dörrie J, Babalija L, Hoyer S, Gerer KF, Schuler G, Heinzerling L, Schaft N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. International Journal of Molecular Sciences. 2018; 19(1):289. https://doi.org/10.3390/ijms19010289
Chicago/Turabian StyleDörrie, Jan, Lek Babalija, Stefanie Hoyer, Kerstin F. Gerer, Gerold Schuler, Lucie Heinzerling, and Niels Schaft. 2018. "BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy" International Journal of Molecular Sciences 19, no. 1: 289. https://doi.org/10.3390/ijms19010289
APA StyleDörrie, J., Babalija, L., Hoyer, S., Gerer, K. F., Schuler, G., Heinzerling, L., & Schaft, N. (2018). BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. International Journal of Molecular Sciences, 19(1), 289. https://doi.org/10.3390/ijms19010289